Long Non-Coding RNA Expression Levels Modulate Cell-Type-Specific Splicing Patterns by Altering Their Interaction Landscape with RNA-Binding Proteins



Abstract

1. Introduction
2. Materials and Methods
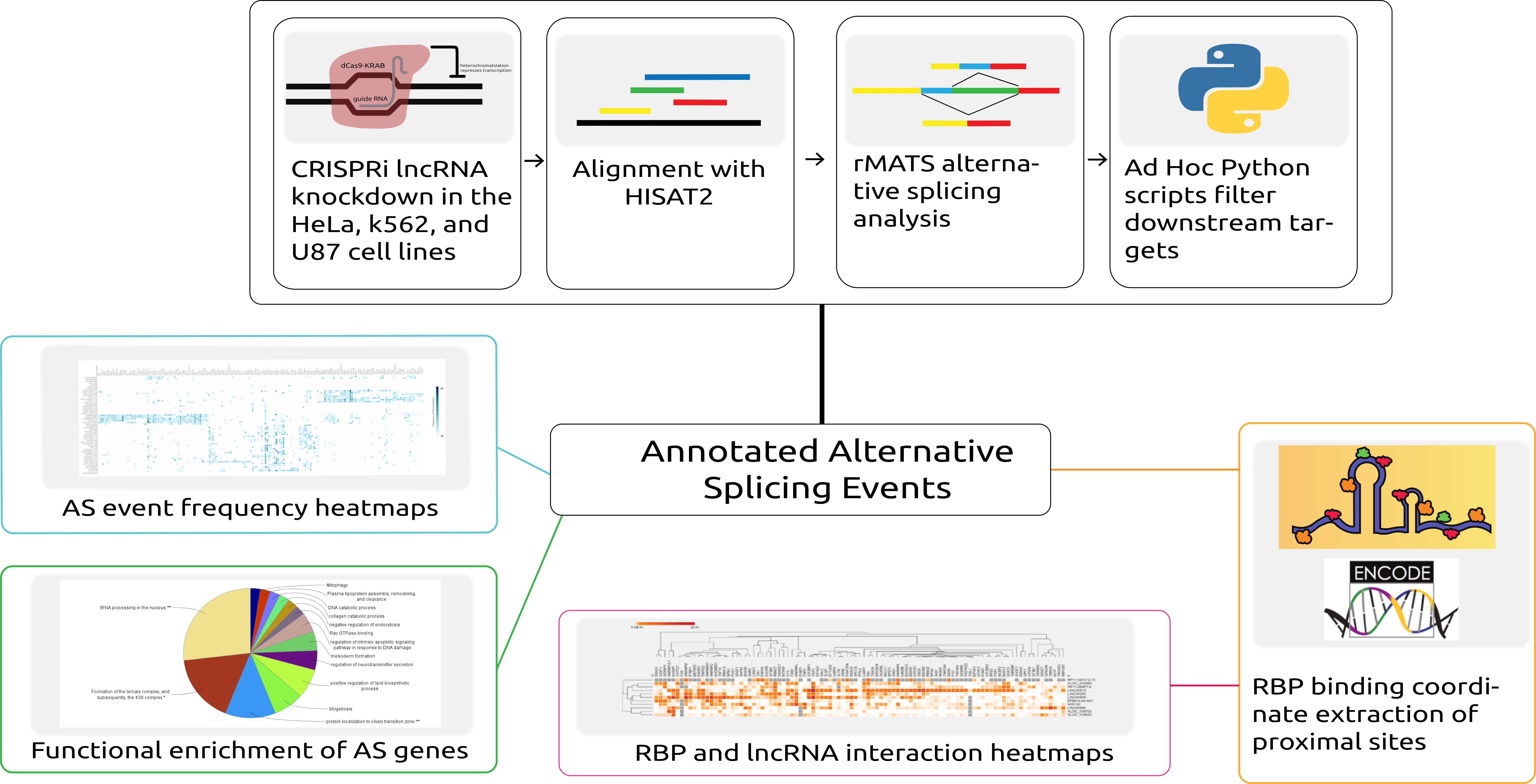
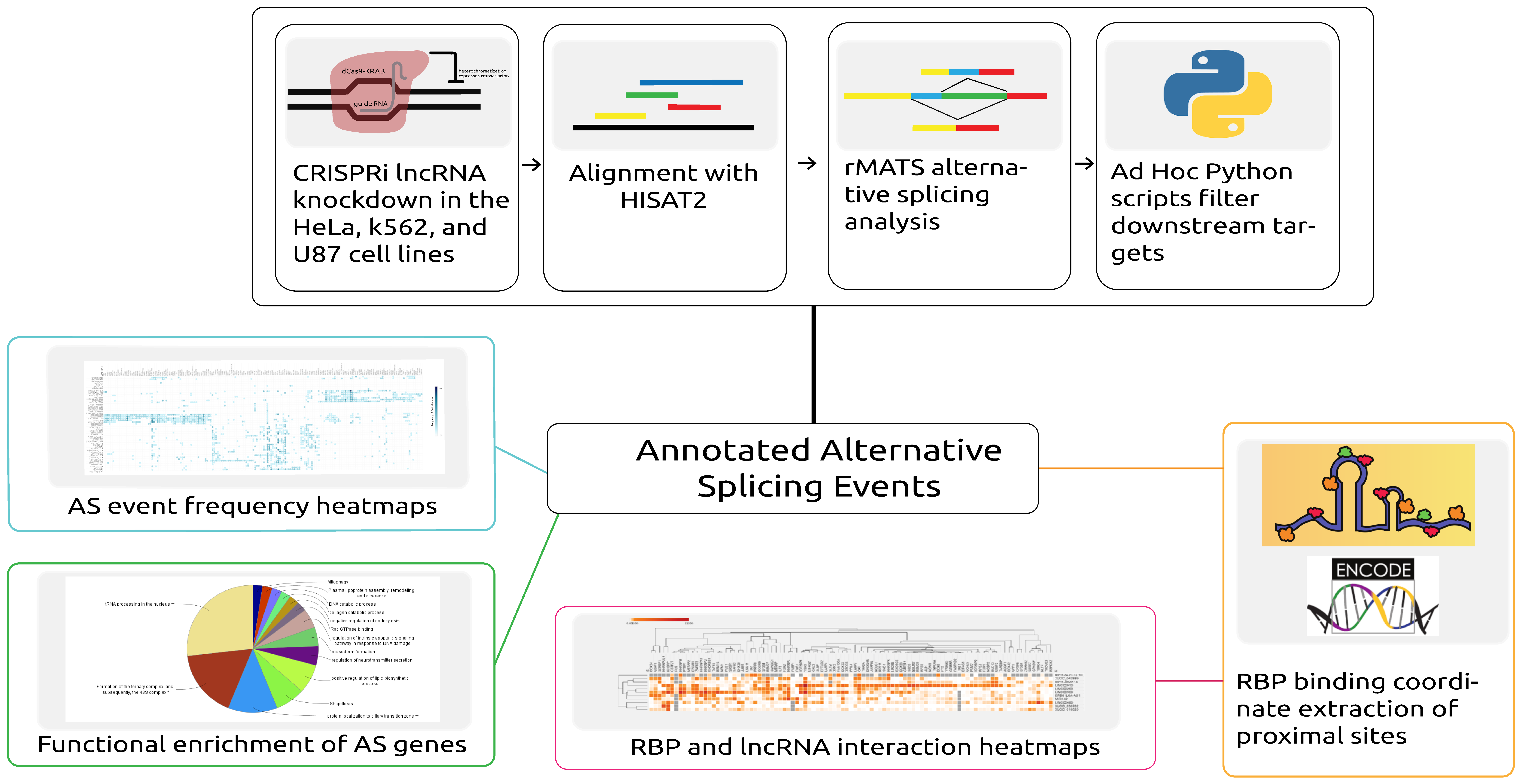
2.1. Sequence Alignment of RNA-Seq Reads Using HISAT
2.2. Identifying Differentially Spliced Events Using Replicate Multivariate Analysis of Transcript Splicing (rMATS)
2.3. Visualizing Incidence of Differentially Spliced Genes in lncRNA Knocked Down Cell Lines
2.4. Functional Enrichment Analysis of Alternatively Spliced Genes
2.5. Extraction of RBP Binding Profiles across AS Events
2.6. Enrichment of RBP Binding over Alternative Splicing Events
2.7. Correlation Analysis of lncRNA Specific RBP Binding Activity and AS Events
3. Results
3.1. Framework for Studying Alternative Splicing Outcomes in lncRNA Knockdown RNA-Sequencing Experiments
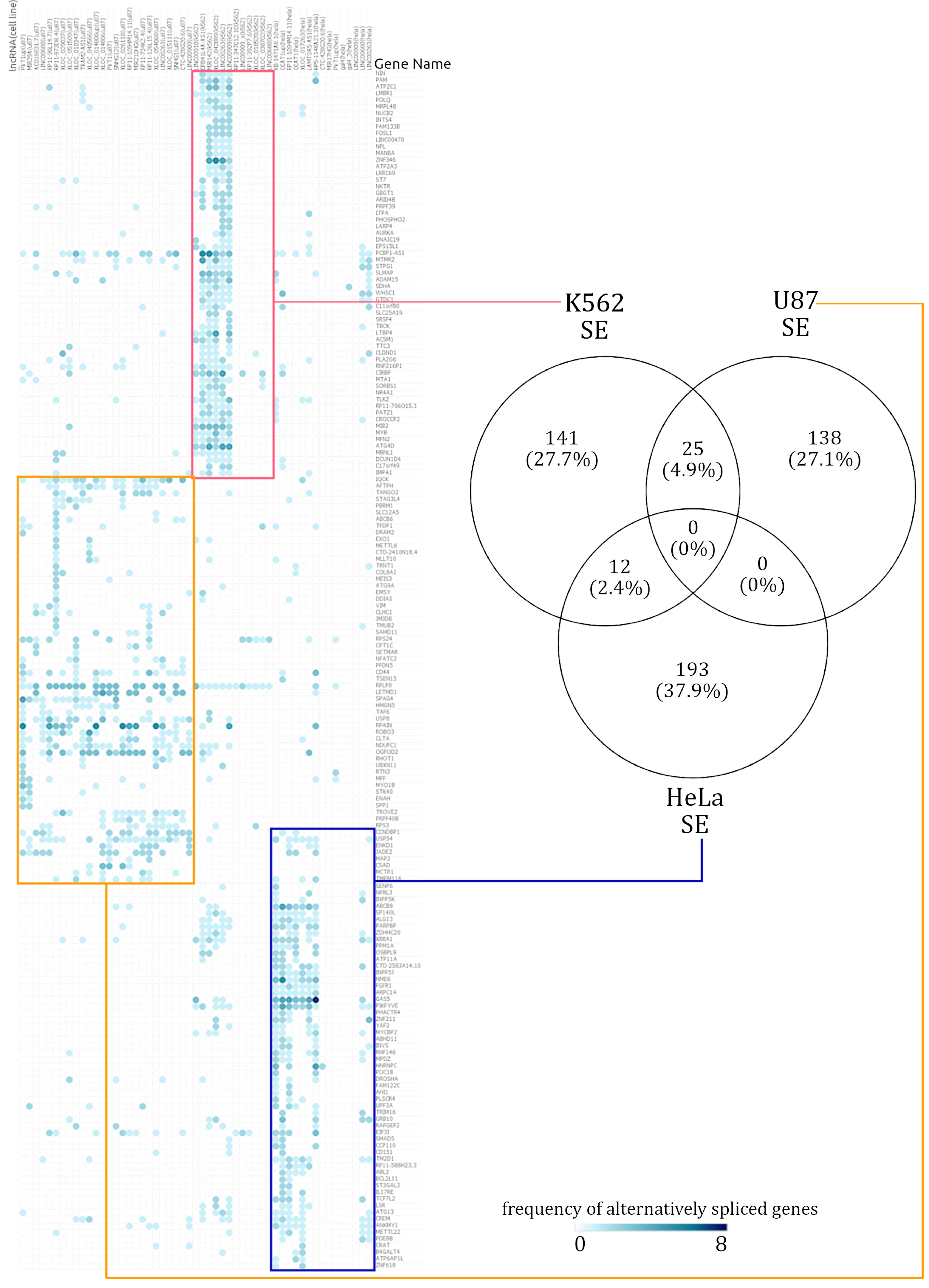
3.2. Skipped Exon Events Followed by Retained Intron Events Are the Most Prominent Alternative Splicing Events Occurring Due to the Loss of lncRNAs across Multiple Human Cell Lines
3.3. Hierarchically Clustered Heatmaps of AS Event Frequency in lncRNA Knockdowns Reveal Cell-Type-Specific Alternative Splicing Signatures
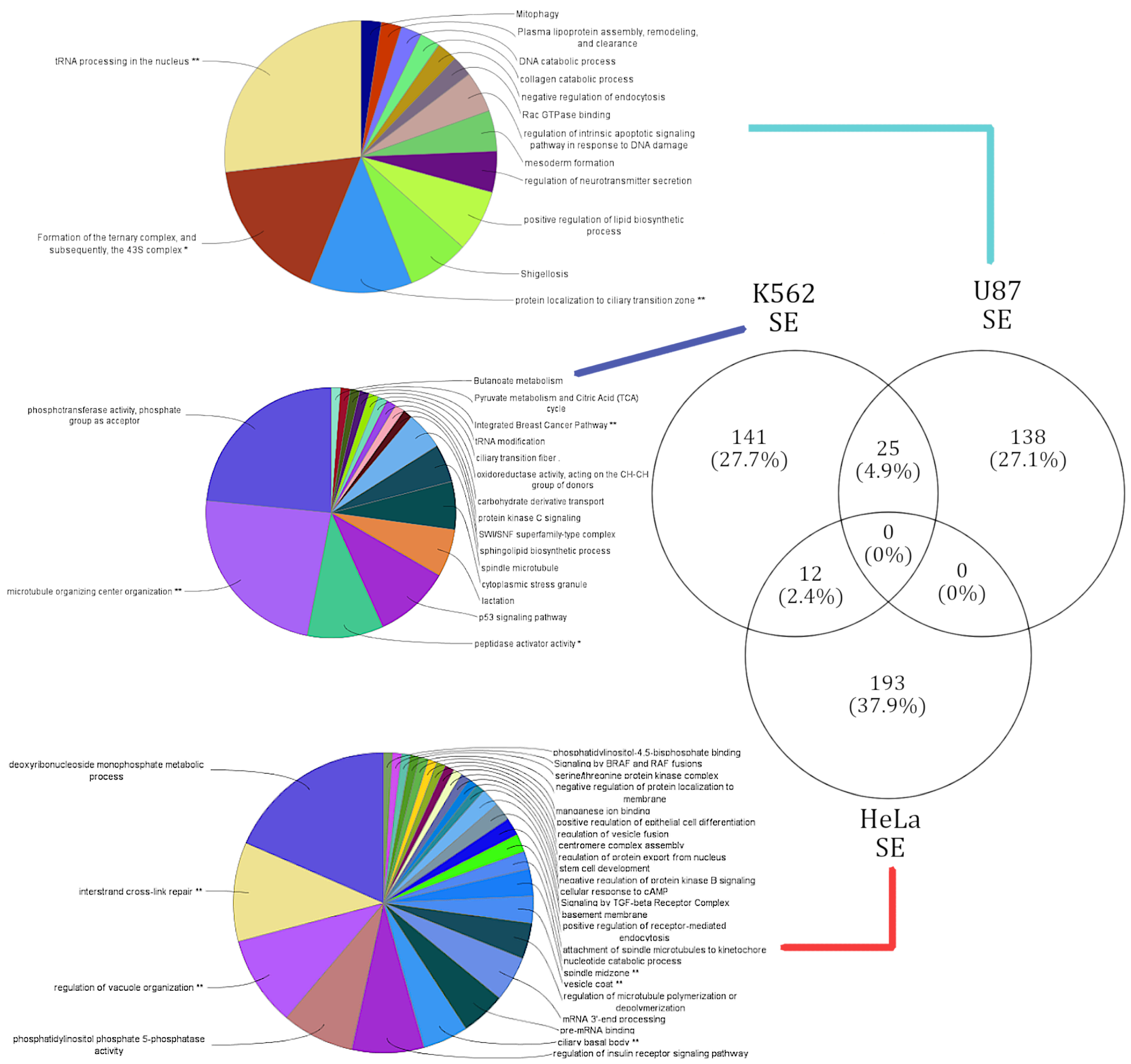
3.4. lncRNA Knockdowns Not only Enriched for Cell-Type-Specific Functions but Occasionally Favor Similar Functions via Varied Alternatively Spliced Gene Sets
3.4.1. Genes Alternatively Spliced via SE Events in U87, HeLa, and K562 Reveal Cell-Type-Specific Functional Enrichment
3.4.2. Genes Alternatively Spliced via RI Events in HeLa and K562 Reveal Cell-Type-Specific Functional Enrichment
3.5. Analysis of lncRNA AS Events Proximal to RBP Binding Sites Reveals Cell-Type-Specific Interactions and Supports a lncRNA–RBP Sponge Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Leone, S.; Santoro, R. Challenges in the analysis of long noncoding RNA functionality. FEBS Lett. 2016, 590, 2342–2353. [Google Scholar] [CrossRef] [PubMed]
- Shukla, C.J.; McCorkindale, A.L.; Gerhardinger, C.; Korthauer, K.D.; Cabili, M.N.; Shechner, D.M.; Irizarry, R.A.; Maass, P.G.; Rinn, J.L. High-throughput identification of RNA nuclear enrichment sequences. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Horlbeck, M.A.; Cho, S.W.; Birk, H.S.; Malatesta, M.; He, D.; Attenello, F.J.; Villalta, J.E.; Cho, M.Y.; Chen, Y.; et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017, 355. [Google Scholar] [CrossRef] [PubMed]
- Zare, K.; Shademan, M.; Ghahramani Seno, M.M.; Dehghani, H. CRISPR/Cas9 Knockout Strategies to Ablate CCAT1 lncRNA Gene in Cancer Cells. Biol. Proced. Online 2018, 20, 21. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Shi, A.; Long, Z.; Xu, L.; Liao, G.; Deng, C.; Yan, M.; Xie, A.; Luo, T.; Huang, J.; et al. Capturing functional long non-coding RNAs through integrating large-scale causal relations from gene perturbation experiments. EBioMedicine 2018, 35, 369–380. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Yu, D.; Zhang, C.; Gui, J. RNA-binding protein HuR promotes bladder cancer progression by competitively binding to the long noncoding HOTAIR with miR-1. OncoTargets Ther. 2017, 10, 2609–2619. [Google Scholar] [CrossRef]
- Hirose, T.; Virnicchi, G.; Tanigawa, A.; Naganuma, T.; Li, R.; Kimura, H.; Yokoi, T.; Nakagawa, S.; Benard, M.; Fox, A.H.; et al. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol. Biol. Cell 2014, 25, 169–183. [Google Scholar] [CrossRef]
- Romero-Barrios, N.; Legascue, M.F.; Benhamed, M.; Ariel, F.; Crespi, M. Splicing regulation by long noncoding RNAs. Nucleic Acids Res. 2018, 46, 2169–2184. [Google Scholar] [CrossRef]
- Bazin, J.; Romero, N.; Rigo, R.; Charon, C.; Blein, T.; Ariel, F.; Crespi, M. Nuclear Speckle RNA Binding Proteins Remodel Alternative Splicing and the Non-coding Arabidopsis Transcriptome to Regulate a Cross-Talk Between Auxin and Immune Responses. Front. Plant. Sci. 2018, 9, 1209. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Jensen, K.; Mele, A.; Darnell, R.B. CLIP: A method for identifying protein-RNA interaction sites in living cells. Methods 2005, 37, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Budak, G.; Srivastava, R.; Janga, S.C. Seten: A tool for systematic identification and comparison of processes, phenotypes, and diseases associated with RNA-binding proteins from condition-specific CLIP-seq profiles. RNA 2017, 23, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xu, G.; Yang, Y.T.; Xu, Z.; Chen, X.; Shi, B.; Xie, D.; Lu, Z.J.; Wang, P. POSTAR2: Deciphering the post-transcriptional regulatory logics. Nucleic Acids Res. 2019, 47, D203–D211. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Neelamraju, Y.; Gonzalez-Perez, A.; Bhat-Nakshatri, P.; Nakshatri, H.; Janga, S.C. Mutational landscape of RNA-binding proteins in human cancers. RNA Biol. 2018, 15, 115–129. [Google Scholar] [CrossRef]
- Van Nostrand, E.L.; Pratt, G.A.; Shishkin, A.A.; Gelboin-Burkhart, C.; Fang, M.Y.; Sundararaman, B.; Blue, S.M.; Nguyen, T.B.; Surka, C.; Elkins, K.; et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 2016, 13, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M.; International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Sbugroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.; Akanni, W.; Amode, M.R.; Barrell, D.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; Fitzgerald, S.; Gil, L.; et al. Ensembl 2016. Nucleic Acids Res. 2016, 44, D710–D716. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute. Morpheus. Available online: https://software.broadinstitute.org/morpheus (accessed on 3 April 2018).
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 28 June 2018).
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Huppertz, I.; Attig, J.; D’Ambrogio, A.; Easton, L.E.; Sibley, C.R.; Sugimoto, Y.; Tajnik, M.; Konig, J.; Ule, J. iCLIP: Protein-RNA interactions at nucleotide resolution. Methods 2014, 65, 274–287. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Naidu, M.D.; Mason, J.M.; Pica, R.V.; Fung, H.; Pena, L.A. Radiation resistance in glioma cells determined by DNA damage repair activity of Ape1/Ref-1. J. Radiat. Res. 2010, 51, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Van der Ven, L.T.; Roholl, P.J.; Gloudemans, T.; Van Buul-Offers, S.C.; Welters, M.J.; Bladergroen, B.A.; Faber, J.A.; Sussenbach, J.S.; Den Otter, W. Expression of insulin-like growth factors (IGFs), their receptors and IGF binding protein-3 in normal, benign and malignant smooth muscle tissues. Br. J. Cancer 1997, 75, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Kim, H.; Chung, K.H.; Hong, N.H.; Shin, B.; Park, W.J.; Jun, Y.; Rhee, S.; Song, W.K. SPIN90 knockdown attenuates the formation and movement of endosomal vesicles in the early stages of epidermal growth factor receptor endocytosis. PLoS ONE 2013, 8, e82610. [Google Scholar] [CrossRef] [PubMed]
- Farrar, J.E.; Quarello, P.; Fisher, R.; O’Brien, K.A.; Aspesi, A.; Parrella, S.; Henson, A.L.; Seidel, N.E.; Atsidaftos, E.; Prakash, S.; et al. Exploiting pre-rRNA processing in Diamond Blackfan anemia gene discovery and diagnosis. Am. J. Hematol. 2014, 89, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Novikova, I.V.; Hennelly, S.P.; Tung, C.S.; Sanbonmatsu, K.Y. Rise of the RNA machines: Exploring the structure of long non-coding RNAs. J. Mol. Biol. 2013, 425, 3731–3746. [Google Scholar] [CrossRef] [PubMed]
- Bond, C.S.; Fox, A.H. Paraspeckles: Nuclear bodies built on long noncoding RNA. J. Cell Biol. 2009, 186, 637–644. [Google Scholar] [CrossRef]
- Lin, Y.; Schmidt, B.F.; Bruchez, M.P.; McManus, C.J. Structural analyses of NEAT1 lncRNAs suggest long-range RNA interactions that may contribute to paraspeckle architecture. Nucleic Acids Res. 2018, 46, 3742–3752. [Google Scholar] [CrossRef]
- Rivas, E.; Clements, J.; Eddy, S.R. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat. Methods 2017, 14, 45–48. [Google Scholar] [CrossRef]
- Chan, J.J.; Tay, Y. Noncoding RNA: RNA Regulatory Networks in Cancer. Int. J. Mol. Sci. 2018, 19, 1310. [Google Scholar] [CrossRef]
- Du, Z.; Sun, T.; Hacisuleyman, E.; Fei, T.; Wang, X.; Brown, M.; Rinn, J.L.; Lee, M.G.; Chen, Y.; Kantoff, P.W.; et al. Integrative analyses reveal a long noncoding RNA-mediated sponge regulatory network in prostate cancer. Nat. Commun. 2016, 7, 10982. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.; Zhao, S.; Jiang, Z.; Wang, S.; Sun, P.; Quan, J.; Yan, D.; Wang, X. MALAT1 sponges miR-106b-5p to promote the invasion and metastasis of colorectal cancer via SLAIN2 enhanced microtubules mobility. EBioMedicine 2019, 41, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Bai, Y.; Yang, X.; Lin, J.; Yang, X.; Wang, D.; He, L.; Zheng, Y.; Zhao, H. Construction and comprehensive analysis of a ceRNA network to reveal potential prognostic biomarkers for hepatocellular carcinoma. Cancer Cell Int. 2019, 19, 90. [Google Scholar] [CrossRef] [PubMed]
- Du, W.W.; Zhang, C.; Yang, W.; Yong, T.; Awan, F.M.; Yang, B.B. Identifying and Characterizing circRNA-Protein Interaction. Theranostics 2017, 7, 4183–4191. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Abdelmohsen, K.; Yang, X.; De, S.; Grammatikakis, I.; Noh, J.H.; Gorospe, M. LncRNA OIP5-AS1/cyrano sponges RNA-binding protein HuR. Nucleic Acids Res. 2016, 44, 2378–2392. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, L.; Ding, Y.; Lu, X.; Zhang, G.; Yang, J.; Zheng, H.; Wang, H.; Jiang, Y.; Xu, L. LncRNA Structural Characteristics in Epigenetic Regulation. Int. J. Mol. Sci. 2017, 18, 2659. [Google Scholar] [CrossRef]
- Srivastava, R.; Budak, G.; Dash, S.; Lachke, S.A.; Janga, S.C. Transcriptome analysis of developing lens reveals abundance of novel transcripts and extensive splicing alterations. Sci. Rep. 2017, 7, 11572. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| lncRNA Symbol | ENSG ID | Cell line/s Perturbed In | Length (bp) | Strand | Gene Type |
|---|---|---|---|---|---|
| AC016831.7 | ENSG00000285106.2 | U87 | 318,897 | + | LncRNA |
| CCAT1 | ENSG00000247844.1 | HeLa | 11,222 | - | LincRNA |
| CTC-428G20.6 | ENSG00000271797.1 | U87, HeLa | 943 | + | Antisense |
| EPB41L4A-AS1 | ENSG00000224032.7 | K562 | 4292 | + | LncRNA |
| GAL4 | ENSG00000282992.2 | HeLa | 11,263 | - | Protein_coding |
| KB-1471A8.1 | ENSG00000245330.1 | HeLa | 7069 | - | LincRNA |
| LAMTOR5-AS1 | ENSG00000224699.9 | HeLa | 96,701 | + | LncRNA |
| LINC00263 | ENSG00000235823.3 | K562, U87, HeLa | 80,944 | + | LncRNA |
| LINC00680 | ENSG00000215190.9 | K562, U87, HeLa | 15,427 | - | Transcribed_unprocessed_pseudogene |
| LINC00909 | ENSG00000264247.2 | K562, U87, HeLa | 8568 | - | LncRNA |
| LINC00910 | ENSG00000188825.14 | K562 | 50,458 | - | LncRNA |
| MIR142 | ENSG00000284353.1 | K562 | 86 | - | miRNA |
| MIR17HG | ENSG00000215417.13 | HeLa | 6759 | + | LncRNA |
| MIR210HG | ENSG00000247095.3 | U87 | 2797 | - | LncRNA |
| MIR29A | ENSG00000284032.1 | U87 | 63 | - | miRNA |
| PVT1 | ENSG00000249859.11 | U87, HeLa | 392,575 | + | LncRNA |
| RP11-1094M14.11 | ENSG00000267321.3 | U87, HeLa | 6824 | + | LncRNA |
| RP11-126L15.4 | ENSG00000236305.1 | U87 | 15,302 | - | LncRNA |
| RP11-347C12.10 | ENSG00000260219.1 | K562 | 1086 | + | LincRNA |
| RP11-392P7.6 | ENSG00000247498.10 | K562 | 56,919 | + | LncRNA |
| RP11-734K2.4 | ENSG00000270344.3 | U87 | 23,411 | + | LncRNA |
| RP11-96L14.7 | ENSG00000236782.1 | U87 | 1874 | - | Antisense |
| RP11-973D8.4 | ENSG00000258554.1 | U87 | 636 | - | Antisense |
| RP5-1148A21.3 | ENSG00000266680.1 | HeLa | 1403 | - | Antisense |
| SNHG1 | ENSG00000255717.7 | U87 | 3969 | - | LncRNA |
| SNHG12 | ENSG00000197989.14 | U87 | 4594 | - | LncRNA |
| TRAM2-AS1 | ENSG00000225791.7 | U87 | 66,271 | + | LncRNA |
| XLOC_010347 | ENSG00000254430.1 | U87 | 699 | - | Unprocessed pseudogene |
| XLOC_014806 | ENSG00000211813.2 | U87 | 607 | + | TR V gene |
| XLOC_015111 | ENSG00000258687.1 | U87 | 17,537 | - | Antisense |
| XLOC_017263 | ENSG00000259656.1 | HeLa | 1473 | + | LincRNA |
| XLOC_018520 | ENSG00000243007.1 | K562 | 495 | - | Processed pseudogene |
| XLOC_026118 | ENSG00000272396.1 | U87 | 8285 | - | Antisense |
| XLOC_029037 | ENSG00000200718.1 | U87 | 319 | + | LncRNA |
| XLOC_038702 | ENSG00000261519.2 | K562 | 1909 | + | LincRNA |
| XLOC_040566 | ENSG00000109686.18 | U87 | 222,881 | - | Protein_coding |
| XLOC_042889 | ENSG00000271862.1 | K562 | 1588 | - | LincRNA |
| XLOC_051509 | ENSG00000253372.5 | U87 | 8771 | + | LncRNA |
| XLOC_054068 | ENSG00000268674.1 | U87 | 509 | + | Protein_coding |
| Genetic Regions | Bound | Unbound |
|---|---|---|
| Regions where an RBP may bind across genes in a cell line | X | Y |
| All proximal splicing regions generated from a lncRNA knockdown in a cell | A | B |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porto, F.W.; Daulatabad, S.V.; Janga, S.C. Long Non-Coding RNA Expression Levels Modulate Cell-Type-Specific Splicing Patterns by Altering Their Interaction Landscape with RNA-Binding Proteins. Genes 2019, 10, 593. https://doi.org/10.3390/genes10080593
Porto FW, Daulatabad SV, Janga SC. Long Non-Coding RNA Expression Levels Modulate Cell-Type-Specific Splicing Patterns by Altering Their Interaction Landscape with RNA-Binding Proteins. Genes. 2019; 10(8):593. https://doi.org/10.3390/genes10080593
Chicago/Turabian StylePorto, Felipe Wendt, Swapna Vidhur Daulatabad, and Sarath Chandra Janga. 2019. "Long Non-Coding RNA Expression Levels Modulate Cell-Type-Specific Splicing Patterns by Altering Their Interaction Landscape with RNA-Binding Proteins" Genes 10, no. 8: 593. https://doi.org/10.3390/genes10080593
APA StylePorto, F. W., Daulatabad, S. V., & Janga, S. C. (2019). Long Non-Coding RNA Expression Levels Modulate Cell-Type-Specific Splicing Patterns by Altering Their Interaction Landscape with RNA-Binding Proteins. Genes, 10(8), 593. https://doi.org/10.3390/genes10080593