SiNPle: Fast and Sensitive Variant Calling for Deep Sequencing Data



Abstract
1. Introduction
2. Materials and Methods
2.1. SiNPle Algorithm
- The likelihood that each base of the variant arises as a sequencing error: There are two possible approaches to estimate it, starting from the baseline error rate and under the approximate assumption that all “non-variant” alleles are correct:or based on the base qualities (or equivalent qualities for indels) of the variant:To obtain a more conservative estimate, we sum the likelihoods from both approaches.
- The likelihood that the variant is actually a PCR artefact generated by a mutation during the PCR: The density of the frequency distribution of mutations arising from a binary exponential PCR amplification process is approximately where is the mutation rate per base during the PCR process. After discretising the distribution over r reads, the likelihood becomes
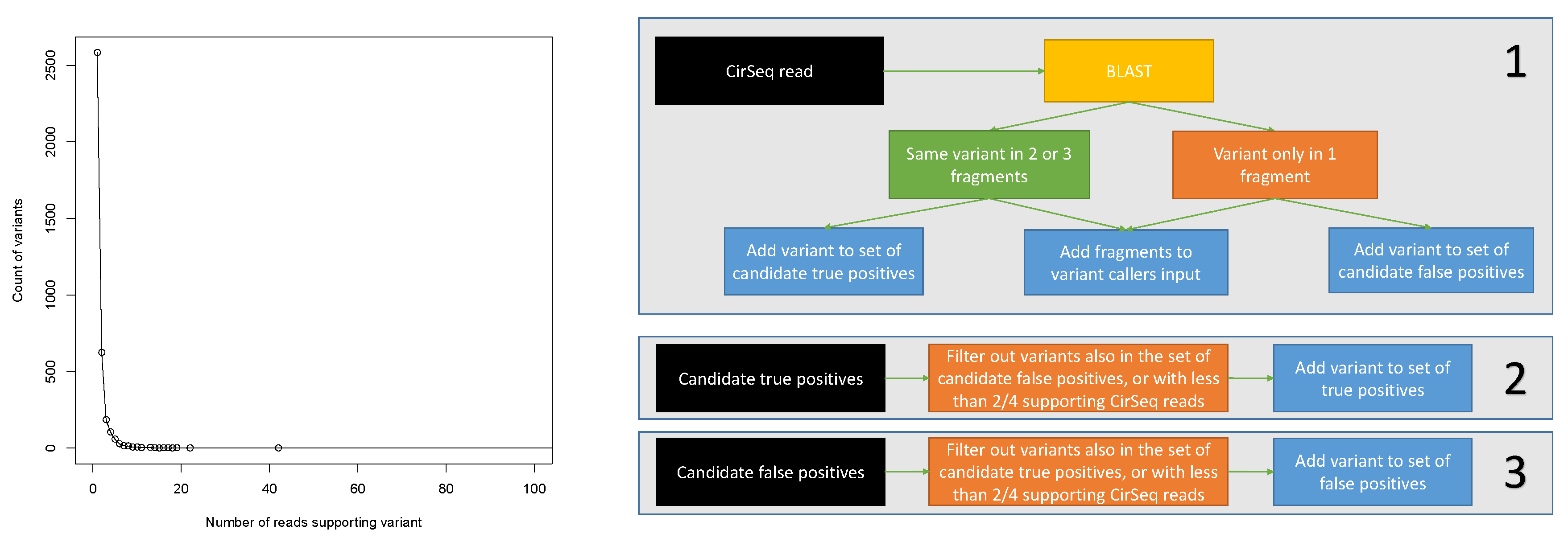
2.2. Programs Setup
2.3. Assessing Specificity and Sensitivity of Variant Calling on Simulated Data
2.4. Assessing Specificity and Sensitivity of Variant Calling on Real Data
3. Results
3.1. Assessing Specificity and Sensitivity of Variant Calling in Simulated Data
3.2. Comparative Assessment of Poliovirus Predictions
3.3. Variant Calling on Different Viruses
3.4. Speed Benchmarks
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SNP | Single nucleotide polymorphism |
| NCBI | National Center for Biotechnology Information |
| SRA | Short Read Archive |
| ROC | Receiver-operator curve |
| IBV | Infectious bronchitis virus |
| HIV | Human immunodeficiency virus. |
References
- Domingo, E. Quasispecies theory in virology. J. Virol. 2002, 76, 463–465. [Google Scholar] [CrossRef]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis ToolKit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Larson, D.E.; Wilson, R.K. Using VarScan 2 for germline variant calling and somatic mutation detection. Curr. Protoc. Bioinform. 2013, 44, 15.4.1–15.4.17. [Google Scholar]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. SOAPsnv: An integrated tool for somatic single-nucleotide variants detection with or without normal tissues in cancer genome. J. Clin. Oncol. 2014, 32, e22086. [Google Scholar] [CrossRef]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Beisel, C.; Rechsteiner, M.; Wild, P.; Schraml, P.; Moch, H.; Beerenwinkel, N. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat. Commun. 2012, 3, 811. [Google Scholar] [CrossRef] [PubMed]
- Carrot-Zhang, J.; Majewski, J. Lolopicker: Detecting low allelic-fraction variants in low-quality cancer samples from whole-exome sequencing data. bioRxiv 2016, 043612. [Google Scholar] [CrossRef]
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: Cambridge, UK, 1983. [Google Scholar]
- Raineri, E.; Ferretti, L.; Esteve-Codina, A.; Nevado, B.; Heath, S.; Pérez-Enciso, M. SNP calling by sequencing pooled samples. BMC Bioinform. 2012, 13, 239. [Google Scholar] [CrossRef] [PubMed]
- Source Code for SiNPle. Available online: https://mallorn.pirbright.ac.uk:4443/gitlab/drcyber/SiNPle (accessed on 8 July 2019).
- Reppell, M.; Boehnke, M.; Zöllner, S. Ftec: A coalescent simulator for modeling faster than exponential growth. Bioinformatics 2012, 28, 1282–1283. [Google Scholar] [CrossRef]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. Art: A next-generation sequencing read simulator. Bioinformatics 2011, 28, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, A.; Brodsky, L.; Andino, R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 2014, 505, 686. [Google Scholar] [CrossRef] [PubMed]
- NCBI SRA. Available online: https://www.ncbi.nlm.nih.gov/sra (accessed on 8 July 2019).
- DiversiTools. Available online: http://josephhughes.github.io/DiversiTools/ (accessed on 8 July 2019).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Dodt, M.; Roehr, J.; Ahmed, R.; Dieterich, C. FLEXBAR — Flexible barcode and adapter processing for next-generation sequencing platforms. Biology 2012, 1, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Marco-Sola, S.; Sammeth, M.; Guigó, R.; Ribeca, P. The GEM mapper: Fast, accurate and versatile alignment by filtration. Nat. Methods 2012, 9, 1185–1188. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coverage Threshold | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| True positives | 1125 | 233 | 96 | 59 | 35 |
| False positives | 10,158 | 4739 | 2386 | 1302 | 739 |
| Position | Genotype | Supporting Reads | LoFreq Default | Sensitive | SiNPle Default | VarScan2 Default | Sensitive |
|---|---|---|---|---|---|---|---|
| 2133 | C | 172 | ✓ | ✓ | ✓ | ✓ | ✓ |
| 4348 | A | 42 | ✓ | ✓ | ✓ | ✓ | |
| 6952 | T | 15 | ✓ | ✓ | |||
| 2456 | C | 13 | ✓ | ✓ | |||
| 4357 | C | 13 | ✓ | ✓ | |||
| 1867 | G | 11 | ✓ | ✓ | |||
| 2547 | A | 10 | ✓ | ✓ | ✓ | ✓ | |
| 4104 | C | 10 | ✓ | ✓ | ✓ | ✓ | |
| 1870 | A | 8 | ✓ | ✓ | |||
| 3255 | G | 8 | ✓ | ✓ | ✓ | ||
| 4994 | G | 8 | ✓ | ✓ | |||
| 5091 | A | 8 | ✓ | ||||
| 5233 | C | 8 | ✓ | ✓ | |||
| 6937 | C | 8 | ✓ | ✓ | |||
| 2009 | T | 7 | ✓ | ✓ | |||
| 3950 | T | 7 | ✓ | ✓ | |||
| 4100 | A | 7 | ✓ | ✓ | ✓ | ✓ | |
| 6224 | T | 7 | ✓ | ✓ | |||
| 3978 | A | 6 | ✓ | ✓ | |||
| 4203 | G | 6 | ✓ | ✓ | |||
| 5143 | T | 6 | ✓ | ✓ | |||
| 5192 | A | 6 | ✓ | ✓ | |||
| 6802 | G | 6 | ✓ | ✓ | |||
| 7029 | T | 6 | ✓ | ✓ | |||
| 2088 | T | 5 | ✓ | ✓ | |||
| 2684 | T | 5 | ✓ | ✓ | |||
| 3690 | C | 5 | ✓ | ✓ | |||
| 4356 | C | 5 | ✓ | ||||
| 6234 | T | 5 | ✓ | ✓ | |||
| 6413 | T | 5 | ✓ | ✓ | |||
| 6477 | A | 5 | ✓ | ||||
| 6508 | G | 5 | ✓ | ✓ | |||
| 7080 | G | 5 | ✓ | ✓ | |||
| 7320 | G | 5 | ✓ | ✓ |
| Dataset | Reads | Pileup | SiNPle | VarScan2 | LoFreq |
|---|---|---|---|---|---|
| HIV | 74,996 | 20.5 | 23.4 | 43.2 | 72.3 |
| Poliovirus | 100,000 | 34.8 | 36.6 | 45.7 | 51.2 |
| IBV | 3,778,012 | 690 | 755 | 910 | 7,324 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferretti, L.; Tennakoon, C.; Silesian, A.; Freimanis, G.; Ribeca, P. SiNPle: Fast and Sensitive Variant Calling for Deep Sequencing Data. Genes 2019, 10, 561. https://doi.org/10.3390/genes10080561
Ferretti L, Tennakoon C, Silesian A, Freimanis G, Ribeca P. SiNPle: Fast and Sensitive Variant Calling for Deep Sequencing Data. Genes. 2019; 10(8):561. https://doi.org/10.3390/genes10080561
Chicago/Turabian StyleFerretti, Luca, Chandana Tennakoon, Adrian Silesian, Graham Freimanis, and Paolo Ribeca. 2019. "SiNPle: Fast and Sensitive Variant Calling for Deep Sequencing Data" Genes 10, no. 8: 561. https://doi.org/10.3390/genes10080561
APA StyleFerretti, L., Tennakoon, C., Silesian, A., Freimanis, G., & Ribeca, P. (2019). SiNPle: Fast and Sensitive Variant Calling for Deep Sequencing Data. Genes, 10(8), 561. https://doi.org/10.3390/genes10080561