Longevity: Lesson from Model Organisms



{kind=link}
Abstract
1. Introduction
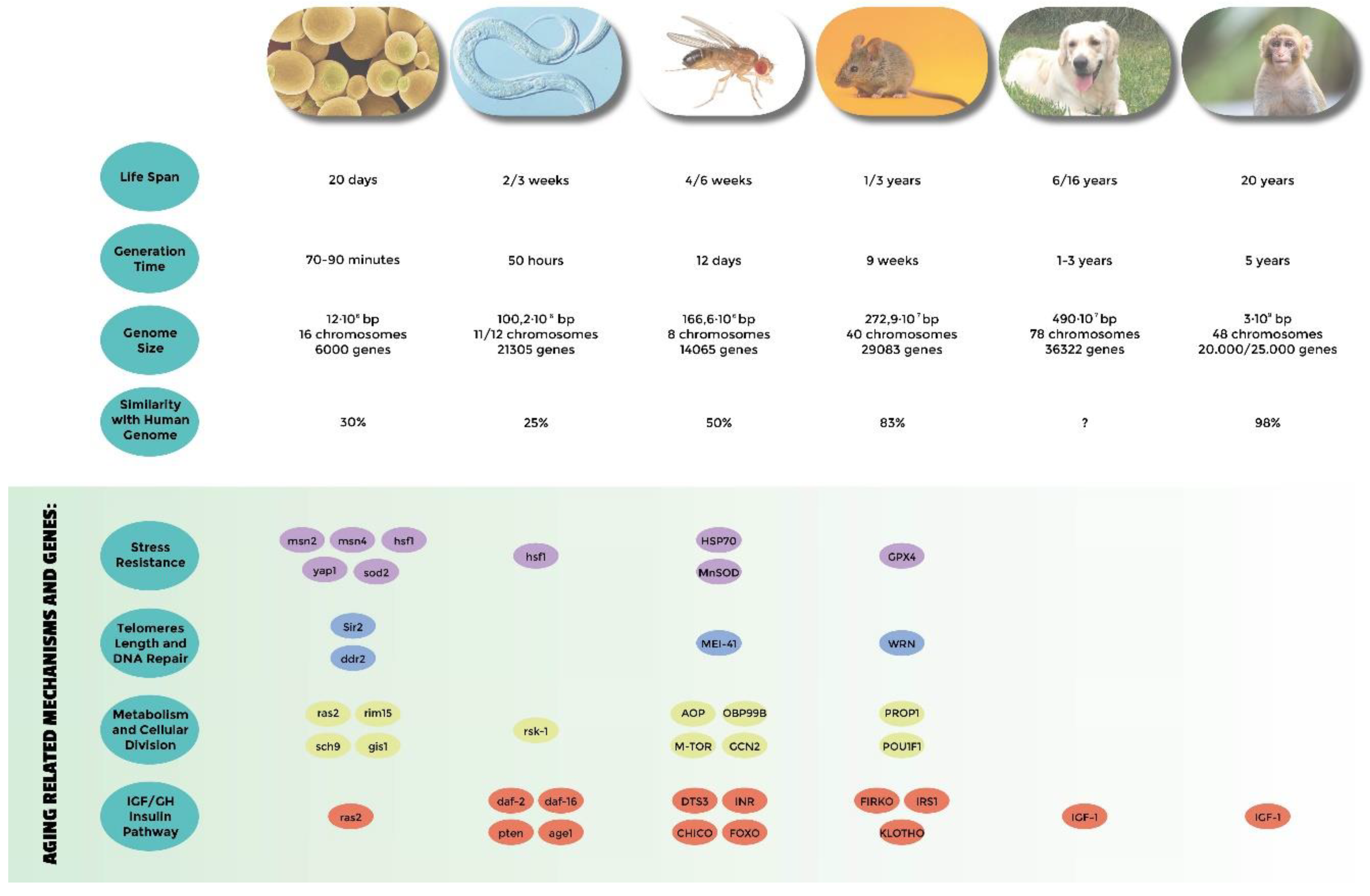
2. The Simplest Eukaryotic Model: Yeast Cells
3. Caenorabtidis elegans
4. Drosophila melanogaster
5. Mouse
6. Domestic Dog
7. Non-Human Primates
8. Discussion
- Genes related to stress resistance: their role in longevity has first been demonstrated in many different model systems [18,24,25,32,33,34,51,54,58,82,83,84,85] and eventually confirmed in centenarians who show a low degree of oxidative stress as well as high antioxidant protection [138,139]. A high level of oxidative stress is also an important risk factor of other age-related diseases such as hypertension, atherosclerosis, and diabetes. SNP (single nucleotide polymorphisms) studies have identified Tp53, coding for tumor suppressor p53 [140,141,142], EXO1 [143], GPX1 (glutathione peroxidase1) [144], SOD2 (manganese superoxide dismutase) [145], heat shock proteins genes HSPA1A, HSPA1B, and HSPA1L [146,147,148], GSTZ1 (glutathione S-transferase zeta 1) [149], NOS1, NOS2 (nitric oxide synthase 1 and 2) [150], and UCPs (uncoupling proteins) [147,151,152] as susceptibility genes.
- Genes involved in telomeres length: they have been found to be associated with human longevity such as TERT and TERC (telomerase reverse transcriptase, telomerase RNA component) [153], SIRT1, and SIRT3 (sirtuins) [154,155]. The first discoveries were made in yeasts and tetrahymena by Elizabeth Blackburn, finding the role of TERT and TERC ([156] and references within). In Caenorhabditis elegans over-expressing a protein involved in telomere length regulation leads to the elongation of telomeres and extends the life span, making the organism more resistant to heat stress [157]. The over-expression of TERT also extends the life span of mice [158]. In yeast, sirtuins promote longevity [159]; in particular, it has been reported that Sir2 mediates life-span extension due by calorie restriction [160]. These findings have been replicated in other model organisms [161], but their role in longevity is not consistent for all species, and therefore is still under debate [162].
- Genes involved in metabolism and cellular division: APOE (apolipoprotein E) [163], TXNRD1 (thioredoxin reductase 1), XDH (xanthine dehydrogenase) [163], MAP3K7 (mitogen-activated protein kinase kinase kinase 7) [149], AKT kinase, and TOR [164]. The association of APOE with human longevity have been replicated in different populations: [165,166,167]. Apolipoprotein E (apoE) exhibits three isoforms: apoE2, apoE3 and apoE4. They are involved in inflammation, elevated lipid levels, and oxidative stress; furthermore, these are risk factors for cardiovascular disease and Alzheimer’s disease, as reported by Huebbe et al. (2011) [168]. APOE2 has been defined as a longevity gene for its putative protective function; it is abundant in long-lived people, while APOE4, that differs from e3 allele at a single aa (112cys), and has been considered a frailty allele [169]. In fact, it increases the risk of Alzheimer’s disease and cardiovascular diseases, maybe for a putative interaction with the β amyloid protein, and it is almost absent in centenarians.
- Genes belonging to the IGF/GH and insulin pathway: mutations in genes belonging to the insulin or insulin-like signaling pathway extend the life span of Caenorhabditis elegans [170,171], Drosophila melanogaster [59,109,172], and mice [69,173]. In humans, it has been observed that insulin sensitivity normally decreases during aging. On the other hand, centenarians are more sensitive to insulin than other people, and often show lower IGF-1 plasma levels [174]. SNP studies have found an association of particular alleles or haplotypes for INS (insulin) [175], INSR (insulin receptor) [176], IGF1 (insulin growth factor 1) [177], IGF1R (insulin growth factor 1 receptor); in fact, a specific haplotype of the IGF-I receptor and the kinase PI3KCB is frequently found in individuals living longer together with low plasma levels of IGF-1 [178], IGF2 (insulin growth factor 2) [179], IGF2R (insulin growth factor 2 receptor) [180], IRS1 (insulin receptor substrate 1) [177], GH1 (growth hormone 1) [177], GHSR (growth hormone secretagogue receptor type 1) [175], FOXO1A (forkhead box protein O1 A), and FOXO3A (forkhead box protein O3 A) transcription factor, which contains alleles that are associated with longevity in multiple Asian and European populations [181,182,183,184,185].
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Williams, G.C. Pleiotropy, natural selection, and the evolution of senescence. Evolution 1957, 11, 398. [Google Scholar] [CrossRef]
- Longo, V.D.; Finch, C.E. Evolutionary Medicine: From Dwarf Model Systems to Healthy Centenarians? Science 2003, 299, 1342. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B. Understanding the odd science of aging. Cell 2005, 120, 437. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Mitteldorf, J.; Skulachev, V.P. Programmed and altruistic ageing. Nat. Rev. Genet. 2005, 6, 866. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H. Nobel Lecture. Yeast and cancer. Biosci. Rep. 2002, 22, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, C.M.; Brodsky, J.L. Use of yeast as a model system to investigate protein confor-mational diseases. Mol. Biotechnol. 2005, 30, 171–180. [Google Scholar] [CrossRef]
- Nakano, A. Yeast Golgi apparatus--Dynamics and sorting. Cell Mol. Life Sci. 2004, 61, 186–191. [Google Scholar] [CrossRef]
- Bowers, K.; Stevens, T.H. Protein transport from the late Golgi to the vacuole in the yeast Sac-charomyces cerevisiae. Biochim. Biophys. Acta 2005, 1744, 438–454. [Google Scholar] [CrossRef]
- Petranovic, D.; Nielsen, J. Can yeast systems biology contribute to the understanding of human disease? Trends Biotechnol. 2008, 26, 584–590. [Google Scholar] [CrossRef]
- Karathia, H.; Vilaprinyo, E.; Sorribas, A.; Alves, R. Saccharomyces cerevisiae as a model or-ganism: A comparative study. PLoS ONE 2011, 6, e16015. [Google Scholar] [CrossRef]
- Oliveira, A.V.; Vilaça, R.; Santos, C.N.; Costa, V.; Menezes, R. Exploring the power of yeast to model aging and age-related neurodegenerative disorders. Biogerontology 2017, 18, 3–34. [Google Scholar] [CrossRef] [PubMed]
- Mirisola, M.G.; Braun, R.J.; Petranovic, D. Approaches to study yeast cell aging and death. FEMS Yeast Res. 2014, 14, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Shadel, G.S.; Kaeberlein, M.; Kennedy, B. Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 2012, 16, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, R.K.; Johnston, J.R. Life span of individual yeast cells. Nature 1959, 183, 1751–1752. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wei, M.; Mirisola, M.G.; Longo, V.D. Assessing chronological aging in Saccharomyces cerevisiae. Methods Mol. Biol. 2013, 965, 463–472. [Google Scholar] [PubMed]
- Mirisola, M.G.; Longo, V.D. Acetic acid and acidification accelerate chronological and replicative aging in yeast. Cell Cycle 2012, 11, 3532–3533. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, P.; Gattazzo, C.; Battistella, L.; Wei, M.; Cheng, C.; McGrew, K.; Longo, V.D. Sir2 blocks extreme life-span extension. Cell 2005, 123, 655–667. [Google Scholar] [CrossRef]
- Longo, V.D. Mutations in signal transduction proteins increase stress resistance and longevity in yeast, nematodes, fruit flies, and mammalian neuronal cells. Neurobiol. Aging 1999, 20, 479–486. [Google Scholar] [CrossRef]
- Longo, V.D.; Gralla, E.B.; Valentine, J.S. Superoxide dismutase activity is essential for sta-tionary phase survival in Saccharomyces cerevisiae. Mitochondrial production of toxic oxygen spe-cies in vivo. J. Biol. Chem. 1996, 271, 12275–12280. [Google Scholar] [CrossRef]
- Dhirendra, K.S.; Dwight, V.N.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar]
- Mirisola, M.G.; Seidita, G.; Verrotti, A.C.; Di Blasi, F.; Fasano, O. Mutagenic alteration of the distal switch II region of RAS blocks CDC25-dependent signaling functions. J. Biol. Chem. 1994, 269, 15740–15748. [Google Scholar] [PubMed]
- Liu, Y.; Yang, F.; Li, S.; Dai, J.; Deng, H. Glutaredoxin Deletion Shortens Chronological Life Span in Saccharomyces cerevisiae via ROS-Mediated Ras/PKA Activation. J. Proteome Res. 2018, 17, 2318–2327. [Google Scholar] [CrossRef] [PubMed]
- Pedruzzi, I.; Burckert, N.; Egger, P.; De Virgilio, C. Saccharomyces cerevisiae Ras/cAMP pathway controls post-diauxic shift element-dependent transcription through the zinc finger protein Gis1. EMBO J. 2000, 19, 2569–2579. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D. The Chronological Life Span of Saccharomyces Cerevisiae. Studies of Superoxide Dismutase, Ras and Bcl-2. Ph.D. Thesis, University of California Los Angeles, Westwood, CA, USA, 1997. [Google Scholar]
- Wei, M.; Fabrizio, P.; Hu, J.; Ge, H.; Cheng, C.; Li, L.; Longo, V.D. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 2008, 4, e13. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Pastor, M.T.; Marchler, G.; Schuller, C.; Marchler-Bauer, A.; Ruis, H.; Estruch, F. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J. 1996, 15, 2227–2235. [Google Scholar] [CrossRef]
- Fabrizio, P.; Pozza, F.; Pletcher, S.D.; Gendron, C.M.; Longo, V.D. Regulation of longevity and stress resistance by Sch9 in yeast. Science 2001, 292, 288–290. [Google Scholar] [CrossRef]
- Hu, J.; Wei, M.; Mirzaei, H.; Madia, F.; Mirisola, M.; Amparo, C.; Chagoury, S.; Kennedy, B.; Longo, V.D. Tor-Sch9 deficiency activates catabolism of the ketone body-like acetic acid to promote trehalose accumulation and longevity. Aging Cell 2014, 13, 457–467. [Google Scholar] [CrossRef]
- Toda, T.; Cameron, S.; Sass, P.; Wigler, M. SCH9, a gene of Saccharomyces cerevisiae that encodes a protein distinct from, but functionally and structurally related to, cAMP-dependent protein kinase catalytic subunits. Genes Dev. 1988, 2, 517–527. [Google Scholar] [CrossRef]
- Thevelein, J.M.; De Winde, J.H. Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 1999, 33, 904–918. [Google Scholar] [CrossRef]
- Mirisola, M.G.; Taormina, G.; Fabrizio, P.; Wei, M.; Hu, J.; Longo, V.D. Serine- and threo-nine/valine-dependent activation of PDK and Tor orthologs converge on Sch9 to promote aging. PLoS Genet. 2014, 10, e1004113. [Google Scholar] [CrossRef]
- Flattery-O’Brien, J.A.; Grant, C.M.; Dawes, I.W. Stationary-phase regulation of the Saccha-romyces cerevisiaeSOD2 gene is dependent on additive effects of HAP2/3/4/5- and STRE-binding elements. Mol. Microbiol. 1997, 23, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Harris, N.; MacLean, M.; Hatzianthis, K.; Panaretou, B.; Piper, P.W. Increasing Saccharomyces cerevisiae stress resistance, through the overactivation of the heat shock response resulting from defects in the Hsp90 chaperone, does not extend replicative life span but can be associated with slower chronological ageing of nondividing cells. Mol. Genet. Genom. 2001, 265, 258–263. [Google Scholar]
- Herker, E.; Jungwirth, H.; Lehmann, K.A.; Maldener, C.; Frohlich, K.U.; Wissing, S.; Buttner, S.; Fehr, M.; Sigrist, S.; Madeo, F. Chronological aging leads to apoptosis in yeast. J. Cell Biol. 2004, 164, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D. The Ras and Sch9 pathways regulate stress resistance and longevity. Exp. Gerontol. 2003, 38, 807–811. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span--From yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Parrella, E.; Longo, V.D. Insulin/IGF-I and related signaling pathways regulate aging in nondividing cells: From yeast to the mammalian brain. Sci. World J. 2010, 10, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Kim, S.K. The early bird catches the worm: New technologies for the Caenorhabditis elegans toolkit. Nat. Rev. Genet. 2011, 12, 793–801. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boulin, T.; Hobert, O. From genes to function: The C. elegans genetic toolbox. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 114–137. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Chou, C.Y.; Ch’ang, L.Y.; Liu, C.S.; Lin, W. Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res. 2000, 10, 703–713. [Google Scholar] [CrossRef]
- Calixto, A.; Ma, C.; Chalfie, M. Conditional gene expression and RNAi using MEC-8-dependent splicing in C. elegans. Nat. Methods 2010, 7, 407–411. [Google Scholar] [CrossRef]
- Tarkhov, A.E.; Alla, R.; Ayyadevara, S.; Pyatnitskiy, M.; Menshikov, L.I.; Reis, R.J.S.; Fedichev, P.O. A universal transcriptomic signature of age reveals the temporal scaling of Caenorhabditis elegans aging trajectories. Sci. Rep. 2019, 9, 7368. [Google Scholar] [CrossRef] [PubMed]
- Son, H.G.; Altintas, O.; Kim, E.J.E.; Kwon, S.; Lee, S.V. Age-dependent changes and biomarkers of aging in Caenorhabditis elegans. Aging Cell 2019, 18, e12853. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R.A.C. C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Riddle, D.L.; Albert, P.S. Genetic and Environmental Regulation of Dauer Larva Development. In C. elegans II, 2nd ed.; Riddle, D.L., Blumenthal, T., Meyer, B.J., Priess, J.R., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1997. [Google Scholar]
- Kimura, K.D.; Tissenbaum, H.A.; Liu, Y.; Ruvkun, G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 1997, 277, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Dorman, J.B.; Rodan, A.; Kenyon, C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 1997, 278, 1319–1322. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T.; Johnson, T.E. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 2001, 11, 1975–1980. [Google Scholar] [CrossRef]
- Lee, R.Y.; Hench, J.; Ruvkun, G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr. Biol. 2001, 11, 1950–1957. [Google Scholar] [CrossRef]
- Hesp, K.; Smant, G.; Kammenga, J.E. Caenorhabditis elegans DAF-16/FOXO transcription factor and its mammalian homologs associate with age-related disease. Exp. Gerontol. 2015, 72, 1–7. [Google Scholar] [CrossRef]
- Morley, J.F.; Morimoto, R.I. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell 2004, 15, 657–664. [Google Scholar] [CrossRef]
- Larsen, P.L.; Albert, P.S.; Riddle, D.L. Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics 1995, 139, 1567–1583. [Google Scholar]
- Dorman, J.B.; Albinder, B.; Shroyer, T.; Kenyon, C. The age-1 and daf-2 genes function in a common pathway to control the lifespan of Caenorhabditis elegans. Genetics 1995, 141, 1399–1406. [Google Scholar] [PubMed]
- Honda, Y.; Honda, S. Oxidative stress and life span determination in the nematode Caenorhabditis elegans. Ann. N. Y. Acad. Sci. 2002, 959, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Branicky, R.; Benard, C.; Hekimi, S. clk-1, mitochondria, and physiological rates. Bioessays 2000, 22, 48–56. [Google Scholar] [CrossRef]
- Piper, M.D.W.; Partridge, L. Drosophila as a model for ageing. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2707–2717. [Google Scholar] [CrossRef] [PubMed]
- Brandt, A.; Vilcinskas, A. The Fruit Fly Drosophila melanogaster as a Model for Aging Research. Adv. Biochem. Eng. Biotechnol. 2013, 135, 63–77. [Google Scholar] [PubMed]
- Orr, W.C.; Sohal, R.S. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 1994, 263, 1128–1130. [Google Scholar] [CrossRef]
- Clancy, D.J.; Gems, D.; Harshman, L.G.; Oldham, S.; Stocker, H.; Hafen, E.; Leevers, S.J.; Partridge, L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 2001, 292, 104–106. [Google Scholar] [CrossRef]
- Sun, J.; Tower, J. FLP recombinase-mediated induction of Cu/Zn-superoxide dismutase transgene expression can extend the life span of adult Drosophila melanogaster flies. Mol. Cell Biol. 1999, 19, 216–228. [Google Scholar] [CrossRef]
- Piper, M.D.; Selman, C.; McElwee, J.J.; Partridge, L. Separating cause from effect: How does insulin/IGF signalling control lifespan in worms, flies and mice? J. Intern. Med. 2008, 263, 179–191. [Google Scholar] [CrossRef]
- Alic, N.; Giannakou, M.E.; Papatheodorou, I.; Hoddinott, M.P.; Andrews, T.D.; Bolukbasi, E.; Partridge, L. Interplay of dFOXO and two ETS-family transcription factors determines lifespan in Drosophila melanogaster. PLoS Genet. 2014, 10, e1004619. [Google Scholar] [CrossRef]
- Kucerova, L.; Kubrak, O.I.; Bengtsson, J.M.; Strnad, H.; Nylin, S.; Theopold, U.; Nassel, D.R. Slowed aging during reproductive dormancy is reflected in genome-wide transcriptome changes in Drosophila melanogaster. BMC Genom. 2016, 17, 50. [Google Scholar] [CrossRef]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010, 11, 35–46. [Google Scholar] [CrossRef]
- Kapahi, P.; Zid, B.M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 2004, 14, 885–890. [Google Scholar] [CrossRef]
- Katewa, S.D.; Kapahi, P. Role of TOR signaling in aging and related biological processes in Drosophila melanogaster. Exp. Gerontol. 2011, 46, 382–390. [Google Scholar] [CrossRef]
- Ulgherait, M.; Rana, A.; Rera, M.; Graniel, J.; Walker, D.W. AMPK modulates tissue and or-ganismal aging in a non-cell-autonomous manner. Cell Rep. 2014, 8, 1767–1780. [Google Scholar] [CrossRef]
- Chapman, T.; Partridge, L. Female fitness in Drosophila melanogaster: An interaction between the effect of nutrition and of encounter rate with males. Proc. Biol. Sci. 1996, 263, 755–759. [Google Scholar]
- Kang, M.J.; Vasudevan, D.; Kang, K.; Kim, K.; Park, J.E.; Zhang, N.; Zeng, X.; Neubert, T.A.; Marr, M.T., 2nd; Ryoo, H.D. 4E-BP is a target of the GCN2-ATF4 pathway during Drosophila development and aging. J. Cell Biol. 2017, 216, 115–129. [Google Scholar] [CrossRef]
- Brown-Borg, H.M.; Borg, K.E.; Meliska, C.J.; Bartke, A. Dwarf mice and the ageing process. Nature 1996, 384, 33. [Google Scholar] [CrossRef]
- Dolle, M.E.; Snyder, W.K.; Vijg, J. Genotyping the Prop-1 mutation in Ames dwarf mice. Mech. Ageing Dev. 2001, 122, 1915–1918. [Google Scholar] [CrossRef]
- Andersen, B.; Pearse, R.V., 2nd; Jenne, K.; Sornson, M.; Lin, S.C.; Bartke, A.; Rosenfeld, M.G. The Ames dwarf gene is required for Pit-1 gene activation. Dev. Biol. 1995, 172, 495–503. [Google Scholar] [CrossRef][Green Version]
- Snell, G.D. Dwarf, a new mendelian recessive character of the house mouse. Proc. Natl. Acad. Sci. USA 1929, 15, 733–734. [Google Scholar] [CrossRef]
- Flurkey, K.; Papaconstantinou, J.; Miller, R.A.; Harrison, D.E. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc. Natl. Acad. Sci. USA 2001, 98, 6736–6741. [Google Scholar] [CrossRef]
- Flurkey, K.; Papaconstantinou, J.; Harrison, D.E. The Snell dwarf mutation Pit1(dw) can in-crease life span in mice. Mech. Ageing Dev. 2002, 123, 121–130. [Google Scholar] [CrossRef]
- Turyn, D.; Dominici, F.P.; Sotelo, A.I.; Bartke, A. Specific interactions of growth hormone (GH) with GH-receptors and GH-binding proteins in vivo in genetically GH-deficient Ames dwarf mice. Growth Horm. IGF Res. 1998, 8, 389–396. [Google Scholar] [CrossRef]
- Bartke, A.; Brown-Borg, H.M.; Bode, A.M.; Carlson, J.; Hunter, W.S.; Bronson, R.T. Does growth hormone prevent or accelerate aging? Exp. Gerontol. 1998, 33, 675–687. [Google Scholar] [CrossRef]
- Hunter, W.S.; Croson, W.B.; Bartke, A.; Gentry, M.V.; Meliska, C.J. Low body temperature in long-lived Ames dwarf mice at rest and during stress. Physiol. Behav. 1999, 67, 433–437. [Google Scholar] [CrossRef]
- Svare, B.; Bartke, A.; Doherty, P.; Mason, I.; Michael, S.D.; Smith, M.S. Hyperprolactinemia suppresses copulatory behavior in male rats and mice. Biol. Reprod. 1979, 21, 529–535. [Google Scholar] [CrossRef]
- Garcia, A.M.; Busuttil, R.A.; Calder, R.B.; Dolle, M.E.; Diaz, V.; McMahan, C.A.; Bartke, A.; Nelson, J.; Reddick, R.; Vijg, J. Effect of Ames dwarfism and caloric restriction on spontaneous DNA mutation frequency in different mouse tissues. Mech. Ageing Dev. 2008, 129, 528–533. [Google Scholar] [CrossRef]
- Alderman, J.M.; Flurkey, K.; Brooks, N.L.; Naik, S.B.; Gutierrez, J.M.; Srinivas, U.; Ziara, K.B.; Jing, L.; Boysen, G.; Bronson, R.; et al. Neuroendocrine inhibition of glucose production and resistance to cancer in dwarf mice. Exp. Gerontol. 2009, 44, 26–33. [Google Scholar] [CrossRef]
- Brown-Borg, H.; Johnson, W.T.; Rakoczy, S.; Romanick, M. Mitochondrial oxidant gen-eration and oxidative damage in Ames dwarf and GH transgenic mice. J. Am. Aging Assoc. 2001, 24, 85–96. [Google Scholar]
- Brown-Borg, H.M.; Bode, A.M.; Bartke, A. Antioxidative mechanisms and plasma growth hormone levels: Potential relationship in the aging process. Endocrine 1999, 11, 41–48. [Google Scholar] [CrossRef]
- Brown-Borg, H.M.; Rakoczy, S.G. Catalase expression in delayed and premature aging mouse models. Exp. Gerontol. 2000, 35, 199–212. [Google Scholar] [CrossRef]
- Bokov, A.F.; Lindsey, M.L.; Khodr, C.; Sabia, M.R.; Richardson, A. Long-lived ames dwarf mice are resistant to chemical stressors. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 819–827. [Google Scholar] [CrossRef]
- Patrick, A.; Seluanov, M.; Hwang, C.; Tam, J.; Khan, T.; Morgenstern, A.; Wiener, L.; Vazquez, J.M.; Zafar, H.; Wen, R.; et al. Sensitivity of primary fibroblasts in culture to atmospheric oxygen does not correlate with species lifespan. Aging 2016, 8, 841–847. [Google Scholar] [CrossRef][Green Version]
- Bluher, M.; Kahn, B.B.; Kahn, C.R. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003, 299, 572–574. [Google Scholar] [CrossRef]
- Bartke, A. Minireview: Role of the growth hormone/insulin-like growth factor system in mammalian aging. Endocrinology 2005, 146, 3718–3723. [Google Scholar] [CrossRef]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef]
- Bektas, A.; Schurman, S.H.; Sharov, A.A.; Carter, M.G.; Dietz, H.C.; Francomano, C.A. Klotho gene variation and expression in 20 inbred mouse strains. Mamm. Genome 2004, 15, 759–767. [Google Scholar] [CrossRef]
- Chang, S. A mouse model of Werner Syndrome: What can it tell us about aging and cancer? Int. J. Biochem. Cell Biol. 2005, 37, 991–999. [Google Scholar] [CrossRef]
- Austad, S.N. Comparative biology of aging. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 199–201. [Google Scholar] [CrossRef]
- Greer, K.A.; Canterberry, S.C.; Murphy, K.E. Statistical analysis regarding the effects of height and weight on life span of the domestic dog. Res. Vet. Sci. 2007, 82, 208–214. [Google Scholar] [CrossRef]
- Bonnett, B.N.; Egenvall, A. Age patterns of disease and death in insured Swedish dogs, cats and horses. J. Comp. Pathol. 2010, 142, S33–S38. [Google Scholar] [CrossRef]
- Fleming, J.M.; Creevy, K.E.; Promislow, D.E. Mortality in north american dogs from 1984 to 2004: An investigation into age-, size-, and breed-related causes of death. J. Vet. Intern. Med. 2011, 25, 187–198. [Google Scholar] [CrossRef]
- Creevy, K.E.; Austad, S.N.; Hoffman, J.M.; O’Neill, D.G.; Promislow, D.E. The Companion Dog as a Model for the Longevity Dividend. Cold Spring Harb. Perspect. Med. 2016, 6, a026633. [Google Scholar] [CrossRef]
- Freeman, L.M. Cachexia and sarcopenia: Emerging syndromes of importance in dogs and cats. J. Vet. Intern. Med. 2012, 26, 3–17. [Google Scholar] [CrossRef]
- Urfer, S.R.; Greer, K.; Wolf, N.S. Age-related cataract in dogs: A biomarker for life span and its relation to body size. Age (Dordr) 2011, 33, 451–460. [Google Scholar] [CrossRef]
- Vite, C.H.; Head, E. Aging in the canine and feline brain. Vet. Clin. N. Am. Small Anim. Pract. 2014, 44, 1113–1129. [Google Scholar] [CrossRef]
- Boyko, A.R.; Quignon, P.; Li, L.; Schoenebeck, J.J.; Degenhardt, J.D.; Lohmueller, K.E.; Zhao, K.; Brisbin, A.; Parker, H.G.; vonHoldt, B.M.; et al. A simple genetic architecture underlies morphological variation in dogs. PLoS Biol. 2010, 8, e1000451. [Google Scholar] [CrossRef]
- Vonholdt, B.M.; Pollinger, J.P.; Lohmueller, K.E.; Han, E.; Parker, H.G.; Quignon, P.; De-genhardt, J.D.; Boyko, A.R.; Earl, D.A.; Auton, A.; et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 2010, 464, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Ostrander, E.A.; Wayne, R.K. The canine genome. Genome Res. 2005, 15, 1706–1716. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, K.M.; Greer, K.A. Why is the dog an ideal model for aging research? Exp. Gerontol. 2015, 71, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Al-Regaiey, K.A.; Masternak, M.M.; Bonkowski, M.; Sun, L.; Bartke, A. Long-lived growth hormone receptor knockout mice: Interaction of reduced insulin-like growth factor i/insulin signaling and caloric restriction. Endocrinology 2005, 146, 851–860. [Google Scholar] [CrossRef]
- Eigenmann, J.E.; Amador, A.; Patterson, D.F. Insulin-like growth factor I levels in proportionate dogs, chondrodystrophic dogs and in giant dogs. Acta Endocrinol. (Copenh) 1988, 118, 105–108. [Google Scholar] [CrossRef]
- Greer, K.A.; Hughes, L.M.; Masternak, M.M. Connecting serum IGF-1, body size, and age in the domestic dog. Age (Dordr) 2011, 33, 475–483. [Google Scholar] [CrossRef]
- Tatar, M.; Bartke, A.; Antebi, A. The endocrine regulation of aging by insulin-like signals. Science 2003, 299, 1346–1351. [Google Scholar] [CrossRef]
- Waters, D.J.; Kengeri, S.S.; Clever, B.; Booth, J.A.; Maras, A.H.; Schlittler, D.L.; Hayek, M.G. Exploring mechanisms of sex differences in longevity: Lifetime ovary exposure and exceptional lon-gevity in dogs. Aging Cell 2009, 8, 752–755. [Google Scholar] [CrossRef]
- Adams, V.J.; Ceccarelli, K.; Watson, P.; Carmichael, S.; Penell, J.; Morgan, D.M. Evidence of longer life; a cohort of 39 labrador retrievers. Vet. Rec. 2018, 182, 408. [Google Scholar] [CrossRef]
- Kealy, R.D.; Lawler, D.F.; Ballam, J.M.; Mantz, S.L.; Biery, D.N.; Greeley, E.H.; Lust, G.; Segre, M.; Smith, G.K.; Stowe, H.D. Effects of diet restriction on life span and age-related changes in dogs. J. Am. Vet. Med. Assoc. 2002, 220, 1315–1320. [Google Scholar] [CrossRef]
- Lawler, D.F.; Larson, B.T.; Ballam, J.M.; Smith, G.K.; Biery, D.N.; Evans, R.H.; Greeley, E.H.; Segre, M.; Stowe, H.D.; Kealy, R.D. Diet restriction and ageing in the dog: Major observations over two decades. Br. J. Nutr. 2008, 99, 793–805. [Google Scholar] [CrossRef]
- Richards, S.E.; Wang, Y.; Claus, S.P.; Lawler, D.; Kochhar, S.; Holmes, E.; Nicholson, J.K. Metabolic phenotype modulation by caloric restriction in a lifelong dog study. J. Proteome Res. 2013, 12, 3117–3127. [Google Scholar] [CrossRef]
- Greeley, E.H.; Spitznagel, E.; Lawler, D.F.; Kealy, R.D.; Segre, M. Modulation of canine im-munosenescence by life-long caloric restriction. Vet. Immunol. Immunopathol. 2006, 111, 287–299. [Google Scholar] [CrossRef]
- Jimenez, A.G.; Winward, J.; Beattie, U.; Cipolli, W. Cellular metabolism and oxidative stress as a possible determinant for longevity in small breed and large breed dogs. PLoS ONE 2018, 13, e0195832. [Google Scholar] [CrossRef]
- Alexander, J.E.; Colyer, A.; Haydock, R.M.; Hayek, M.G.; Park, J. Understanding How Dogs Age: Longitudinal Analysis of Markers of Inflammation, Immune Function, and Oxidative Stress. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 720–728. [Google Scholar] [CrossRef]
- Gibbs, R.A.; Rogers, J.; Katze, M.G.; Bumgarner, R.; Weinstock, G.M.; Mardis, E.R.; Remington, K.A.; Strausberg, R.L.; Venter, J.C.; Wilson, R.K.; et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science 2007, 316, 222–234. [Google Scholar]
- Zimin, A.V.; Cornish, A.S.; Maudhoo, M.D.; Gibbs, R.M.; Zhang, X.; Pandey, S.; Meehan, D.T.; Wipfler, K.; Bosinger, S.E.; Johnson, Z.P.; et al. A new rhesus macaque assembly and annotation for next-generation sequencing analyses. Biol. Direct 2014, 9, 20. [Google Scholar] [CrossRef]
- Yates, A.; Akanni, W.; Amode, M.R.; Barrell, D.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; Fitzgerald, S.; Gil, L.; et al. Ensembl 2016. Nucleic Acids Res. 2016, 44, D710–D716. [Google Scholar] [CrossRef]
- Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 2005, 437, 69–87. [Google Scholar] [CrossRef]
- Colman, R.J. Non-human primates as a model for aging. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2733–2741. [Google Scholar] [CrossRef]
- Uno, H. Age-related pathology and biosenescent markers in captive rhesus macaques. Age (Omaha) 1997, 20, 1–13. [Google Scholar] [CrossRef]
- Colman, R.J.; McKiernan, S.H.; Aiken, J.M.; Weindruch, R. Muscle mass loss in Rhesus monkeys: Age of onset. Exp. Gerontol. 2005, 40, 573–581. [Google Scholar] [CrossRef]
- Colman, R.J.; Kemnitz, J.W.; Lane, M.A.; Abbott, D.H.; Binkley, N. Skeletal effects of aging and menopausal status in female rhesus macaques. J. Clin. Endocrinol. Metab. 1999, 84, 4144–4148. [Google Scholar] [CrossRef]
- Bodkin, N.L.; Alexander, T.M.; Ortmeyer, H.K.; Johnson, E.; Hansen, B.C. Mortality and morbidity in laboratory-maintained Rhesus monkeys and effects of long-term dietary restriction. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, 212–219. [Google Scholar] [CrossRef]
- Mattison, J.A.; Roth, G.S.; Beasley, T.M.; Tilmont, E.M.; Handy, A.M.; Herbert, R.L.; Longo, D.L.; Allison, D.B.; Young, J.E.; Bryant, M.; et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 2012, 489, 318–321. [Google Scholar] [CrossRef]
- Ramsey, J.J.; Colman, R.J.; Binkley, N.C.; Christensen, J.D.; Gresl, T.A.; Kemnitz, J.W.; Weindruch, R. Dietary restriction and aging in rhesus monkeys: The University of Wisconsin study. Exp. Gerontol. 2000, 35, 1131–1149. [Google Scholar] [CrossRef]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef]
- Colman, R.J.; Beasley, T.M.; Kemnitz, J.W.; Johnson, S.C.; Weindruch, R.; Anderson, R.M. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 2014, 5, 3557. [Google Scholar] [CrossRef]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; De Cabo, R.; Anderson, R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017, 8, 14063. [Google Scholar] [CrossRef]
- Vaughan, K.L.; Kaiser, T.; Peaden, R.; Anson, R.M.; De Cabo, R.; Mattison, J.A. Caloric Re-striction Study Design Limitations in Rodent and Nonhuman Primate Studies. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 73, 48–53. [Google Scholar] [CrossRef]
- De Magalhaes, J.P.; Church, G.M. Analyses of human-chimpanzee orthologous gene pairs to explore evolutionary hypotheses of aging. Mech. Ageing Dev. 2007, 128, 355–364. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blalock, E.M.; Grondin, R.; Chen, K.C.; Thibault, O.; Thibault, V.; Pandya, J.D.; Dowling, A.; Zhang, Z.; Sullivan, P.; Porter, N.M.; et al. Aging-related gene expression in hippocampus proper compared with dentate gyrus is selectively associated with metabolic syndrome variables in rhesus monkeys. J. NeuroSci. 2010, 30, 6058–6071. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Han, D.; Yan, Z.; Boyd-Kirkup, J.D.; Green, C.D.; Khaitovich, P.; Han, J.D. Stress-associated H3K4 methylation accumulates during postnatal development and aging of rhesus ma-caque brain. Aging Cell 2012, 11, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Kumar, V.; Lackner, A.A.; Alvarez, X. Dysregulated miR-34a-SIRT1-acetyl p65 axis is a potential mediator of immune activation in the colon during chronic simian immunodeficiency virus infection of rhesus macaques. J. Immunol. 2015, 194, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Taormina, G.; Mirisola, M.G. Longevity: Epigenetic and biomolecular aspects. BioMol. Concepts 2015, 6, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Paolisso, G.; Tagliamonte, M.R.; Rizzo, M.R.; Manzella, D.; Gambardella, A.; Varricchio, M. Oxidative stress and advancing age: Results in healthy centenarians. J. Am. Geriatr. Soc. 1998, 46, 833–838. [Google Scholar] [CrossRef]
- Mecocci, P.; Polidori, M.C.; Troiano, L.; Cherubini, A.; Cecchetti, R.; Pini, G.; Straatman, M.; Monti, D.; Stahl, W.; Sies, H.; et al. Plasma antioxidants and longevity: A study on healthy centenarians. Free Radic. Biol. Med. 2000, 28, 1243–1248. [Google Scholar] [CrossRef]
- Di Pietro, F.; Dato, S.; Carpi, F.M.; Corneveaux, J.J.; Serfaustini, S.; Maoloni, S.; Mignini, F.; Huentelman, M.J.; Passarino, G.; Napolioni, V. TP53*P72 allele influences negatively female life expectancy in a population of central Italy: Cross-sectional study and genetic-demographic approach analysis. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 68, 539–545. [Google Scholar] [CrossRef]
- Van Heemst, D.; Mooijaart, S.P.; Beekman, M.; Schreuder, J.; De Craen, A.J.; Brandt, B.W.; Slagboom, P.E.; Westendorp, R.G. Variation in the human TP53 gene affects old age survival and cancer mortality. Exp. Gerontol. 2005, 40, 11–15. [Google Scholar] [CrossRef]
- Altilia, S.; Santoro, A.; Malagoli, D.; Lanzarini, C.; Álvarez, J.A.B.; Galazzo, G.; Porter, D.C.; Crocco, P.; Rose, G.; Passarino, G.; et al. TP53 codon 72 polymorphism affects accumulation of mtDNA damage in human cells. Aging 2012, 4, 28–39. [Google Scholar] [CrossRef]
- Nebel, A.; Flachsbart, F.; Till, A.; Caliebe, A.; Blanché, H.; Arlt, A.; Häsler, R.; Jacobs, G.; Kleindorp, R.; Franke, A.; et al. A functional EXO1 promoter variant is associated with prolonged life expectancy in centenarians. Mech. Ageing Dev. 2009, 130, 691–699. [Google Scholar] [CrossRef]
- Soerensen, M.; Christensen, K.; Stevnsner, T.; Christiansen, L. The Mn-superoxide dismutase single nucleotide polymorphism rs4880 and the glutathione peroxidase 1 single nucleotide poly-morphism rs1050450 are associated with aging and longevity in the oldest old. Mech. Ageing Dev. 2009, 130, 308–314. [Google Scholar] [CrossRef]
- Lunetta, K.L.; D’Agostino, R.B., Sr.; Karasik, D.; Benjamin, E.J.; Guo, C.Y.; Govindaraju, R.; Kiel, D.P.; Kelly-Hayes, M.; Massaro, J.M.; Pencina, M.J.; et al. Genetic correlates of longevity and selected age-related phenotypes: A genome-wide association study in the Framingham Study. BMC Med. Genet. 2007, 8, S13. [Google Scholar] [CrossRef]
- Singh, R.; Kolvraa, S.; Bross, P.; Christensen, K.; Gregersen, N.; Tan, Q.; Jensen, U.B.; Eiberg, H.; Rattan, S.I. Heat-shock protein 70 genes and human longevity: A view from Denmark. Ann. N. Y. Acad. Sci. 2006, 1067, 301–308. [Google Scholar] [CrossRef]
- Ross, O.A.; Curran, M.D.; Crum, K.A.; Rea, I.M.; Barnett, Y.A.; Middleton, D. Increased frequency of the 2437T allele of the heat shock protein 70-Hom gene in an aged Irish population. Exp. Gerontol. 2003, 38, 561–565. [Google Scholar] [CrossRef]
- Altomare, K.; Greco, V.; Bellizzi, D.; Berardelli, M.; Dato, S.; DeRango, F.; Garasto, S.; Rose, G.; Feraco, E.; Mari, V.; et al. The allele (A)(-110) in the promoter region of the HSP70-1 gene is unfavorable to longevity in women. Biogerontology 2003, 4, 215–220. [Google Scholar] [CrossRef]
- Di Cianni, F.; Campa, D.; Tallaro, F.; Rizzato, C.; De Rango, F.; Barale, R.; Passarino, G.; Canzian, F.; Gemignani, F.; Montesanto, A.; et al. MAP3K7 and GSTZ1 are associated with human longevity: A two-stage case-control study using a multilocus genotyping. Age (Dordr) 2013, 35, 1357–1366. [Google Scholar] [CrossRef]
- Montesanto, A.; Crocco, P.; Tallaro, F.; Pisani, F.; Mazzei, B.; Mari, V.; Corsonello, A.; Lattanzio, F.; Passarino, G.; Rose, G. Common polymorphisms in nitric oxide synthase (NOS) genes influence quality of aging and longevity in humans. Biogerontology 2013, 14, 177–186. [Google Scholar] [CrossRef]
- Rose, G.; Crocco, P.; De Rango, F.; Montesanto, A.; Passarino, G. Further support to the un-coupling-to-survive theory: The genetic variation of human UCP genes is associated with longevity. PLoS ONE 2011, 6, e29650. [Google Scholar] [CrossRef]
- Crocco, P.; Montesanto, A.; Passarino, G.; Rose, G. A common polymorphism in the UCP3 promoter influences hand grip strength in elderly people. Biogerontology 2011, 12, 265–271. [Google Scholar] [CrossRef]
- Soerensen, M.; Thinggaard, M.; Nygaard, M.; Dato, S.; Tan, Q.; Hjelmborg, J.; Andersen-Ranberg, K.; Stevnsner, T.; Bohr, V.A.; Kimura, M.; et al. Genetic variation in TERT and TERC and human leukocyte telomere length and longevity: A cross-sectional and longitudinal analysis. Aging Cell 2012, 11, 223–227. [Google Scholar] [CrossRef]
- Kim, S.; Bi, X.; Czarny-Ratajczak, M.; Dai, J.; Welsh, D.A.; Myers, L.; Welsch, M.A.; Cherry, K.E.; Arnold, J.; Poon, L.W.; et al. Telomere maintenance genes SIRT1 and XRCC6 impact age-related decline in telomere length but only SIRT1 is associated with human longevity. Bi-ogerontology 2012, 13, 119–131. [Google Scholar] [CrossRef]
- Atzmon, G.; Cho, M.; Cawthon, R.M.; Budagov, T.; Katz, M.; Yang, X.; Siegel, G.; Bergman, A.; Huffman, D.M.; Schechter, C.B.; et al. Evolution in health and medicine Sackler colloquium: Genetic variation in human telomerase is associated with telomere length in Ashkenazi centenarians. Proc. Natl. Acad. Sci. USA 2010, 107, 1710–1717. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Greider, C.W.; Szostak, J.W. Telomeres and telomerase: The path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 2006, 12, 1133–1138. [Google Scholar] [CrossRef]
- Joeng, K.S.; Song, E.J.; Lee, K.J.; Lee, J. Long lifespan in worms with long telomeric DNA. Nat. Genet. 2004, 36, 607–611. [Google Scholar] [CrossRef]
- Tomas-Loba, A.; Flores, I.; Fernandez-Marcos, P.J.; Cayuela, M.L.; Maraver, A.; Tejera, A.; Borras, C.; Matheu, A.; Klatt, P.; Flores, J.M.; et al. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell 2008, 135, 609–622. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef]
- Lin, S.J.; Defossez, P.A.; Guarente, L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 2000, 289, 2126–2128. [Google Scholar] [CrossRef]
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caeno-rhabditis elegans. Nature 2001, 410, 227–230. [Google Scholar] [CrossRef]
- Park, S.; Mori, R.; Shimokawa, I. Do sirtuins promote mammalian longevity? A critical review on its relevance to the longevity effect induced by calorie restriction. Mol. Cells 2013, 35, 474–480. [Google Scholar] [CrossRef]
- Soerensen, M.; Dato, S.; Tan, Q.; Thinggaard, M.; Kleindorp, R.; Beekman, M.; Suchiman, H.E.; Jacobsen, R.; McGue, M.; Stevnsner, T.; et al. Evidence from case-control and longitudinal studies supports associations of genetic variation in APOE, CETP, and IL6 with human longevity. Age (Dordr) 2013, 35, 487–500. [Google Scholar] [CrossRef]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Kervinen, K.; Savolainen, M.J.; Salokannel, J.; Hynninen, A.; Heikkinen, J.; Ehnholm, C.; Koistinen, M.J.; Kesaniemi, Y.A. Apolipoprotein E and B polymorphisms--Longevity factors assessed in nonagenarians. Atherosclerosis 1994, 105, 89–95. [Google Scholar] [CrossRef]
- Schachter, F.; Faure-Delanef, L.; Guenot, F.; Rouger, H.; Froguel, P.; Lesueur-Ginot, L.; Cohen, D. Genetic associations with human longevity at the APOE and ACE loci. Nat. Genet. 1994, 6, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Huebbe, P.; Nebel, A.; Siegert, S.; Moehring, J.; Boesch-Saadatmandi, C.; Most, E.; Pallauf, J.; Egert, S.; Muller, M.J.; Schreiber, S.; et al. APOE epsilon4 is associated with higher vitamin D levels in targeted replacement mice and humans. FASEB J. 2011, 25, 3262–3270. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, L.U.; Jeune, B.; Ranberg, K.A.; Nybo, H.; Vaupel, J.W. Estimation of apolipoprotein E genotype-specific relative mortality risks from the distribution of genotypes in centenarians and middle-aged men: Apolipoprotein E gene is a “frailty gene,” not a “longevity gene”. Genet. Epidemiol. 2000, 19, 202–210. [Google Scholar] [CrossRef]
- Vanfleteren, J.R.; Braeckman, B.P. Mechanisms of life span determination in Caenorhabditis elegans. Neurobiol. Aging 1999, 20, 487–502. [Google Scholar] [CrossRef]
- Murakami, S.; Johnson, T.E. A genetic pathway conferring life extension and resistance to UV stress in Caenorhabditis elegans. Genetics 1996, 143, 1207–1218. [Google Scholar]
- Tatar, M.; Kopelman, A.; Epstein, D.; Tu, M.P.; Yin, C.M.; Garofalo, R.S. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 2001, 292, 107–110. [Google Scholar] [CrossRef]
- Steger, R.W.; Bartke, A.; Cecim, M. Premature ageing in transgenic mice expressing different growth hormone genes. J. Reprod. Fertil. Suppl. 1993, 46, 61–75. [Google Scholar]
- Paolisso, G.; Gambardella, A.; Ammendola, S.; D’Amore, A.; Balbi, V.; Varricchio, M.; D’Onofrio, F. Glucose tolerance and insulin action in healthy centenarians. Am. J. Physiol. 1996, 270, E890–E894. [Google Scholar]
- Soerensen, M.; Dato, S.; Tan, Q.; Thinggaard, M.; Kleindorp, R.; Beekman, M.; Jacobsen, R.; Suchiman, H.E.; De Craen, A.J.; Westendorp, R.G.; et al. Human longevity and variation in GH/IGF-1/insulin signaling, DNA damage signaling and repair and pro/antioxidant pathway genes: Cross sectional and longitudinal studies. Exp. Gerontol. 2012, 47, 379–387. [Google Scholar] [CrossRef]
- Kojima, T.; Kamei, H.; Aizu, T.; Arai, Y.; Takayama, M.; Nakazawa, S.; Ebihara, Y.; Inagaki, H.; Masui, Y.; Gondo, Y.; et al. Association analysis between longevity in the Japanese population and polymorphic variants of genes involved in insulin and insulin-like growth factor 1 signaling pathways. Exp. Gerontol. 2004, 39, 1595–1598. [Google Scholar] [CrossRef]
- Van Heemst, D.; Beekman, M.; Mooijaart, S.P.; Heijmans, B.T.; Brandt, B.W.; Zwaan, B.J.; Slagboom, P.E.; Westendorp, R.G. Reduced insulin/IGF-1 signalling and human longevity. Aging Cell 2005, 4, 79–85. [Google Scholar] [CrossRef]
- Bonafe, M.; Barbieri, M.; Marchegiani, F.; Olivieri, F.; Ragno, E.; Giampieri, C.; Mugianesi, E.; Centurelli, M.; Franceschi, C.; Paolisso, G. Polymorphic variants of insulin-like growth factor I (IGF-I) receptor and phosphoinositide 3-kinase genes affect IGF-I plasma levels and human longevity: Cues for an evolutionarily conserved mechanism of life span control. J. Clin. Endocrinol. Metab. 2003, 88, 3299–3304. [Google Scholar] [CrossRef]
- Stessman, J.; Maaravi, Y.; Hammerman-Rozenberg, R.; Cohen, A.; Nemanov, L.; Gritsenko, I.; Gruberman, N.; Ebstein, R.P. Candidate genes associated with ageing and life expectancy in the Jerusalem longitudinal study. Mech. Ageing Dev. 2005, 126, 333–339. [Google Scholar] [CrossRef]
- Rose, G.; Crocco, P.; D’Aquila, P.; Montesanto, A.; Bellizzi, D.; Passarino, G. Two variants lo-cated in the upstream enhancer region of human UCP1 gene affect gene expression and are correlated with human longevity. Exp. Gerontol. 2011, 46, 897–904. [Google Scholar] [CrossRef]
- Li, Y.; Wang, W.J.; Cao, H.; Lu, J.; Wu, C.; Hu, F.Y.; Guo, J.; Zhao, L.; Yang, F.; Zhang, Y.X.; et al. Genetic association of FOXO1A and FOXO3A with longevity trait in Han Chinese populations. Hum. Mol. Genet. 2009, 18, 4897–4904. [Google Scholar] [CrossRef]
- Willcox, B.J.; Donlon, T.A.; He, Q.; Chen, R.; Grove, J.S.; Yano, K.; Masaki, K.H.; Willcox, D.C.; Rodriguez, B.; Curb, J.D. FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. USA 2008, 105, 13987–13992. [Google Scholar] [CrossRef]
- Anselmi, C.V.; Malovini, A.; Roncarati, R.; Novelli, V.; Villa, F.; Condorelli, G.; Bellazzi, R.; Puca, A.A. Association of the FOXO3A locus with extreme longevity in a southern Italian centenar-ian study. Rejuvenation Res. 2009, 12, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Flachsbart, F.; Caliebe, A.; Kleindorp, R.; Blanche, H.; Von Eller-Eberstein, H.; Nikolaus, S.; Schreiber, S.; Nebel, A. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc. Natl. Acad. Sci. USA 2009, 106, 2700–2705. [Google Scholar] [CrossRef] [PubMed]
- Soerensen, M.; Dato, S.; Christensen, K.; McGue, M.; Stevnsner, T.; Bohr, V.A.; Christiansen, L. Replication of an association of variation in the FOXO3A gene with human longevity using both case-control and longitudinal data. Aging Cell 2010, 9, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, M.; Bonafe, M.; Franceschi, C.; Paolisso, G. Insulin/IGF-I-signaling pathway: An evo-lutionarily conserved mechanism of longevity from yeast to humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1064–E1071. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, S.; Mirisola, M.G.; Mazzoni, C. Cell Stress, Metabolic Reprogramming, and Cancer. Front. Oncol. 2018, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; Powers, R.W., 3rd; Steffen, K.K.; Westman, E.A.; Hu, D.; Dang, N.; Kerr, E.O.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005, 310, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- McElwee, J.J.; Schuster, E.; Blanc, E.; Piper, M.D.; Thomas, J.H.; Patel, D.S.; Selman, C.; Withers, D.J.; Thornton, J.M.; Partridge, L.; et al. Evolutionary conservation of regulated lon-gevity assurance mechanisms. Genome Biol. 2007, 8, R132. [Google Scholar] [CrossRef] [PubMed]
- Tullet, J.M.; Hertweck, M.; An, J.H.; Baker, J.; Hwang, J.Y.; Liu, S.; Oliveira, R.P.; Baumeister, R.; Blackwell, T.K. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 2008, 132, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Taormina, G.; Mirisola, M.G. Calorie restriction in mammals and simple model organisms. BioMed Res. Int. 2014, 2014, 308690. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-Aguirre, J.; Wan, J.; et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taormina, G.; Ferrante, F.; Vieni, S.; Grassi, N.; Russo, A.; Mirisola, M.G. Longevity: Lesson from Model Organisms. Genes 2019, 10, 518. https://doi.org/10.3390/genes10070518
Taormina G, Ferrante F, Vieni S, Grassi N, Russo A, Mirisola MG. Longevity: Lesson from Model Organisms. Genes. 2019; 10(7):518. https://doi.org/10.3390/genes10070518
Chicago/Turabian StyleTaormina, Giusi, Federica Ferrante, Salvatore Vieni, Nello Grassi, Antonio Russo, and Mario G. Mirisola. 2019. "Longevity: Lesson from Model Organisms" Genes 10, no. 7: 518. https://doi.org/10.3390/genes10070518
APA StyleTaormina, G., Ferrante, F., Vieni, S., Grassi, N., Russo, A., & Mirisola, M. G. (2019). Longevity: Lesson from Model Organisms. Genes, 10(7), 518. https://doi.org/10.3390/genes10070518