The cGMP Pathway and Inherited Photoreceptor Degeneration: Targets, Compounds, and Biomarkers

 and
and 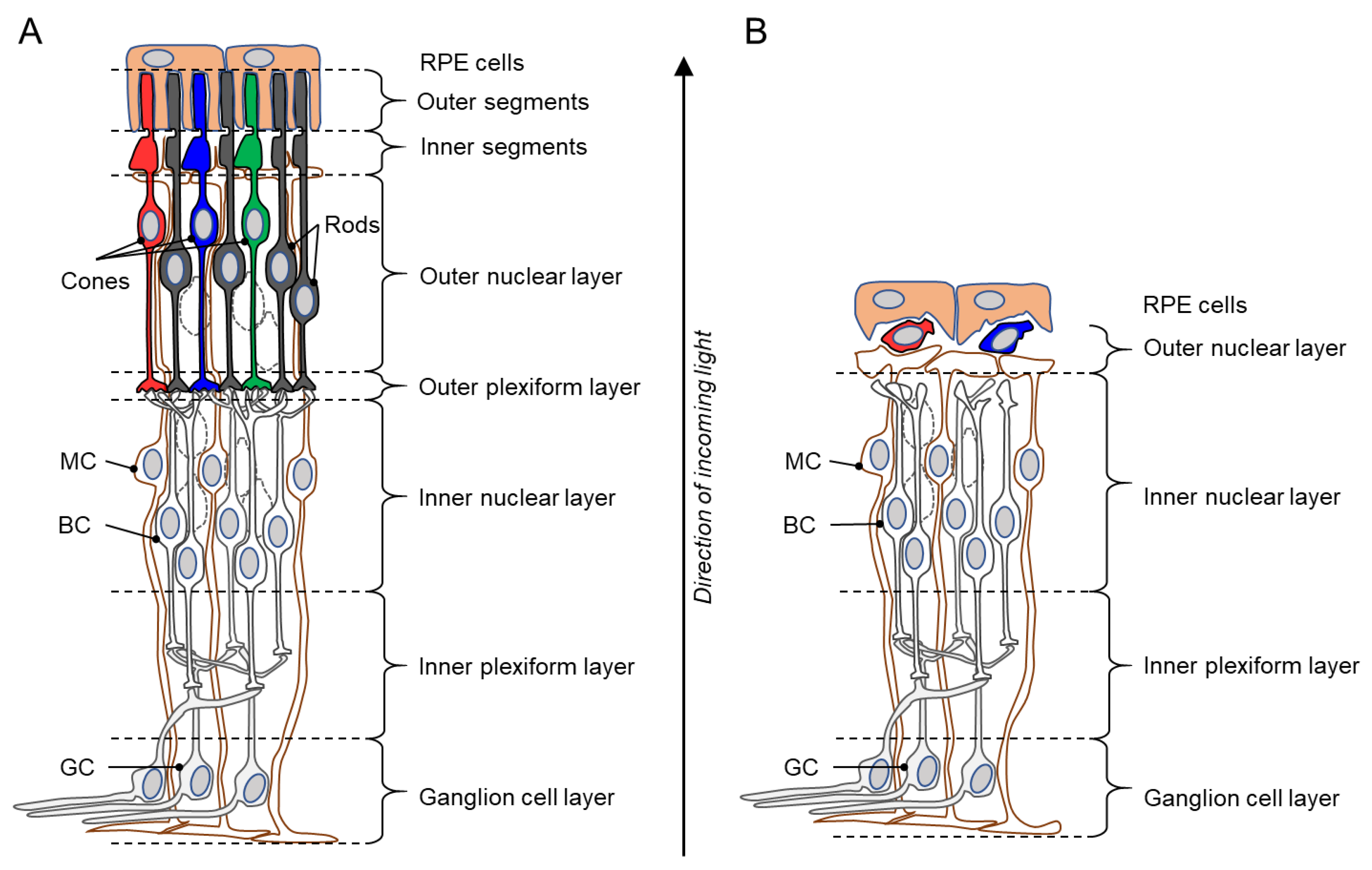{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. The Retina and Inherited Photoreceptor Degeneration
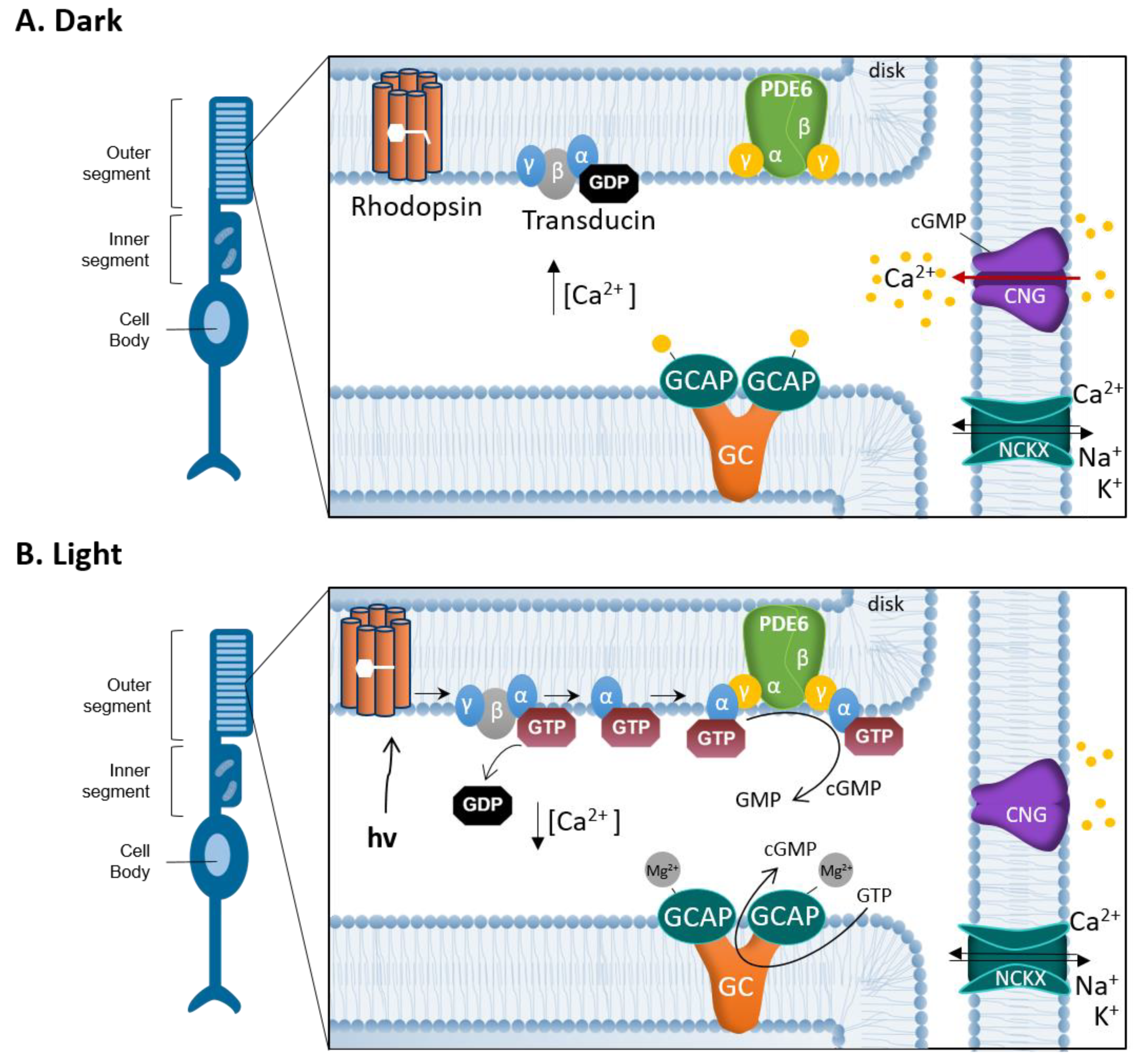
2. cGMP-Signaling in Phototransduction and Degeneration
3. Targeting cGMP-Signaling
3.1. Regulation of Photoreceptor cGMP Synthesis
3.2. The Role of PKG in Normal Physiology
3.3. Effects of PKG Inhibition
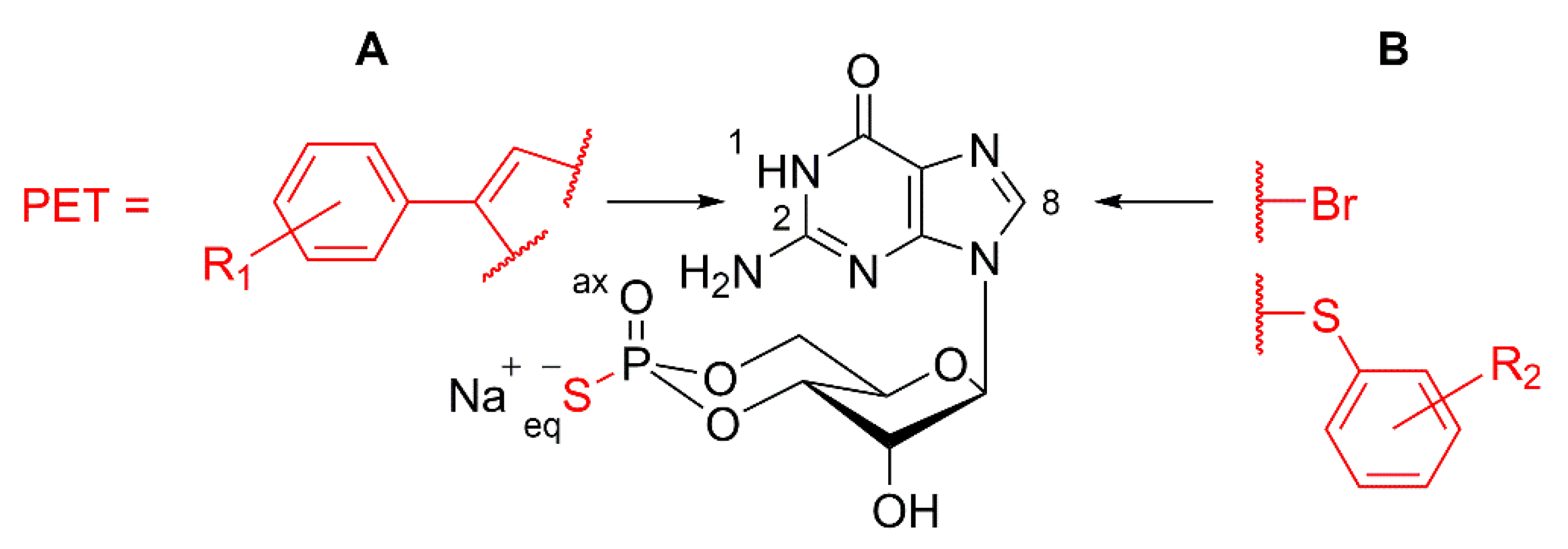
3.4. Design of cGMP Analogues to Inhibit PKG
4. Pre-Clinical and Clinical Biomarkers for Inherited Retinal Degeneration
4.1. Review of Recent Retinal Biomarker Developments
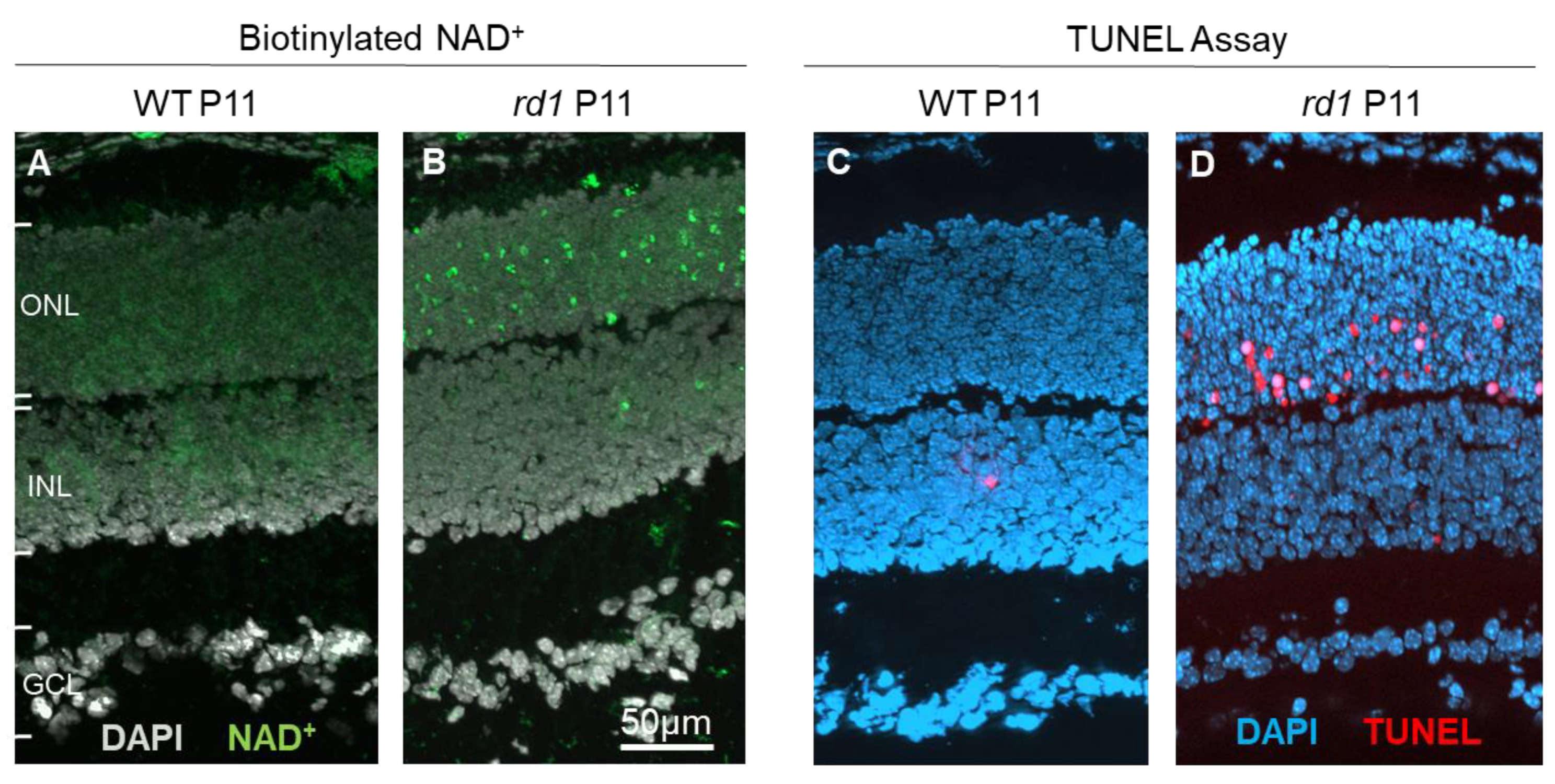
4.2. Using the cGMP Pathway for Biomarker Development
5. Delivery of Compounds to the Retinal Photoreceptors
5.1. Drug Delivery Systems (DDSs) for the Retina
5.2. Retinal Drug Delivery: Local vs. Systemic Administration
6. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoon, M.; Okawa, H.; Della Santina, L.; Wong, R.O. Functional architecture of the retina: Development and disease. Prog. Retin. Eye Res. 2014, 42, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Chizzolini, M.; Galan, A.; Milan, E.; Sebastiani, A.; Costagliola, C.; Parmeggiani, F. Good epidemiologic practice in retinitis pigmentosa: From phenotyping to biobanking. Curr. Genom. 2011, 12, 260–266. [Google Scholar] [CrossRef]
- Kennan, A.; Aherne, A.; Humphries, P. Light in retinitis pigmentosa. Trends Genet. 2005, 21, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C.P. Cone rod dystrophies. Orphanet. J. Rare. Dis. 2007, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, D.; Sahaboglu, A.; Kaur, J.; Mencl, S.; Zrenner, E.; Ueffing, M.; Arango-Gonzalez, B.; Paquet-Durand, F. Neuroprotective Strategies for the Treatment of Inherited Photoreceptor Degeneration. Curr. Mol. Med. 2012, 12, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Gargini, C.; Terzibasi, E.; Mazzoni, F.; Strettoi, E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: A morphological and ERG study. J. Comp. Neurol. 2007, 500, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Carter-Dawson, L.D.; LaVail, M.M.; Sidman, R.L. Differential effect of the rd mutation on rods and cones in the mouse retina. Investig. Ophthalmol. Vis. Sci. 1978, 17, 489–498. [Google Scholar]
- Stryer, L. Cyclic GMP cascade of vision. Annu. Rev. Neurosci. 1986, 9, 87–119. [Google Scholar] [CrossRef]
- Farber, D.B.; Lolley, R.N. Cyclic guanosine monophosphate: Elevation in degenerating photoreceptor cells of the C3H mouse retina. Science 1974, 186, 449–451. [Google Scholar] [CrossRef]
- Arango-Gonzalez, B.; Trifunovic, D.; Sahaboglu, A.; Kranz, K.; Michalakis, S.; Farinelli, P.; Koch, S.; Koch, F.; Cottet, S.; Janssen Bienhold, U.; et al. Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PLoS ONE 2014, 9, e112142. [Google Scholar] [CrossRef] [PubMed]
- Field, G.D.; Uzzell, V.; Chichilnisky, E.J.; Rieke, F. Temporal resolution of single-photon responses in primate rod photoreceptors and limits imposed by cellular noise. J. Neurophysiol. 2019, 121, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Becirovic, E.; Biel, M. Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. Int. J. Mol. Sci. 2018, 19, 749. [Google Scholar] [CrossRef] [PubMed]
- Vinberg, F.; Chen, J.; Kefalov, V.J. Regulation of calcium homeostasis in the outer segments of rod and cone photoreceptors. Prog. Retin. Eye Res. 2018, 67, 87–101. [Google Scholar] [CrossRef]
- Kulkarni, M.; Trifunovic, D.; Schubert, T.; Euler, T.; Paquet-Durand, F. Calcium dynamics change in degenerating cone photoreceptors. Hum. Mol. Genet. 2016, ddw219. [Google Scholar] [CrossRef] [PubMed]
- Lolley, R.N.; Farber, D.B.; Rayborn, M.E.; Hollyfield, J.G. Cyclic GMP accumulation causes degeneration of photoreceptor cells: Simulation of an inherited disease. Science 1977, 196, 664–666. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, M.E.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat. Genet. 1993, 4, 130–134. [Google Scholar] [CrossRef]
- Huang, S.H.; Pittler, S.J.; Huang, X.; Oliveira, L.; Berson, E.L.; Dryja, T.P. Autosomal recessive retinitis pigmentosa caused by mutations in the alpha subunit of rod cGMP phosphodiesterase. Nat. Genet. 1995, 11, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Paquet-Durand, F.; Beck, S.; Michalakis, S.; Goldmann, T.; Huber, G.; Muhlfriedel, R.; Trifunovic, D.; Fischer, M.D.; Fahl, E.; Duetsch, G.; et al. A key role for cyclic nucleotide gated (CNG) channels in cGMP-related retinitis pigmentosa. Hum. Mol. Genet. 2011, 20, 941–947. [Google Scholar] [CrossRef]
- Ramamurthy, V.; Niemi, G.A.; Reh, T.A.; Hurley, J.B. Leber congenital amaurosis linked to AIPL1: A mouse model reveals destabilization of cGMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 2004, 101, 13897–13902. [Google Scholar] [CrossRef]
- Sato, S.; Peshenko, I.V.; Olshevskaya, E.V.; Kefalov, V.J.; Dizhoor, A.M. GUCY2D Cone-Rod Dystrophy-6 Is a “Phototransduction Disease” Triggered by Abnormal Calcium Feedback on Retinal Membrane Guanylyl Cyclase 1. J. Neurosci. 2018, 38, 2990–3000. [Google Scholar] [CrossRef] [PubMed]
- Paquet-Durand, F.; Hauck, S.M.; van Veen, T.; Ueffing, M.; Ekstrom, P. PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J. Neurochem. 2009, 108, 796–810. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.F.; Moritz, O.L.; Williams, D.S. Molecular basis for photoreceptor outer segment architecture. Prog. Retin. Eye Res. 2016, 55, 52–81. [Google Scholar] [CrossRef] [PubMed]
- Marigo, V.; Ekström, P.; Schwede, F.; Rentsch, A.; Paquet-Durand, F. Modulation of cGMP-signaling to prevent retinal degeneration. In Therapies for Retinal Degeneration: Targeting Common Processes; De la Rosa, E.J., Cotter, T.G., Eds.; Royal Society of Chemistry: Cambridge, UK, 2019; p. 88. [Google Scholar]
- Feil, R.; Kemp-Harper, B. cGMP signaling: From bench to bedside. Conference on cGMP generators, effectors and therapeutic implications. EMBO Rep. 2006, 7, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Pannbacker, R.G.; Fleischman, D.E.; Reed, D.W. Cyclic nucleotide phosphodiesterase: High activity in a mammalian photoreceptor. Science 1972, 175, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Roseman, G.; Peshenko, I.; Manchala, G.; Cudia, D.; Dizhoor, A.M.; Millhauser, G.; Ames, J.B. Retinal guanylyl cyclase activating protein 1 forms a functional dimer. PLoS ONE 2018, 13, e0193947. [Google Scholar] [CrossRef]
- Dizhoor, A.M.; Olshevskaya, E.V.; Peshenko, I.V. The R838S Mutation in Retinal Guanylyl Cyclase 1 (RetGC1) Alters Calcium Sensitivity of cGMP Synthesis in the Retina and Causes Blindness in Transgenic Mice. J. Biol. Chem. 2016, 291, 24504–24516. [Google Scholar] [CrossRef]
- Makino, C.L.; Wen, X.H.; Olshevskaya, E.V.; Peshenko, I.V.; Savchenko, A.B.; Dizhoor, A.M. Enzymatic relay mechanism stimulates cyclic GMP synthesis in rod photoresponse: Biochemical and physiological study in guanylyl cyclase activating protein 1 knockout mice. PLoS ONE 2012, 7, e47637. [Google Scholar] [CrossRef]
- Olshevskaya, E.V.; Ermilov, A.N.; Dizhoor, A.M. Factors that affect regulation of cGMP synthesis in vertebrate photoreceptors and their genetic link to human retinal degeneration. Mol. Cell. Biochem. 2002, 230, 139–147. [Google Scholar] [CrossRef]
- Fox, D.A.; Poblenz, A.T.; He, L. Calcium overload triggers rod photoreceptor apoptotic cell death in chemical-induced and inherited retinal degenerations. Ann. N. Y. Acad. Sci. 1999, 893, 282–285. [Google Scholar] [CrossRef]
- Frasson, M.; Sahel, J.A.; Fabre, M.; Simonutti, M.; Dreyfus, H.; Picaud, S. Retinitis pigmentosa: Rod photoreceptor rescue by a calcium-channel blocker in the rd mouse. Nat. Med. 1999, 5, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Nakazawa, M.; Mizukoshi, S. Systemic administration of nilvadipine delays photoreceptor degeneration of heterozygous retinal degeneration slow (rds) mouse. Exp. Eye Res. 2008, 86, 60–69. [Google Scholar] [CrossRef]
- Pearce-Kelling, S.E.; Aleman, T.S.; Nickle, A.; Laties, A.M.; Aguirre, G.D.; Jacobson, S.G.; Acland, G.M. Calcium channel blocker D-cis-diltiazem does not slow retinal degeneration in the PDE6B mutant rcd1 canine model of retinitis pigmentosa. Mol. Vis. 2001, 7, 42–47. [Google Scholar]
- Pawlyk, B.S.; Li, T.; Scimeca, M.S.; Sandberg, M.A.; Berson, E.L. Absence of photoreceptor rescue with D-cis-diltiazem in the rd mouse. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1912–1915. [Google Scholar]
- Barabas, P.; Cutler, P.C.; Krizaj, D. Do calcium channel blockers rescue dying photoreceptors in the Pde6b (rd1) mouse? Adv. Exp. Med. Biol. 2010, 664, 491–499. [Google Scholar] [CrossRef]
- Hofmann, F.; Feil, R.; Kleppisch, T.; Schlossmann, J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol. Rev. 2006, 86, 1–23. [Google Scholar] [CrossRef]
- Hofmann, F.; Bernhard, D.; Lukowski, R.; Weinmeister, P. cGMP regulated protein kinases (cGK). Handb. Exp. Pharmacol. 2009, 137–162. [Google Scholar]
- Piwkowska, A.; Rogacka, D.; Audzeyenka, I.; Kasztan, M.; Angielski, S.; Jankowski, M. Intracellular calcium signaling regulates glomerular filtration barrier permeability: The role of the PKGIalpha-dependent pathway. FEBS Lett. 2016, 590, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.; Zimmermann, P.; Knorn, A.; Brummer, S.; Schlossmann, J.; Hofmann, F.; Feil, R. Distribution of cGMP-dependent protein kinase type I and its isoforms in the mouse brain and retina. Neuroscience 2005, 135, 863–868. [Google Scholar] [CrossRef]
- Ekstrom, P.A.; Ueffing, M.; Zrenner, E.; Paquet-Durand, F. Novel in situ activity assays for the quantitative molecular analysis of neurodegenerative processes in the retina. Curr. Med. Chem. 2014, 21, 3478–3493. [Google Scholar] [CrossRef]
- Pfeifer, A.; Klatt, P.; Massberg, S.; Ny, L.; Sausbier, M.; Hirneiss, C.; Wang, G.X.; Korth, M.; Aszodi, A.; Andersson, K.E.; et al. Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 1998, 17, 3045–3051. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Bernhard, D.; Lukowski, R.; Weinmeister, P.; Worner, R.; Wegener, J.W.; Valtcheva, N.; Feil, S.; Schlossmann, J.; Hofmann, F.; et al. Rescue of cGMP kinase I knockout mice by smooth muscle specific expression of either isozyme. Circ. Res. 2007, 101, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Jaumann, M.; Dettling, J.; Gubelt, M.; Zimmermann, U.; Gerling, A.; Paquet-Durand, F.; Feil, S.; Wolpert, S.; Franz, C.; Varakina, K.; et al. cGMP-Prkg1 signaling and Pde5 inhibition shelter cochlear hair cells and hearing function. Nat. Med. 2012, 18, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Oster, H.; Werner, C.; Magnone, M.C.; Mayser, H.; Feil, R.; Seeliger, M.W.; Hofmann, F.; Albrecht, U. cGMP-dependent protein kinase II modulates mPer1 and mPer2 gene induction and influences phase shifts of the circadian clock. Curr. Biol. 2003, 13, 725–733. [Google Scholar] [CrossRef]
- Fallahian, F.; Karami-Tehrani, F.; Salami, S.; Aghaei, M. Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor-positive and -negative breast cancer cell lines. FEBS J. 2011, 278, 3360–3369. [Google Scholar] [CrossRef] [PubMed]
- Browning, D.D. Protein kinase G as a therapeutic target for the treatment of metastatic colorectal cancer. Expert Opin. Ther. Targets 2008, 12, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.L.; Wong, J.C.; Johlfs, M.G.; Tsang, B.K.; Fiscus, R.R. Protein kinase G type Ialpha activity in human ovarian cancer cells significantly contributes to enhanced Src activation and DNA synthesis/cell proliferation. Mol. Cancer Res. 2010, 8, 578–591. [Google Scholar] [CrossRef]
- Canals, S.; Casarejos, M.J.; de, B.S.; Rodriguez-Martin, E.; Mena, M.A. Nitric oxide triggers the toxicity due to glutathione depletion in midbrain cultures through 12-lipoxygenase. J. Biol. Chem. 2003, 278, 21542–21549. [Google Scholar] [CrossRef]
- Canzoniero, L.M.; Adornetto, A.; Secondo, A.; Magi, S.; Dell’aversano, C.; Scorziello, A.; Amoroso, S.; Di, R.G. Involvement of the nitric oxide/protein kinase G pathway in polychlorinated biphenyl-induced cell death in SH-SY 5Y neuroblastoma cells. J. Neurosci. Res. 2006, 84, 692–697. [Google Scholar] [CrossRef]
- Wang, T.; Tsang, S.H.; Chen, J. Two pathways of rod photoreceptor cell death induced by elevated cGMP. Hum. Mol. Genet. 2017, 26, 2299–2306. [Google Scholar] [CrossRef]
- Dostmann, W.R.; Taylor, M.S.; Nickl, C.K.; Brayden, J.E.; Frank, R.; Tegge, W.J. Highly specific, membrane-permeant peptide blockers of cGMP-dependent protein kinase Ialpha inhibit NO-induced cerebral dilation. Proc. Natl. Acad. Sci. USA 2000, 97, 14772–14777. [Google Scholar] [CrossRef] [PubMed]
- Wolfertstetter, S.; Huettner, J.P.; Schlossmann, J. cGMP-Dependent Protein Kinase Inhibitors in Health and Disease. Pharmaceuticals (Basel) 2013, 6, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Falk, J.; Drinjakovic, J.; Leung, K.M.; Dwivedy, A.; Regan, A.G.; Piper, M.; Holt, C.E. Electroporation of cDNA/Morpholinos to targeted areas of embryonic CNS in Xenopus. BMC. Dev. Biol. 2007, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Babykutty, S.; Suboj, P.; Srinivas, P.; Nair, A.S.; Chandramohan, K.; Gopala, S. Insidious role of nitric oxide in migration/invasion of colon cancer cells by upregulating MMP-2/9 via activation of cGMP-PKG-ERK signaling pathways. Clin. Exp. Metastasis 2012, 29, 471–492. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, M.; Glazova, M.; Gambaryan, S.; Vollkommer, T.; Butt, E.; Bader, B.; Heermeier, K.; Lincoln, T.M.; Walter, U.; Palmetshofer, A. KT5823 inhibits cGMP-dependent protein kinase activity in vitro but not in intact human platelets and rat mesangial cells. J. Biol. Chem. 2000, 275, 33536–33541. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, H.; Kobayashi, R. Pharmacology of protein kinase inhibitors. Annu. Rev. Pharmacol. Toxicol. 1992, 32, 377–397. [Google Scholar] [CrossRef] [PubMed]
- Gambaryan, S.; Butt, E.; Kobsar, A.; Geiger, J.; Rukoyatkina, N.; Parnova, R.; Nikolaev, V.O.; Walter, U. The oligopeptide DT-2 is a specific PKG I inhibitor only in vitro, not in living cells. Br. J. Pharmacol. 2012, 167, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, S.M.; Vaandrager, A.B.; Smolenski, A.; Walter, U.; De Jonge, H.R. Distinct and specific functions of cGMP-dependent protein kinases. Trends Biochem. Sci. 1997, 22, 307–312. [Google Scholar] [CrossRef]
- Schwede, F.; Maronde, E.; Genieser, H.; Jastorff, B. Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol. Ther. 2000, 87, 199–226. [Google Scholar] [CrossRef]
- Hoffmann, D.; Rentsch, A.; Vighi, E.; Bertolotti, E.; Comitato, A.; Schwede, F.; Genieser, H.G.; Marigo, V. New dimeric cGMP analogues reduce proliferation in three colon cancer cell lines. Eur. J. Med. Chem. 2017, 141, 61–72. [Google Scholar] [CrossRef]
- Vighi, E.; Rentsch, A.; Henning, P.; Comitato, A.; Hoffmann, D.; Bertinetti, D.; Bertolotti, E.; Schwede, F.; Herberg, F.W.; Genieser, H.G.; et al. New cGMP analogues restrain proliferation and migration of melanoma cells. Oncotarget 2018, 9, 5301–5320. [Google Scholar] [CrossRef]
- Zhao, J.; Trewhella, J.; Corbin, J.; Francis, S.; Mitchell, R.; Brushia, R.; Walsh, D. Progressive cyclic nucleotide-induced conformational changes in the cGMP-dependent protein kinase studied by small angle X-ray scattering in solution. J. Biol. Chem. 1997, 272, 31929–31936. [Google Scholar] [CrossRef]
- Zimmerman, A.L.; Yamanaka, G.; Eckstein, F.; Baylor, D.A.; Stryer, L. Interaction of hydrolysis-resistant analogs of cyclic GMP with the phosphodiesterase and light-sensitive channel of retinal rod outer segments. Proc. Natl. Acad. Sci. USA 1985, 82, 8813–8817. [Google Scholar] [CrossRef] [PubMed]
- Vighi, E.; Trifunovic, D.; Veiga-Crespo, P.; Rentsch, A.; Hoffmann, D.; Sahaboglu, A.; Strasser, T.; Kulkarni, M.; Bertolotti, E.; van den Heuvel, A.; et al. Combination of cGMP analogue and drug delivery system provides functional protection in hereditary retinal degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E2997–E3006. [Google Scholar] [CrossRef] [PubMed]
- Butt, E.; Pohler, D.; Genieser, H.G.; Huggins, J.P.; Bucher, B. Inhibition of cyclic GMP-dependent protein kinase-mediated effects by (Rp)-8-bromo-PET-cyclic GMPS. Br. J. Pharmacol. 1995, 116, 3110–3116. [Google Scholar] [CrossRef] [PubMed]
- Butt, E.; Eigenthaler, M.; Genieser, H.G. (Rp)-8-pCPT-cGMPS, a novel cGMP-dependent protein kinase inhibitor. Eur. J. Pharmacol. 1994, 269, 265–268. [Google Scholar] [CrossRef]
- Nguyen, C.T.O.; Hui, F.; Charng, J.; Velaedan, S.; van Koeverden, A.K.; Lim, J.K.H.; He, Z.; Wong, V.H.Y.; Vingrys, A.J.; Bui, B.V.; et al. Retinal biomarkers provide "insight" into cortical pharmacology and disease. Pharmacol. Ther. 2017, 175, 151–177. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, M.F.; Normando, E.M.; Cardoso, M.J.; Miodragovic, S.; Jeylani, S.; Davis, B.M.; Guo, L.; Ourselin, S.; A’Hern, R.; Bloom, P.A. Real-time imaging of single neuronal cell apoptosis in patients with glaucoma. Brain 2017, 140, 1757–1767. [Google Scholar] [CrossRef]
- Smith, B.A.; Smith, B.D. Biomarkers and molecular probes for cell death imaging and targeted therapeutics. Bioconjug. Chem. 2012, 23, 1989–2006. [Google Scholar] [CrossRef]
- Camara, M.-F.d.l.; Salom, D.; Sequedo, M.D.; Hervas, D.; Marin-Lambies, C.; Aller, E.; Jaijo, T.; Diaz-Llopis, M.; Millan, J.M.; Rodrigo, R. Altered antioxidant-oxidant status in the aqueous humor and peripheral blood of patients with retinitis pigmentosa. PLoS ONE 2013, 8, e74223. [Google Scholar] [CrossRef]
- Kjellstrom, U.; Veiga-Crespo, P.; Andreasson, S.; Ekstrom, P. Increased Plasma cGMP in a Family With Autosomal Recessive Retinitis Pigmentosa Due to Homozygous Mutations in the PDE6A Gene. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6048–6057. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.Y.; Cringle, S.J. Retinal degeneration and local oxygen metabolism. Exp. Eye Res. 2005, 80, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Cleveland, D.W. Neuronal intermediate filaments. Annu. Rev. Neurosci. 1996, 19, 187–217. [Google Scholar] [CrossRef]
- Petzold, A.; Junemann, A.; Rejdak, K.; Zarnowski, T.; Thaler, S.; Grieb, P.; Kruse, F.E.; Zrenner, E.; Rejdak, R. A novel biomarker for retinal degeneration: Vitreous body neurofilament proteins. J. Neural. Transm. (Vienna) 2009, 116, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, M.F.; Guo, L.; Coxon, K.M.; Duggan, J.; Nizari, S.; Normando, E.M.; Sensi, S.L.; Sillito, A.M.; Fitzke, F.W.; Salt, T.E.; et al. Imaging multiple phases of neurodegeneration: A novel approach to assessing cell death in vivo. Cell Death. Dis. 2010, 1, e3. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Pelluz, J.; Arango-Gonzalez, B.; Kustermann, S.; Romero, F.J.; van Veen, T.; Zrenner, E.; Ekstrom, P.; Paquet-Durand, F. Photoreceptor cell death mechanisms in inherited retinal degeneration. Mol. Neurobiol. 2008, 38, 253–269. [Google Scholar] [CrossRef]
- Ma, H.; Butler, M.R.; Thapa, A.; Belcher, J.; Yang, F.; Baehr, W.; Biel, M.; Michalakis, S.; Ding, X.Q. cGMP/Protein Kinase G Signaling Suppresses Inositol 1,4,5-Trisphosphate Receptor Phosphorylation and Promotes Endoplasmic Reticulum Stress in Photoreceptors of Cyclic Nucleotide-gated Channel-deficient Mice. J. Biol. Chem. 2015, 290, 20880–20892. [Google Scholar] [CrossRef]
- Sancho-Pelluz, J.; Alavi, M.; Sahaboglu, A.; Kustermann, S.; Farinelli, P.; Azadi, S.; van Veen, T.; Romero, F.J.; Paquet-Durand, F.; Ekstrom, P. Excessive HDAC activation is critical for neurodegeneration in the rd1 mouse. Cell Death Dis. 2010, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Paquet-Durand, F.; Johnson, L.; Ekstrom, P. Calpain activity in retinal degeneration. J. Neurosci. Res. 2007, 85, 693–702. [Google Scholar] [CrossRef]
- Farinelli, P.; Perera, A.; Arango-Gonzalez, B.; Trifunovic, D.; Wagner, M.; Carell, T.; Biel, M.; Zrenner, E.; Michalakis, S.; Paquet-Durand, F.; et al. DNA methylation and differential gene regulation in photoreceptor cell death. Cell Death Dis. 2014, 5, e1558. [Google Scholar] [CrossRef] [PubMed]
- Paquet-Durand, F.; Silva, J.; Talukdar, T.; Johnson, L.E.; Azadi, S.; van Veen, T.; Ueffing, M.; Hauck, S.M.; Ekstrom, P.A. Excessive activation of poly(ADP-ribose) polymerase contributes to inherited photoreceptor degeneration in the retinal degeneration 1 mouse. J. Neurosci. 2007, 27, 10311–10319. [Google Scholar] [CrossRef] [PubMed]
- Sahaboglu, A.; Barth, M.; Secer, E.; Amo, E.M.; Urtti, A.; Arsenijevic, Y.; Zrenner, E.; Paquet-Durand, F. Olaparib significantly delays photoreceptor loss in a model for hereditary retinal degeneration. Sci. Rep. 2016, 6, 39537. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Ozaki, E.; Humphries, P. Systemic delivery of therapeutics to neuronal tissues: A barrier modulation approach. Expert. Opin. Drug Deliv. 2010, 7, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.; Kurup, S.K.; Wang, R.C.; Foster, C.S.; Noronha, G.; Nguyen, Q.D.; Do, D.V.; Team, D.S. Suprachoroidal Injection of Triamcinolone Acetonide, CLS-TA, for macular edema due to noninfectious uveitis: A Randomized, Phase 2 Study (DOGWOOD). Retina 2018. [Google Scholar] [CrossRef] [PubMed]
- Ochakovski, G.A.; Peters, T.; Michalakis, S.; Wilhelm, B.; Wissinger, B.; Biel, M.; Bartz-Schmidt, K.U.; Fischer, M.D.; Consortium, R.-C. Subretinal Injection for Gene Therapy Does Not Cause Clinically Significant Outer Nuclear Layer Thinning in Normal Primate Foveae. Invest. Ophthalmol. Vis. Sci. 2017, 58, 4155–4160. [Google Scholar] [CrossRef] [PubMed]
- Ohira, A.; Hara, K.; Johannesson, G.; Tanito, M.; Asgrimsdottir, G.M.; Lund, S.H.; Loftsson, T.; Stefansson, E. Topical dexamethasone gamma-cyclodextrin nanoparticle eye drops increase visual acuity and decrease macular thickness in diabetic macular oedema. Acta Ophthalmol. 2015, 93, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.H.; Krohne, T.U.; Charbel, I.P.; Liu, Z.; Holz, F.G. Routes for Drug Delivery to the Eye and Retina: Intravitreal Injections. Dev. Ophthalmol. 2016, 55, 63–70. [Google Scholar] [CrossRef] [PubMed]
- MacLaren, R.E.; Groppe, M.; Barnard, A.R.; Cottriall, C.L.; Tolmachova, T.; Seymour, L.; Clark, K.R.; During, M.J.; Cremers, F.P.; Black, G.C.; et al. Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. Lancet 2014, 383, 1129–1137. [Google Scholar] [CrossRef]
- Apte, R.S. Gene Therapy for Retinal Degeneration. Cell 2018, 173, 5. [Google Scholar] [CrossRef]
- Solomon, S.D.; Lindsley, K.; Vedula, S.S.; Krzystolik, M.G.; Hawkins, B.S. Anti-vascular endothelial growth factor for neovascular age-related macular degeneration. Cochrane Database Syst. Rev. 2019, 3, CD005139. [Google Scholar] [CrossRef] [PubMed]
- Himawan, E.; Ekström, P.; Buzgo, M.; Gaillard, P.; Stefansson, E.; Marigo, V.; Loftsson, T.; Paquet-Durand, F. Drug delivery to retinal photoreceptors. Drug Discov. Today 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Del Amo, E.M.; Rimpela, A.K.; Heikkinen, E.; Kari, O.K.; Ramsay, E.; Lajunen, T.; Schmitt, M.; Pelkonen, L.; Bhattacharya, M.; Richardson, D.; et al. Pharmacokinetic aspects of retinal drug delivery. Prog. Retin. Eye Res. 2017, 57, 134–185. [Google Scholar] [CrossRef] [PubMed]
- Reichel, F.F.; Peters, T.; Wilhelm, B.; Biel, M.; Ueffing, M.; Wissinger, B.; Bartz-Schmidt, K.U.; Klein, R.; Michalakis, S.; Fischer, M.D.; et al. Humoral Immune Response After Intravitreal But Not After Subretinal AAV8 in Primates and Patients. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1910–1915. [Google Scholar] [CrossRef] [PubMed]
- Rayess, N.; Rahimy, E.; Shah, C.P.; Wolfe, J.D.; Chen, E.; DeCroos, F.C.; Storey, P.; Garg, S.J.; Hsu, J. Incidence and clinical features of post-injection endophthalmitis according to diagnosis. Br. J. Ophthalmol. 2015, 100, 1058–1061. [Google Scholar] [CrossRef]
- Coulson, R.; Baraniak, J.; Stec, W.J.; Jastorff, B. Transport and metabolism of N6- and C8-substituted analogs of adenosine 3’,5’-cyclic monophosphate and adenosine 3’5’-cyclic phosphorothioate by the isolated perfused rat kidney. Life Sci. 1983, 32, 1489–1498. [Google Scholar] [CrossRef]
- Maussang, D.; Rip, J.; van Kregten, J.; van den Heuvel, A.; van der Pol, S.; van der Boom, B.; Reijerkerk, A.; Chen, L.; de Boer, M.; Gaillard, P.; et al. Glutathione conjugation dose-dependently increases brain-specific liposomal drug delivery in vitro and in vivo. Drug Discov. Today Technol. 2016, 20, 59–69. [Google Scholar] [CrossRef]
- Williams, M.L.; Coleman, J.E.; Haire, S.E.; Aleman, T.S.; Cideciyan, A.V.; Sokal, I.; Palczewski, K.; Jacobson, S.G.; Semple-Rowland, S.L. Lentiviral expression of retinal guanylate cyclase-1 (RetGC1) restores vision in an avian model of childhood blindness. PLoS Med. 2006, 3, e201. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolone, A.; Belhadj, S.; Rentsch, A.; Schwede, F.; Paquet-Durand, F. The cGMP Pathway and Inherited Photoreceptor Degeneration: Targets, Compounds, and Biomarkers. Genes 2019, 10, 453. https://doi.org/10.3390/genes10060453
Tolone A, Belhadj S, Rentsch A, Schwede F, Paquet-Durand F. The cGMP Pathway and Inherited Photoreceptor Degeneration: Targets, Compounds, and Biomarkers. Genes. 2019; 10(6):453. https://doi.org/10.3390/genes10060453
Chicago/Turabian StyleTolone, Arianna, Soumaya Belhadj, Andreas Rentsch, Frank Schwede, and François Paquet-Durand. 2019. "The cGMP Pathway and Inherited Photoreceptor Degeneration: Targets, Compounds, and Biomarkers" Genes 10, no. 6: 453. https://doi.org/10.3390/genes10060453
APA StyleTolone, A., Belhadj, S., Rentsch, A., Schwede, F., & Paquet-Durand, F. (2019). The cGMP Pathway and Inherited Photoreceptor Degeneration: Targets, Compounds, and Biomarkers. Genes, 10(6), 453. https://doi.org/10.3390/genes10060453