Characterization of the Complete Mitochondrial Genome of a Whipworm Trichuris skrjabini (Nematoda: Trichuridae)

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Parasite Collection and Isolation of DNA
2.2. Trichuris skrjabini ITS-2 Amplification
2.3. Amplification of Short and Long Fragments by PCR
2.4. Gene Annotation and Sequence Analysis
2.5. Phylogenetic Analysis
3. Results
3.1. ITS-2 Analysis
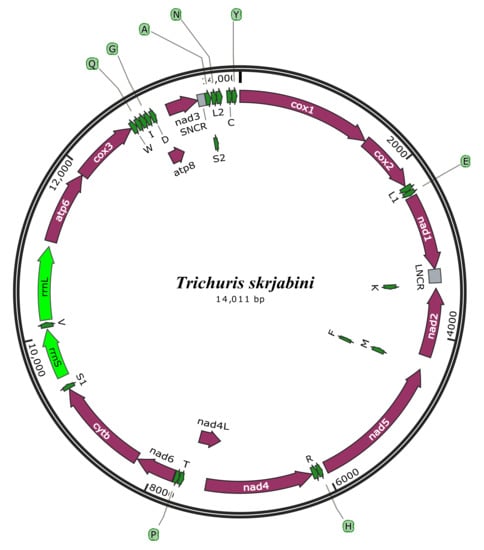
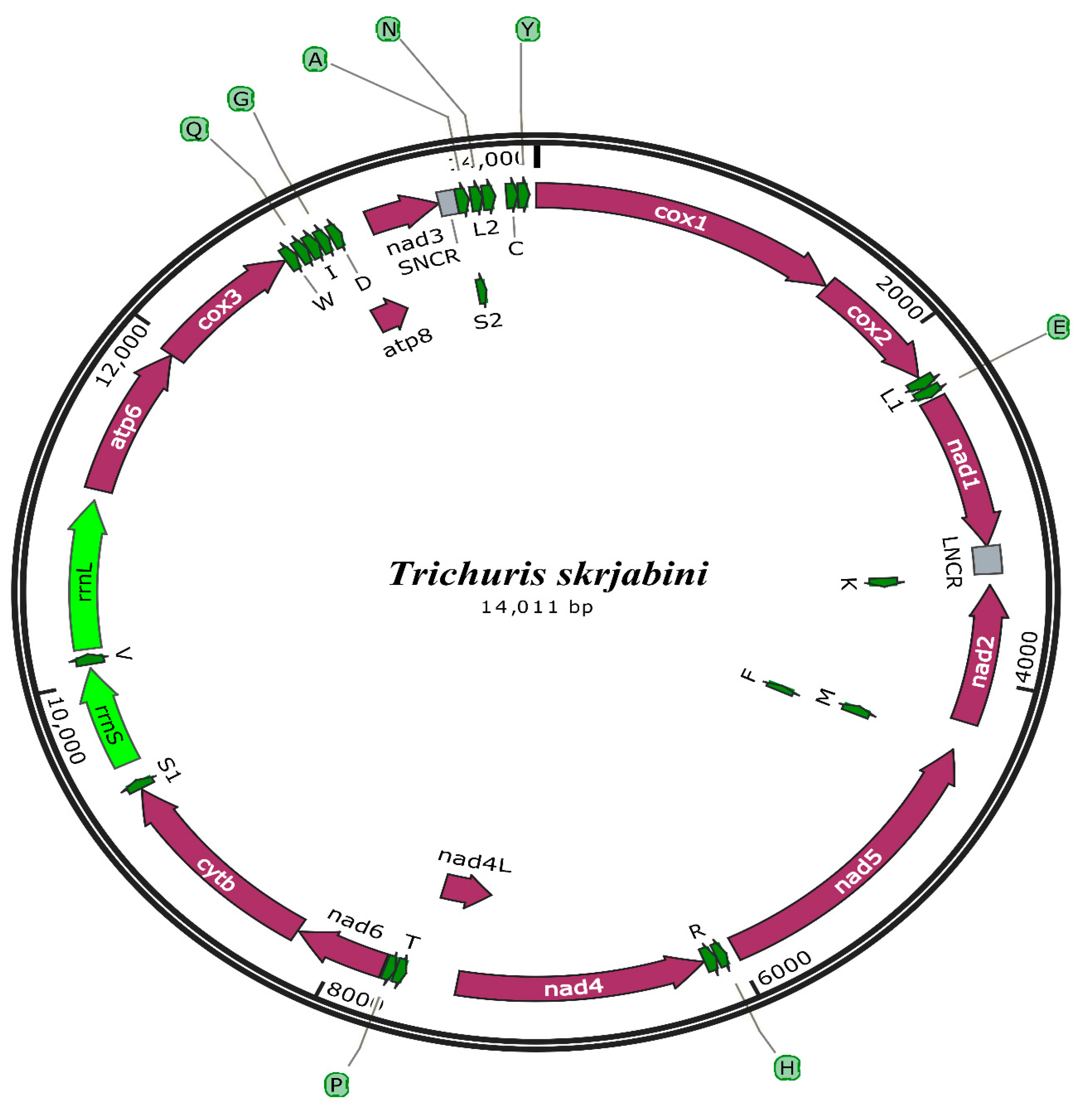
3.2. mt Genome Features
3.3. Annotation
3.4. Phylogenetic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hotez, P.J.; Fenwick, A.; Savioli, L.; Molyneux, D.H. Rescuing the bottom billion through control of neglected tropical diseases. Lancet 2009, 373, 1570–1575. [Google Scholar] [CrossRef]
- Pullan, L.R.; Jennifer, L.S.; Rashmi, J.; Simon, J.B. Global numbers of infection and disease burden of soil transmitted helminth infections in 2010. Parasit. Vectors. 2014, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, R.J.; McGrane, P.; Powell, C.; Grantham-McGregor, S. Effects of a parasitic infection on cognitive functioning. J. Exp. Psychol. Appl. 1997, 3, 67–76. [Google Scholar] [CrossRef]
- Bethony, J.; Brooker, S.; Albonico, M.; Geiger, S.M.; Loukas, A.; Diemert, D.; Hotez, P.J. Soil-transmitted helminth infections: Ascariasis, trichuriasis, and hookworm. Lancet 2006, 367, 1521–1532. [Google Scholar] [CrossRef]
- Hotez, P.J.; Savioli, L.; Fenwick, A. Neglected tropical diseases of the Middle East and North Africa: Review of their prevalence, distribution, and opportunities for control. PLoS Negl. Trop. Dis. 2012, 6, e1475. [Google Scholar] [CrossRef] [PubMed]
- Cutillas, C.; De Rojas, M.; Ariza, C.; Ubeda, J.M.; Guevara, D. Molecular identification of Trichuris vulpis and Trichuris suis isolated from different hosts. Parasitol. Res. 2007, 100, 383–389. [Google Scholar] [CrossRef]
- Khalafalla, R.E.; Elseify, M.A.; Elbahy, N.M. Seasonal prevalence of gastrointestinal nematode parasites of sheep in Northern region of Nile Delta, Egypt. Parasitol. Res. 2011, 108, 337–340. [Google Scholar] [CrossRef]
- Bundy, D.A.P.; Cooper, E.S. Trichuris and Trichuriasis in Humans. Adv. Parasitol. 1989, 28, 107–173. [Google Scholar]
- Jex, A.R.; Lim, Y.A.L.; Bethony, J.M.; Hotez, P.J.; Young, N.D.; Gasser, R.B. Soil-transmitted helminths of humans in Southeast Asia-towards integrated control. Adv. Parasitol. 2011, 74, 231–235. [Google Scholar]
- Roepstorff, A.; Mejer, H.; Nejsum, P.; Thamsborg, S.M. Helminth parasites in pigs: New challenges in pig production and current research highlights. Vet. Parasitol. 2011, 180, 72–81. [Google Scholar] [CrossRef]
- Cutillas, C.; German, P.; Arias, P.; Guevara, D. Trichuris ovis and Trichuris globulosa: Morphological, biometrical, and genetic studies. Exp. Parasitol. 1995, 81, 621–625. [Google Scholar] [CrossRef] [PubMed]
- del Rosario Robles, M. New Species of Trichuris (Nematoda: Trichuridae) from Akodon montensis Thomas, 1913, of the Paranaense Forest in Argentina. J. Parasitol. 2011, 97, 319–327. [Google Scholar] [CrossRef] [PubMed]
- del Rosario Robles, M.; Navone, G.T.; Notarnicola, J. A new species of Trichuris (Nematoda: Trichuridae) from Phyllotini rodents in Argentina. J. Parasitol. 2006, 92, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Callejón, R.; De Rojas, M.; Ariza, C.; Ubeda, J.M.; Guevara, D.C.; Cutillas, C. Cytochrome oxidase subunit 1 and mitochondrial 16S rDNA sequences of Trichuris skrjabini (Tricocephalida: Trichuridae). Parasitol. Res. 2009, 104, 715–716. [Google Scholar] [CrossRef] [PubMed]
- Cutillas, C.; German, P.; Arias, P.; Guevara, D. Characterization of Trichuris skrjabini by isoenzyme gel electrophoresis: Comparative study with Trichuris ovis. Acta Trop. 1996, 62, 63–69. [Google Scholar] [CrossRef]
- Gasser, R.B. Mutation scanning methods for the analysis of parasite genes. Int. J. Parasitol. 1997, 27, 449–463. [Google Scholar] [CrossRef]
- Gasser, R.B.; Zhu, X.; Beveridge, I.; Chilton, N. Mutation scanning analysis of sequence heterogenity in the second internal transcribed spacer (rDNA) within some members of the Hypodontus macropi (Nematoda: Strongyloidea) complex. Electrophoresis 2001, 22, 1076–1085. [Google Scholar] [CrossRef]
- Saccone, C. The evolution of mitochondrial DNA. Curr. Opin. Genet. Dev. 1994, 4, 875–881. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Zhao, G.H.; Li, J.; Song, H.Q.; Li, X.Y.; Chen, F.; Lin, R.Q.; Yuan, Z.G.; Weng, Y.B.; Hu, M.; Zou, F.C.; et al. A specific PCR assay for the identification and differentiation of Schistosoma japonicum geographical isolates in mainland China based on analysis of mitochondrial genome sequences. Infect. Genet. Evol. 2012, 12, 1027–1036. [Google Scholar] [CrossRef]
- Nadler, S.A.; De Len, G.P.P. Integrating molecular and morphological approaches for characterizing parasite cryptic species: Implications for parasitology. Parasitology 2011, 138, 1688–1709. [Google Scholar] [CrossRef] [PubMed]
- Ross, H.A.; Murugan, S.; Li, W.L. Testing the reliability of genetic methods of species identification via simulation. Syst. Biol. 2008, 57, 216–230. [Google Scholar] [CrossRef] [PubMed]
- International Helminth Genomes Consortium: Comparative genomics of the major parasitic worms. Nat. Genet. 2019, 51, 163–174. [CrossRef] [PubMed]
- Liu, G.H.; Gasser, R.B.; Su, A.; Nejsum, P.; Peng, L.; Lin, R.Q.; Li, M.W.; Xu, M.J.; Zhu, X.Q. Clear genetic distinctiveness between human- and pig-derived trichuris based on analyses of mitochondrial datasets. PLoS Negl. Trop. Dis. 2012, 6, e1539. [Google Scholar] [CrossRef] [PubMed]
- Callejón, R.; Halajian, A.; de Rojas, M.; Marrugal, A.; Guevara, D.; Cutillas, C. 16S partial gene mitochondrial DNA and internal transcribed spacers ribosomal DNA as differential markers of Trichuris discolor populations. Vet. Parasitol. 2012, 186, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Anshary, H.; Sriwulan; Freeman, M.A.; Ogawa, K. Occurrence and molecular identification of Anisakis dujardin, 1845 from marine fish in southern Makassar Strait, Indonesia. Korean J. Parasitol. 2014, 52, 9–19. [Google Scholar] [CrossRef]
- Liu, G.H.; Wu, C.Y.; Song, H.Q.; Wei, S.J.; Xu, M.J.; Lin, R.Q.; Zhao, G.H.; Huang, S.Y.; Zhu, X.Q. Comparative analyses of the complete mitochondrial genomes of Ascaris lumbricoides and Ascaris suum from humans and pigs. Gene 2012, 492, 110–116. [Google Scholar] [CrossRef]
- Wasimuddin; Bryja, J.; Ribas, A.; Baird, S.J.E.; Piálek, J.; Goüy de Bellocq, J. Testing parasite “intimacy”: The whipworm Trichuris muris in the European house mouse hybrid zone. Ecol. Evol. 2016, 6, 2688–2701. [Google Scholar] [CrossRef]
- Webb, K.M.; Rosenthal, B.M. Next-generation sequencing of the Trichinella murrelli mitochondrial genome allows comprehensive comparison of its divergence from the principal agent of human trichinellosis, Trichinella spiralis. Infect. Genet. Evol. 2011, 11, 116–123. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Brown, W.M. Trichinella spiralis mtDNA: A nematode mitochondrial genome that encodes a putative atp8 and normally structured tRNAs and has a gene arrangement relatable to those of coelomate metazoans. Genetics 2001, 157, 621–637. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Chilton, N.B.; Gasser, R.B. The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea). Int. J. Parasitol. 2002, 32, 145–158. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.M.; Liu, G.H.; Ando, K.; Woo, H.C.; Ma, J.; Sohn, W.M.; Sugiyama, H.; Zhu, X.Q. Complete mitochondrial genomes of Gnathostoma nipponicum and Gnathostoma sp., and their comparison with other Gnathostoma species. Infect. Genet. Evol. 2017, 48, 109–115. [Google Scholar]
- Liu, G.H.; Nadler, S.A.; Liu, S.S.; Podolska, M.; D’Amelio, S.; Shao, R.; Gasser, R.B.; Zhu, X.Q. Mitochondrial phylogenomics yields strongly supported hypotheses for ascaridomorph nematodes. Sci. Rep. 2016, 6, 39248. [Google Scholar] [CrossRef]
- Liu, G.H.; Wang, Y.; Xu, M.J.; Zhou, D.H.; Ye, Y.G.; Li, J.Y.; Song, H.Q.; Lin, R.Q.; Zhu, X.Q. Characterization of the complete mitochondrial genomes of two whipworms Trichuris ovis and Trichuris discolor (Nematoda: Trichuridae). Infect. Genet. Evol. 2012, 12, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Yang, X.; Zhang, T.; Wang, C.; Zhou, C.; Yan, X.; Hassan, M.; Ikram, M.; Hu, M. Characterization of the complete mitochondrial genome of Ostertagia trifurcata of small ruminants and its phylogenetic associations for the Trichostrongyloidea superfamily. Genes 2019, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Jones, J.; Armstrong, M.; Lamberti, F.; Moens, M. The mitochondrial genome of Xiphinema americanum sensu stricto (Nematoda: Enoplea): Considerable economization in the length and structural features of encoded genes. J. Mol. Evol. 2005, 61, 819–833. [Google Scholar] [CrossRef]
- Andi, E.T.R.; Ucchini, V.I.L.; Rewitt, T.A.R.A.A.R.; Imball, R.E.T.K.; Raun, E.D.L.B.; Igon, J.D.A.L.; Randi, E.; Lucchini, V.; Armijo-Prewitt, T.; Kimball, R.T.; et al. Mitochondrial DNA phylogeny and speciation in the tragopans. Auk 2000, 117, 1003–1015. [Google Scholar]
- Špakulová, M. Discriminant analysis as a method for the numerical evaluation of taxonomic characters in male trichurid nematodes. Syst. Parasitol. 1994, 29, 113–119. [Google Scholar] [CrossRef]
- Yevstafieva, V.A.; Yuskiv, I.D.; Melnychuk, V.V.; Yasnolob, I.O.; Kovalenko, V.A.; Horb, K.O. Nematodes of the genus Trichuris (Nematoda, Trichuridae), parasitizing sheep in central and South-Eastern regions of Ukraine. Vestn. Zool. 2018, 52, 193–204. [Google Scholar] [CrossRef][Green Version]
- Lin, R.Q.; Qiu, L.L.; Liu, G.H.; Wu, X.Y.; Weng, Y.B.; Xie, W.Q.; Hou, J.; Pan, H.; Yuan, Z.G.; Zou, F.C.; et al. Characterization of the complete mitochondrial genomes of five Eimeria species from domestic chickens. Gene 2011, 480, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gao, J.; Liu, H.; Liu, H.; Liang, A.; Zhou, X.; Cai, W. The architecture and complete sequence of mitochondrial genome of an assassin bug agriosphodrus dohrni (Hemiptera: Reduviidae). Int. J. Biol. Sci. 2011, 7, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, H.; Cao, L.; Shi, A.; Yang, H.; Cai, W. The complete mitochondrial genome of the damsel bug Alloeorhynchus bakeri (Hemiptera: Nabidae). Int. J. Biol. Sci. 2011, 8, 93–107. [Google Scholar] [CrossRef]
- Liu, G.-H.; Lin, R.-Q.; Li, M.-W.; Liu, W.; Liu, Y.; Yuan, Z.-G.; Song, H.-Q.; Zhao, G.-H.; Zhang, K.-X.; Zhu, X.-Q. The complete mitochondrial genomes of three cestode species of Taenia infecting animals and humans. Mol. Biol. Rep. 2011, 38, 2249–2256. [Google Scholar] [CrossRef]
- Liu, G.H.; Li, C.; Li, J.Y.; Zhou, D.H.; Xiong, R.C.; Lin, R.Q.; Zou, F.C.; Zhu, X.Q. Characterization of the complete mitochondrial genome sequence of Spirometra erinaceieuropaei (Cestoda: Diphyllobothriidae) from China. Int. J. Biol. Sci. 2012, 8, 640–649. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.R.; Zhao, G.H.; Gao, J.F.; Li, M.W.; Zhu, X.Q. The complete mitochondrial genome of Orientobilharzia turkestanicum supports its affinity with African Schistosoma spp. Infect. Genet. Evol. 2011, 11, 1964–1970. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Primers | Sequence (5′ to 3′) |
|---|---|
| ITS-2 | |
| NC5 | GTAGGTGAACCTGCGGAAGGAT |
| NC2 | TTAGTTTCTTTTCCTCCGCT |
| Short PCR | |
| Trichuris_cox1_F | GAAAGTGTTGGGGYAKAAAAGTTA |
| Trichuris_cox1_R | CAGGAAATCACAAAAAAATTGG |
| NAD-5F | CAAGGATTTTTTTGAGATCTTTTTC |
| NAD-5R | TAAACCGAATTGGAGATTTTTGTTT |
| Cytb-F | GAGTAATTTTTATAATACGAGAAGT |
| Cytb-R | AATTTTCAGGGTCTCTGCTTCAATA |
| Long PCR | |
| Cox1-F | CCTTGAAGCTTTGACACCTCC |
| Nad5-R | TGAGCTTTACGCAAGTTATGC |
| Nad5-F | CTAGCGTAAAAACTAGTGAGGTAAC |
| Cytb-R | GAGTGTGGGAACCAAGTGGA |
| Cytb-F | CTCTATGTGAAGCTTTTTGGGG |
| Cox1-R | TGGCAACTGCATGAGCAGATA |
| Gene/Codons | Position and Sequence Length of Nt | Amino Acids | Start/Stop Codons | Anticodons |
|---|---|---|---|---|
| cox1 | 1–1545 (1545) | 514 | ATG/TAA | |
| cox2 | 1550–2233 (684) | 227 | ATG/TAA | |
| trnL1 (UUR) | 2241–2302 (62) | TAA | ||
| trnE | 2304–2360 (57) | TTC | ||
| nad1 | 2376–3245 (870) | 289 | ATA/TGA | |
| LNCR | 3246–3401 (156) | |||
| trnK | 3402–3464 (63) | TTT | ||
| nad2 | 3463–4245 (783) | 260 | ATG/TAA | |
| trnM | 4357–4419 (63) | CAT | ||
| trnF | 4416–4471 (56) | GAA | ||
| nad5 | 4410–5996 (1587) | 528 | ATG/TAA | |
| trnH | 6042–6095 (54) | GTG | ||
| trnR | 6096–6159 (64) | TCG | ||
| nad4 | 6161–7396 (1236) | 411 | ATG/TAA | |
| nad4L | 7293–7601 (309) | 102 | ATG/TAG | |
| trnT | 7654–7708 (55) | TGT | ||
| trnP | 7709–7762 (54) | TGG | ||
| nad6 | 7772–8230 (459) | 152 | ATG/TAA | |
| cytb | 8239–9351 (1113) | 370 | ATG/TAA | |
| trnS1 (AGN) | 9359–9411 (53) | GCT | ||
| rrnS | 9515–10,080 (566) | |||
| trnV | 10,103–10,159 (57) | TAC | ||
| rrnL | 10,189–11,028 (840) | |||
| atp6 | 11,089–11,931 (843) | 280 | ATG/TAA | |
| cox3 | 11,933–12,706 (774) | 257 | ATG/TAA | |
| trnW | 12,709–12,770 (62) | TCA | ||
| trnQ | 12,778–12,834 (57) | TTG | ||
| trnI | 12,836–12,898 (63) | GAT | ||
| trnG | 12,899–12,955 (57) | TCC | ||
| trnD | 12,966–13,022 (57) | GTC | ||
| atp8 | 12,951–13,169 (219) | 72 | ATA/TAA | |
| nad3 | 13,169–13,531 (363) | 120 | ATA/TAG | |
| SNCR | 13,532–13,619 (88) | |||
| trnA | 13,620–13,680 (61) | TGC | ||
| trnS2 (UGN) | 13,638–13,692 (55) | TGA | ||
| trnN | 13,691–13,747 (57) | GTT | ||
| trnL2 (CUA) | 13,749–13,811 (63) | TAG | ||
| trnC | 13,866–13,922 (57) | GCA | ||
| trnY | 13,923–13,979 (57) | GTA |
| Gene | A | G | C | T | A + T (%) | AT skew | GC skew |
|---|---|---|---|---|---|---|---|
| cox1 | 27.50 | 16.18 | 22.78 | 33.52 | 61.02 | −0.09 | 0.16 |
| cox2 | 34.06 | 13.74 | 23.09 | 29.09 | 63.12 | 0.07 | 0.25 |
| nad1 | 28.96 | 13.21 | 20.45 | 37.35 | 66.31 | −0.12 | 0,21 |
| nad2 | 40.99 | 16.34 | 12.00 | 30.65 | 71.64 | 0.14 | −0.15 |
| nad5 | 41.14 | 14.36 | 13.35 | 31.12 | 72.26 | 0.13 | −0.03 |
| nad4 | 44.82 | 13.34 | 13.75 | 28.07 | 72.89 | 0.22 | 0.01 |
| nad4L | 42.71 | 10.67 | 17.79 | 28.80 | 71.51 | 0.19 | 0.25 |
| nad6 | 23.09 | 12.85 | 13.28 | 50.76 | 73.85 | −0.37 | 0.01 |
| cytb | 27.76 | 15.36 | 15.81 | 41.06 | 68.82 | −0.19 | 0.01 |
| atp6 | 33.33 | 18.62 | 18.62 | 38.19 | 71.52 | −0.06 | 0.30 |
| cox3 | 28.16 | 19.89 | 19.89 | 36.69 | 64.85 | −0.13 | 0.13 |
| atp8 | 30.59 | 16.43 | 16.43 | 42.92 | 73.51 | −0.16 | 0.24 |
| nad3 | 32.23 | 12.94 | 15.15 | 39.66 | 71.89 | −0.10 | 0.07 |
| rrnS | 35.68 | 13.78 | 13.42 | 37.10 | 72.78 | −0.01 | −0.01 |
| rrnL | 38.33 | 12.73 | 13.80 | 35.11 | 73.44 | 0.04 | 0.04 |
| LNCR | 34.61 | 16.02 | 13.46 | 35.89 | 70.50 | −0.01 | −0.08 |
| SNCR | 47.72 | 14.54 | 13.63 | 34.09 | 81.81 | 0.16 | 0.50 |
| Overall | 34.75 | 13.88 | 16.38 | 34.96 | 69.71 | −0.00 | 0.08 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, A.A.; Shabbir, M.A.B.; Xin, Y.; Ikram, M.; Hafeez, M.A.; Wang, C.; Zhang, T.; Zhou, C.; Yan, X.; Hassan, M.; et al. Characterization of the Complete Mitochondrial Genome of a Whipworm Trichuris skrjabini (Nematoda: Trichuridae). Genes 2019, 10, 438. https://doi.org/10.3390/genes10060438
Ahmad AA, Shabbir MAB, Xin Y, Ikram M, Hafeez MA, Wang C, Zhang T, Zhou C, Yan X, Hassan M, et al. Characterization of the Complete Mitochondrial Genome of a Whipworm Trichuris skrjabini (Nematoda: Trichuridae). Genes. 2019; 10(6):438. https://doi.org/10.3390/genes10060438
Chicago/Turabian StyleAhmad, Awais Ali, Muhammad Abu Bakr Shabbir, Yang Xin, Muhammad Ikram, Mian Abdul Hafeez, Chunqun Wang, Ting Zhang, Caixian Zhou, Xingrun Yan, Mubashar Hassan, and et al. 2019. "Characterization of the Complete Mitochondrial Genome of a Whipworm Trichuris skrjabini (Nematoda: Trichuridae)" Genes 10, no. 6: 438. https://doi.org/10.3390/genes10060438
APA StyleAhmad, A. A., Shabbir, M. A. B., Xin, Y., Ikram, M., Hafeez, M. A., Wang, C., Zhang, T., Zhou, C., Yan, X., Hassan, M., & Hu, M. (2019). Characterization of the Complete Mitochondrial Genome of a Whipworm Trichuris skrjabini (Nematoda: Trichuridae). Genes, 10(6), 438. https://doi.org/10.3390/genes10060438