A Missense Variant in SCN8A in Alpine Dachsbracke Dogs Affected by Spinocerebellar Ataxia

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. DNA Samples and Single Nucleotide Variant Genotyping
2.3. Pathology
2.4. Linkage and Autozygosity Analyses
2.5. Whole-Genome Resequencing
2.6. Targeted Genotyping
2.7. Protein Predictions
2.8. Availability of Data and Material
3. Results
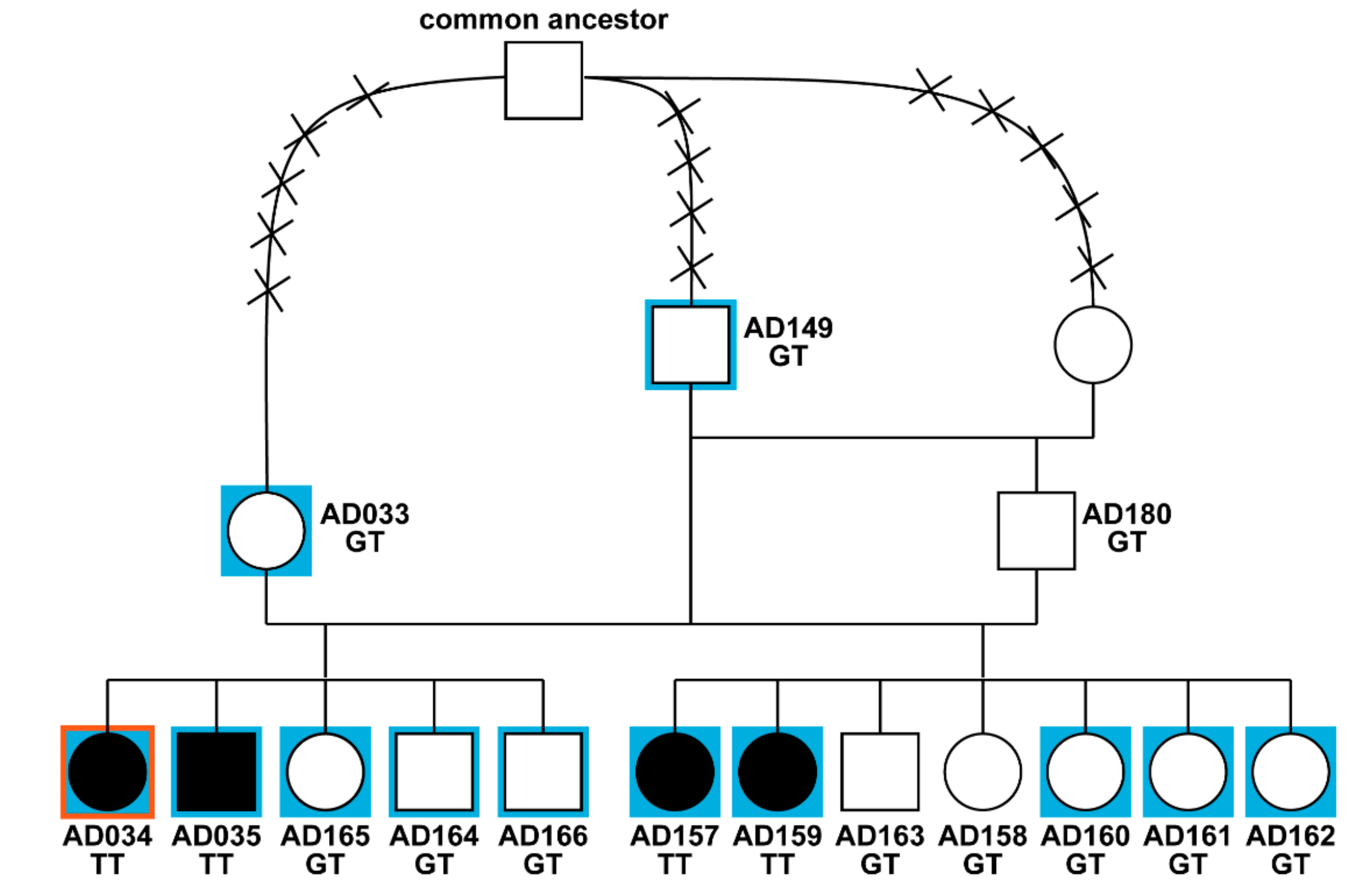
3.1. Phenotype and Pedigree Analysis
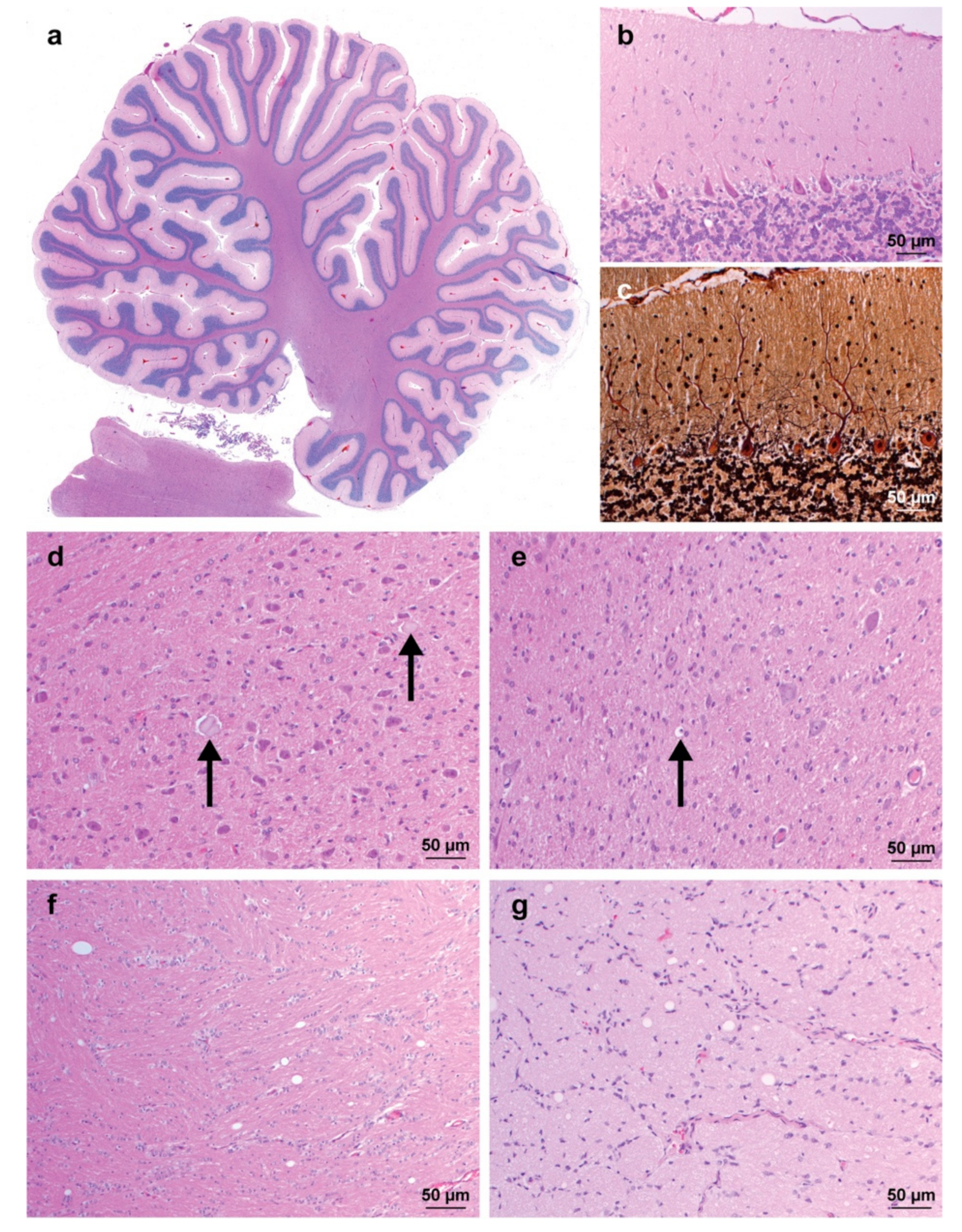
3.2. Pathological Findings
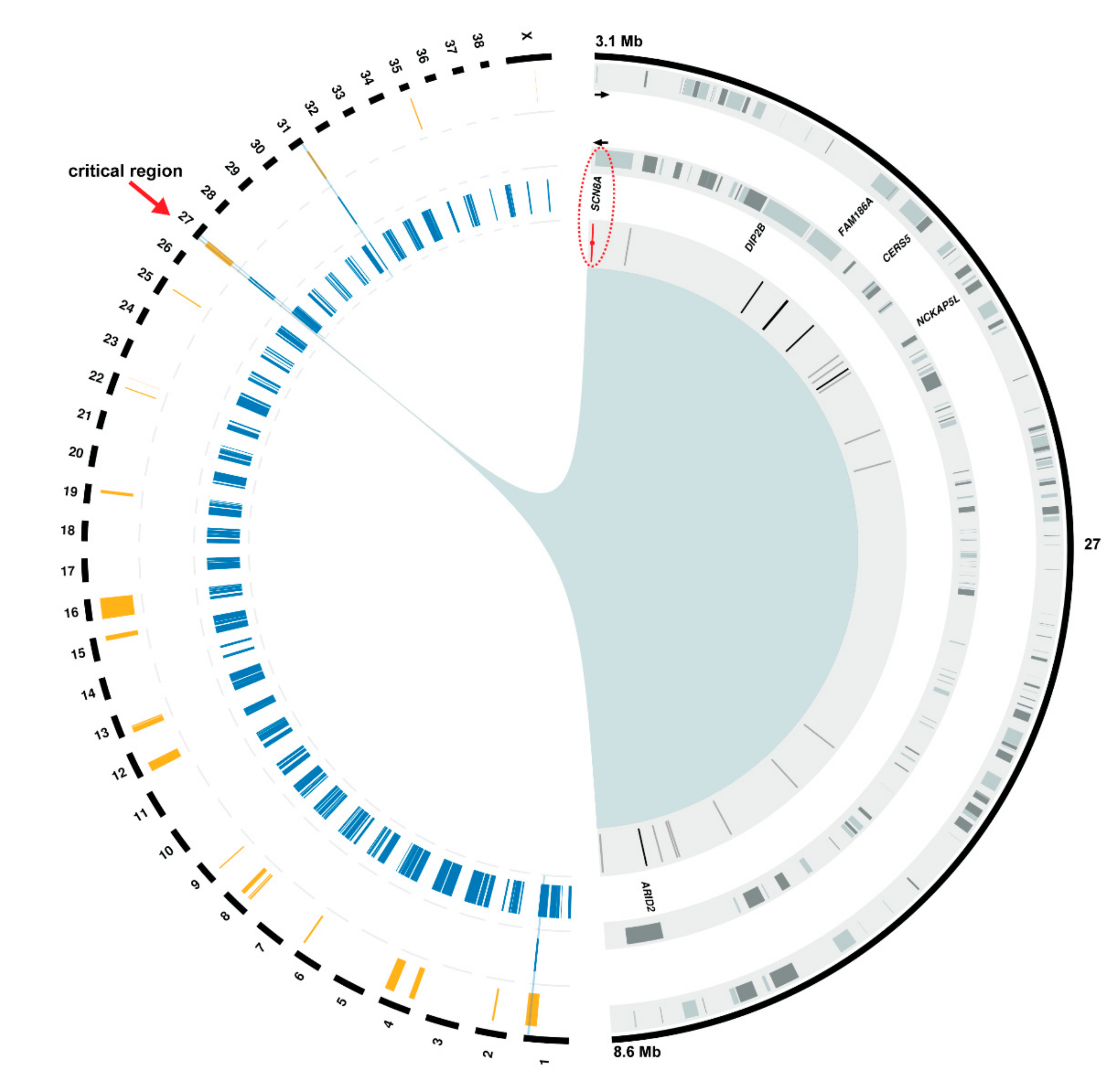
3.3. Positional Cloning of the Spinocerebellar Ataxia-Associated Locus
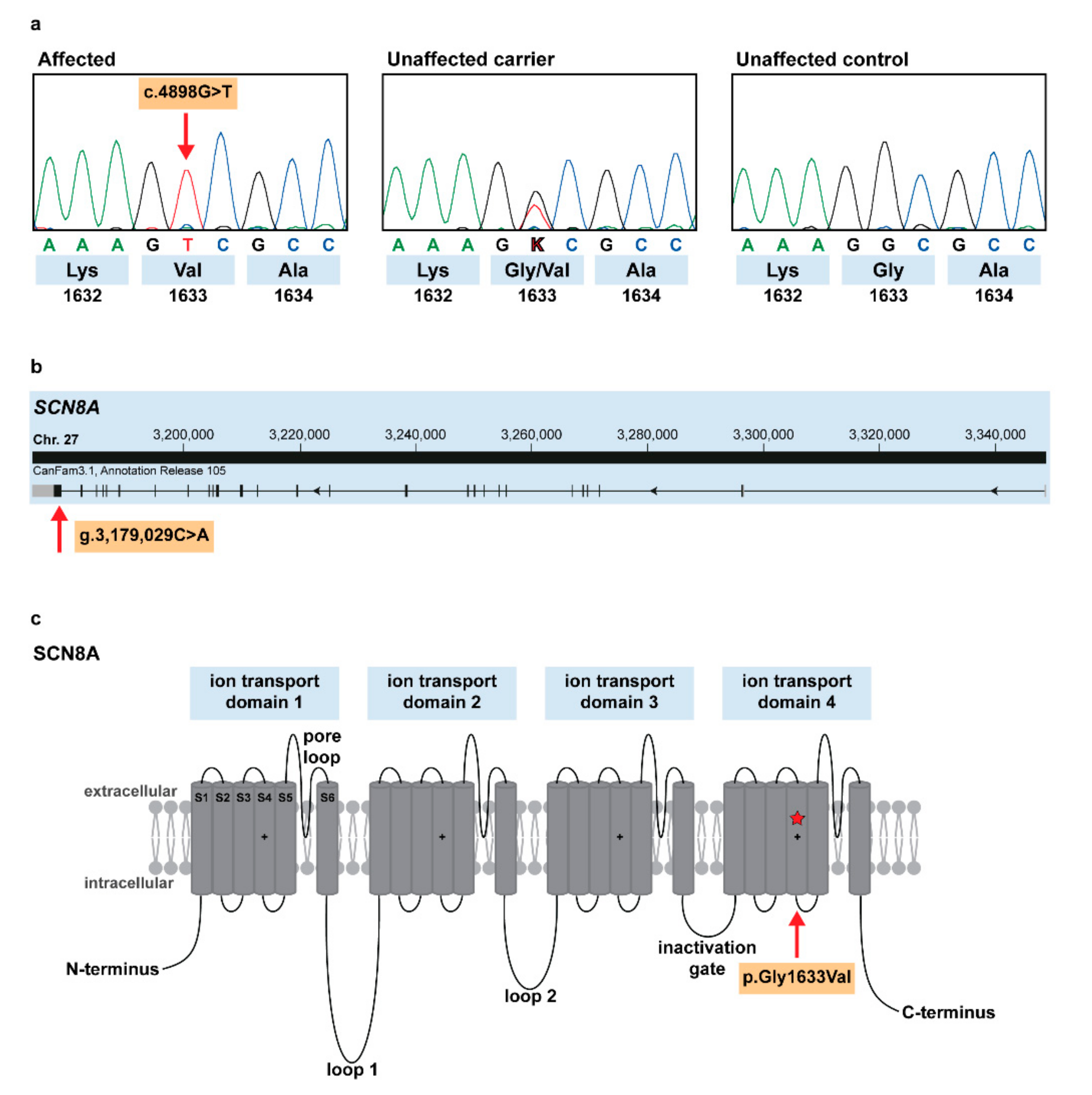
3.4. Identification of the Causative Variant
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manto, M.; Marmolino, D. Cerebellar ataxias. Curr. Opin. Neurol. 2009, 22, 419–429. [Google Scholar] [CrossRef]
- Jayadev, S.; Bird, T.D. Hereditary ataxias: Overview. Genet. Med. 2013, 15, 673–683. [Google Scholar] [CrossRef]
- Sandford, E.; Burmeister, M. Genes and genetic testing in hereditary ataxias. Genes 2014, 5, 586–603. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, T.; Paulson, H. Milestones in Ataxia. Mov. Disord. 2011, 26, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Urkasemsin, G.; Olby, N.J. Canine Hereditary Ataxia. Vet. Clin. N. Am. Small Anim. Pract. 2014, 44, 1075–1089. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 1–16. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.; Cardon, L.R. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002, 30, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yan, C.; Hsu, C.H.; Chen, Q.R.; Niu, K.; Komatsoulis, G.A.; Meerzaman, D. OmicCircos: A simple-to-use R package for the circular visualization of multidimensional Omics data. Cancer Inform. 2014, 13, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Mauri, N.; Kleiter, M.; Leschnik, M.; Högler, S.; Dietschi, E.; Wiedmer, M.; Dietrich, J.; Henke, D.; Steffen, F.; Schuller, S.; et al. A Missense Variant in KCNJ10 in Belgian Shepherd Dogs Affected by Spongy Degeneration with Cerebellar Ataxia (SDCA1). G3 Genes Genomes Genetics 2017, 7, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Niroula, A.; Urolagin, S.; Vihinen, M. PON-P2: Prediction method for fast and reliable identification of harmful variants. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. MutPred2: Inferring the molecular and phenotypic impact of amino acid variants. bioRxiv 2017, 134981. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar] [CrossRef]
- Catterall, W.A. International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef]
- Catterall, W.A. Sodium Channels. Encycl. Life Sci. 2006, 65–71. [Google Scholar] [CrossRef]
- Kearney, J.A.; Buchner, D.A.; De Haan, G.; Adamska, M.; Levin, S.I.; Furay, A.R.; Albin, R.L.; Jones, J.M.; Montal, M.; Stevens, M.J.; et al. Molecular and pathological effects of a modifier gene on deficiency of the sodium channel Scn8a (Na(v)1.6). Hum. Mol. Genet. 2002, 11, 2765–2775. [Google Scholar] [CrossRef]
- Hu, W.; Tian, C.; Li, T.; Yang, M.; Hou, H.; Shu, Y. Distinct contributions of Nav1.6 and Nav1.2 in action potential initiation and backpropagation. Nat. Neurosci. 2009, 12, 996–1002. [Google Scholar] [CrossRef]
- Liu, Y.; Schubert, J.; Sonnenberg, L.; Helbig, K.L.; Hoei-hansen, C.E.; Koko, M.; Rannap, M.; Lauxmann, S.; Huq, M.; Schneider, M.C.; et al. Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain 2019, 376–390. [Google Scholar] [CrossRef]
- Wagnon, J.L.; Meisler, M.H. Recurrent and non-recurrent mutations of SCN8A in epileptic encephalopathy. Front. Neurol. 2015, 6, 1–7. [Google Scholar] [CrossRef]
- Chen, K.; Godfrey, D.A.; Ilyas, O.; Xu, J.; Preston, T.W. Cerebellum-related characteristics of Scn8a-Mutant mice. Cerebellum 2009, 8, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Kohrman, D.C.; Smith, M.R.; Goldin, A.L.; Harris, J.; Meisler, M.H. A missense mutation in the sodium channel Scn8a is responsible for cerebellar ataxia in the mouse mutant jolting. J. Neurosci. 1996, 16, 5993–5999. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Status | G/G | G/T | T/T |
|---|---|---|---|
| Affected (n = 4) | 0 | 0 | 4 |
| Non-affected (n = 212) | 193 | 19 1 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Letko, A.; Dietschi, E.; Nieburg, M.; Jagannathan, V.; Gurtner, C.; Oevermann, A.; Drögemüller, C. A Missense Variant in SCN8A in Alpine Dachsbracke Dogs Affected by Spinocerebellar Ataxia. Genes 2019, 10, 362. https://doi.org/10.3390/genes10050362
Letko A, Dietschi E, Nieburg M, Jagannathan V, Gurtner C, Oevermann A, Drögemüller C. A Missense Variant in SCN8A in Alpine Dachsbracke Dogs Affected by Spinocerebellar Ataxia. Genes. 2019; 10(5):362. https://doi.org/10.3390/genes10050362
Chicago/Turabian StyleLetko, Anna, Elisabeth Dietschi, Marco Nieburg, Vidhya Jagannathan, Corinne Gurtner, Anna Oevermann, and Cord Drögemüller. 2019. "A Missense Variant in SCN8A in Alpine Dachsbracke Dogs Affected by Spinocerebellar Ataxia" Genes 10, no. 5: 362. https://doi.org/10.3390/genes10050362
APA StyleLetko, A., Dietschi, E., Nieburg, M., Jagannathan, V., Gurtner, C., Oevermann, A., & Drögemüller, C. (2019). A Missense Variant in SCN8A in Alpine Dachsbracke Dogs Affected by Spinocerebellar Ataxia. Genes, 10(5), 362. https://doi.org/10.3390/genes10050362