Dissecting the Brain/Islet Axis in Metabesity


 ,
,  ,
, 
 ,
, 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Brain and Pancreatic Islets in Glucose Homeostasis: Who Is in Control?
2.1. The Classical Pancreatic Endocrine Model
2.2. The Centralized, Brain-Based Model
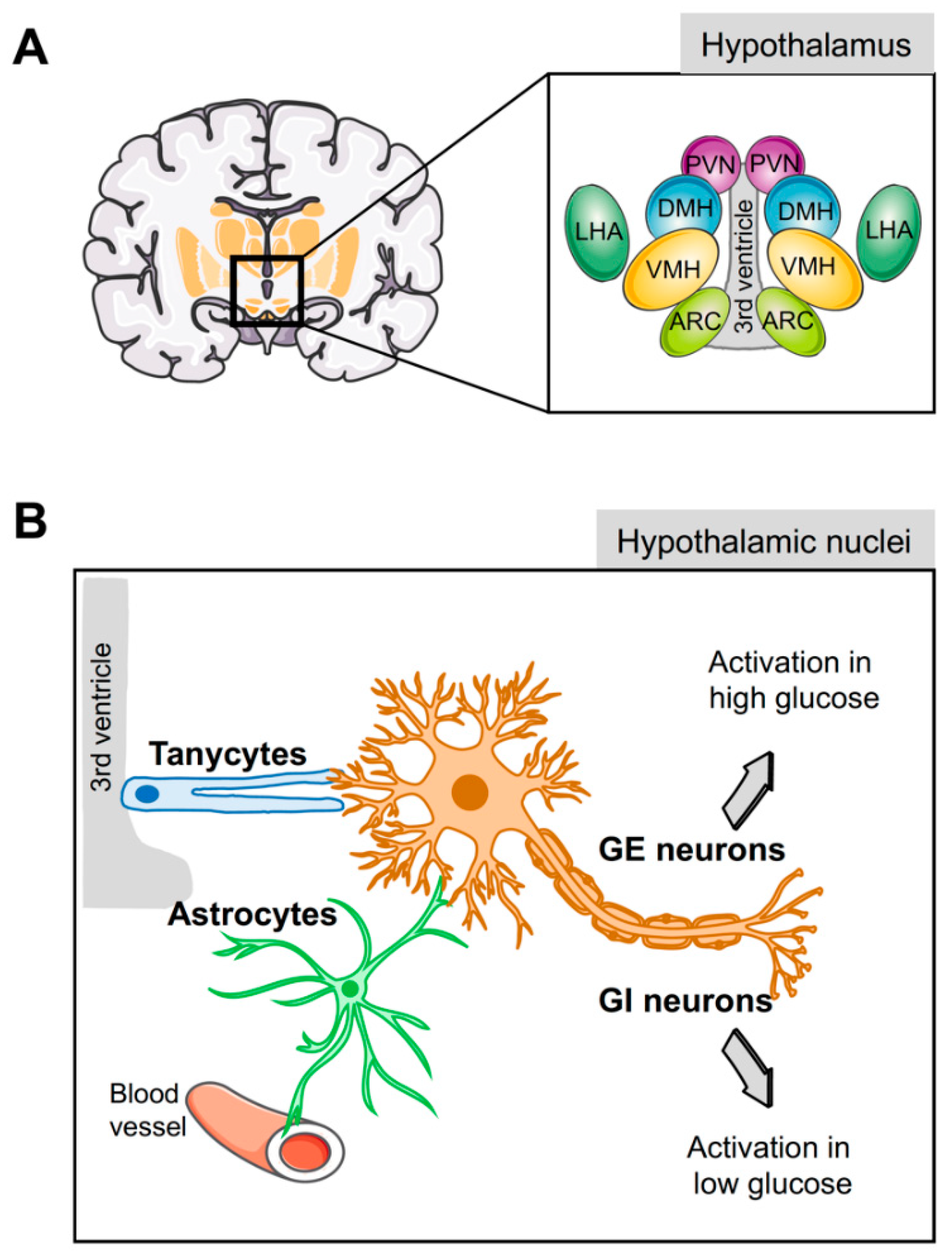
3. Cell Types of the Hypothalamus Implicated in Energy Homeostasis
3.1. Hypothalamic Neurons
3.2. Hypothalamic Glial Cells
4. Astrocytes are Central to Glucose Metabolism and Homeostasis
4.1. Hormonal Input Implicated in Astrocyte-Mediated Glucose Homeostasis
4.2. Astrocytes and Neuroinflammation
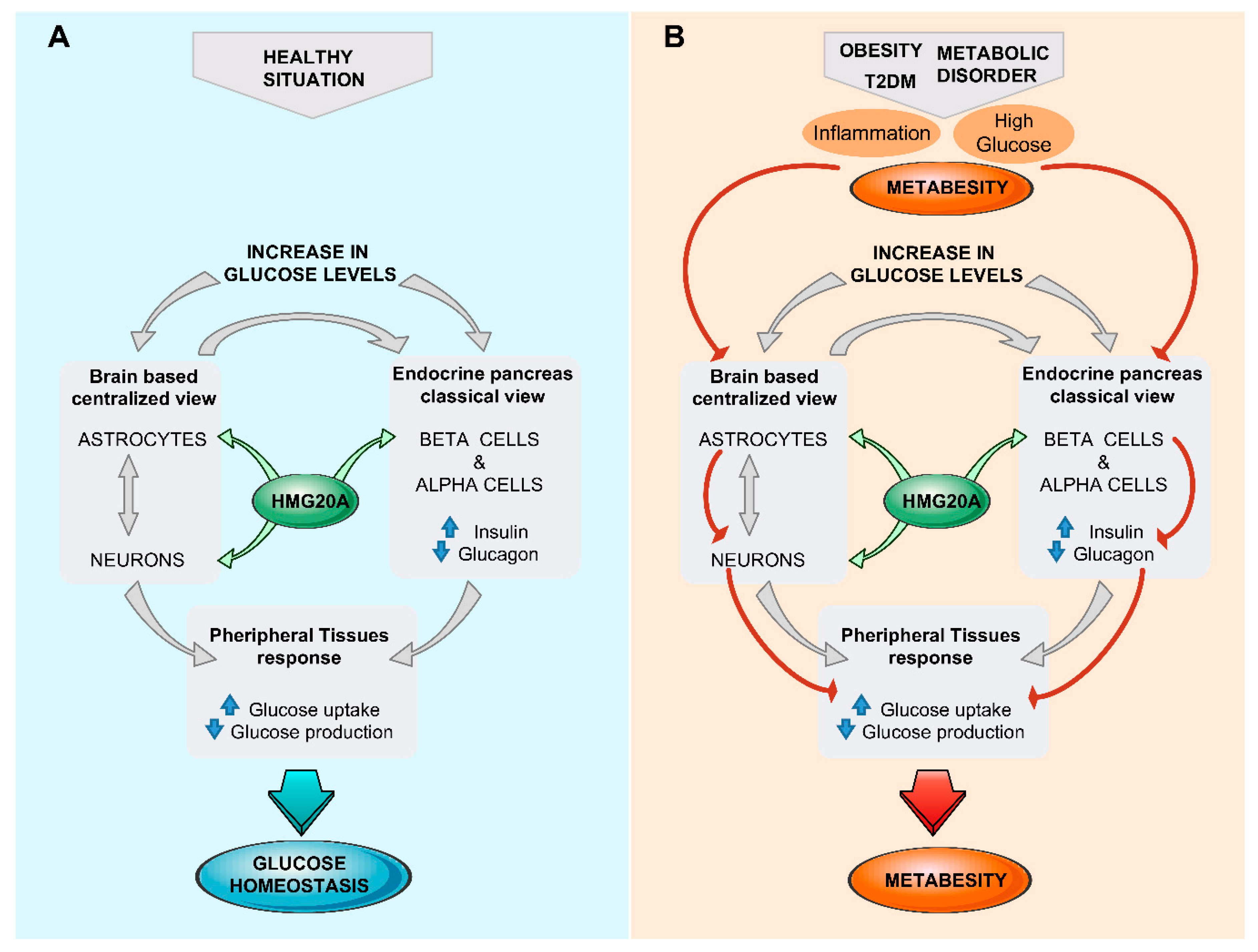
5. Brain/Islet Glucose Homeostasis Axis Orchestrated by HMG20A?
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- IDF Diabetes Atlas. Available online: http://www.diabetesatlas.org (accessed on 9 January 2019).
- Velloso, L.A.; Araujo, E.P.; de Souza, C.T. Diet-induced inflammation of the hypothalamus in obesity. Neuroimmunomodulation 2008, 15, 189–193. [Google Scholar] [CrossRef] [PubMed]
- van Greevenbroek, M.M.; Schalkwijk, C.G.; Stehouwer, C.D. Obesity-associated low-grade inflammation in type 2 diabetes mellitus: Causes and consequences. Neth. J. Med. 2013, 71, 174–187. [Google Scholar] [PubMed]
- Lontchi-Yimagou, E.; Sobngwi, E.; Matsha, T.E.; Kengne, A.P. Diabetes mellitus and inflammation. Curr. Diab. Rep. 2013, 13, 435–444. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef]
- Douglass, J.D.; Dorfman, M.D.; Fasnacht, R.; Shaffer, L.D.; Thaler, J.P. Astrocyte IKKβ/NF-κB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol. Metab. 2017, 6, 366–373. [Google Scholar] [CrossRef]
- Fruhwurth, S.; Vogel, H.; Schurmann, A.; Williams, K.J. Novel Insights into How overnutrition disrupts the hypothalamic actions of leptin. Front. Endocrinol. (Lausanne) 2018, 9, 89. [Google Scholar] [CrossRef]
- Ono, H. Molecular mechanisms of hypothalamic insulin resistance. Int. J. Mol. Sci. 2019, 20, 1317. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Report on Diabetes. World Health Organization. 2016. Available online: http://apps.who.int/iris/bitstream/10665/204871/1/9789241565257_eng.pdf (accessed on 13 March 2019).
- Davies, M.J.; D’Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2018, 61, 2461–2498. [Google Scholar] [CrossRef]
- Roglic, G.; Unwin, N. Mortality attributable to diabetes: Estimates for the year 2010. Diabetes Res. Clin. Pract. 2010, 87, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Carr, R.D.; Bosco, D.; Zuellig, R.A.; Berney, T.; Donath, M.Y. Sulfonylurea induced β-cell apoptosis in cultured human islets. J. Clin. Endocrinol. Metab. 2005, 90, 501–506. [Google Scholar] [CrossRef]
- Munir, K.M.; Davis, S.N. Are SGLT2 inhibitors or GLP-1 receptor agonists more appropriate as a second-line therapy in type 2 diabetes? Expert Opin. Pharmacother. 2018, 19, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a type 3 diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gambero, A.J.; Martinez, F.; Salazar, K.; Cifuentes, M.; Nualart, F. Brain glucose-sensing mechanism and energy homeostasis. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Marty, N.; Dallaporta, M.; Thorens, B. Brain glucose sensing, counterregulation, and energy homeostasis. Physiology 2007, 22, 241–251. [Google Scholar] [CrossRef]
- Rorsman, P.; Salehi, S.A.; Abdulkader, F.; Braun, M.; MacDonald, P.E. KATP-channels and glucose-regulated glucagon secretion. Trends Endocrinol. Metab. 2008, 19, 277–284. [Google Scholar] [CrossRef]
- Prentki, M.; Matschinsky, F.M.; Madiraju, S.R. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013, 18, 162–185. [Google Scholar] [CrossRef]
- Ohneda, M.; Johnson, J.H.; Inman, L.R.; Unger, R.H. GLUT-2 function in glucose-unresponsive β cells of dexamethasone-induced diabetes in rats. J. Clin. Investig. 1993, 92, 1950–1956. [Google Scholar] [CrossRef]
- Gat-Yablonski, G.; Shalitin, S.; Phillip, M. Maturity onset diabetes of the young-review. Pediatr Endocrinol. Rev. 2006, 3, 514–520. [Google Scholar]
- Soria, B.; Martin, F. Cytosolic calcium oscillations and insulin release in pancreatic islets of Langerhans. Diabetes Metab. 1998, 24, 37–40. [Google Scholar] [PubMed]
- Gauthier, B.R.; Wollheim, C.B. Synaptotagmins bind calcium to release insulin. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1279–E1286. [Google Scholar] [CrossRef]
- Franklin, I.; Gromada, J.; Gjinovci, A.; Theander, S.; Wollheim, C.B. β-cell secretory products activate α-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes 2005, 54, 1808–1815. [Google Scholar] [CrossRef]
- Rorsman, P.; Berggren, P.O.; Bokvist, K.; Ericson, H.; Mohler, H.; Ostenson, C.G.; Smith, P.A. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature 1989, 341, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chibalina, M.V.; Bengtsson, M.; Groschner, L.N.; Ramracheya, R.; Rorsman, N.J.; Leiss, V.; Nassar, M.A.; Welling, A.; Gribble, F.M.; et al. Na+ current properties in islet α- and β-cells reflect cell-specific Scn3a and Scn9a expression. J. Physiol. 2014, 592, 4677–4696. [Google Scholar] [CrossRef]
- Zhang, Q.; Ramracheya, R.; Lahmann, C.; Tarasov, A.; Bengtsson, M.; Braha, O.; Braun, M.; Brereton, M.; Collins, S.; Galvanovskis, J.; et al. Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 2013, 18, 871–882. [Google Scholar] [CrossRef]
- Lawlor, N.; Khetan, S.; Ucar, D.; Stitzel, M.L. Genomics of islet (Dys)function and type 2 diabetes. Trends Genet. 2017, 33, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Schuit, F.C.; Huypens, P.; Heimberg, H.; Pipeleers, D.G. Glucose sensing in pancreatic β-cells: A model for the study of other glucose-regulated cells in gut, pancreas, and hypothalamus. Diabetes 2001, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Iezzi, M.; Theander, S.; Antinozzi, P.A.; Gauthier, B.R.; Halban, P.A.; Wollheim, C.B. Suppression of Pdx-1 perturbs proinsulin processing, insulin secretion and GLP-1 signalling in INS-1 cells. Diabetologia 2005, 48, 720–731. [Google Scholar] [CrossRef]
- Gauthier, B.R.; Wiederkehr, A.; Baquie, M.; Dai, C.; Powers, A.C.; Kerr-Conte, J.; Pattou, F.; MacDonald, R.J.; Ferrer, J.; Wollheim, C.B. PDX1 deficiency causes mitochondrial dysfunction and defective insulin secretion through TFAM suppression. Cell Metab. 2009, 10, 110–118. [Google Scholar] [CrossRef]
- Brun, T.; Gauthier, B.R. A focus on the role of Pax4 in mature pancreatic islet β-cell expansion and survival in health and disease. J. Mol. Endocrinol. 2008, 40, 37–45. [Google Scholar] [CrossRef]
- Brun, T.; Hu, K.H.; Lupi, R.; Boehm, B.; Wojtusciszyn, A.; Sauter, N.; Donath, M.; Marchetti, P.; Maedler, K.; Gauthier, B.R. The diabetes-linked transcription factor Pax4 is expressed in human pancreatic islets and is activated by mitogens and GLP-1. Hum. Mol. Genet. 2008, 17, 478–489. [Google Scholar] [CrossRef]
- Lorenzo, P.I.; Fuente-Martin, E.; Brun, T.; Cobo-Vuilleumier, N.; Jimenez-Moreno, C.M.; Gomez, I.G.H.; Lopez Noriega, L.; Mellado-Gil, J.M.; Martin-Montalvo, A.; Soria, B.; et al. PAX4 defines an expandable β-cell subpopulation in the adult pancreatic islet. Sci. Rep. 2015, 5, 15672. [Google Scholar] [CrossRef]
- Mellado-Gil, J.M.; Jimenez-Moreno, C.M.; Martin-Montalvo, A.; Alvarez-Mercado, A.I.; Fuente-Martin, E.; Cobo-Vuilleumier, N.; Lorenzo, P.I.; Bru-Tari, E.; Herrera-Gomez Ide, G.; Lopez-Noriega, L.; et al. PAX4 preserves endoplasmic reticulum integrity preventing β cell degeneration in a mouse model of type 1 diabetes mellitus. Diabetologia 2016, 59, 755–765. [Google Scholar] [CrossRef]
- Lorenzo, P.I.; Juarez-Vicente, F.; Cobo-Vuilleumier, N.; Garcia-Dominguez, M.; Gauthier, B.R. The diabetes-linked transcription factor PAX4: from gene to functional consequences. Genes 2017, 8, 101. [Google Scholar] [CrossRef]
- Lorenzo, P.I.; Cobo-Vuilleumier, N.; Gauthier, B. Therapeutic potential of pancreatic PAX4-regulated pathways in treating diabetes mellitus. Curr. Opin. Pharmacol. 2018, 43, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Jonas, J.C.; Laybutt, D.R. Mechanisms of β-cell dedifferentiation in diabetes: Recent findings and future research directions. J. Endocrinol. 2018, 236, R109–R143. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Chavez, M.; Rivero, S.; Garcia-Gutierrez, P.; Rodriguez-Paredes, M.; Garcia-Dominguez, M.; Bhattacharya, S.; Reyes, J.C. Control of neuronal differentiation by sumoylation of BRAF35, a subunit of the LSD1-CoREST histone demethylase complex. Proc. Natl. Acad. Sci. USA 2012, 109, 8085–8090. [Google Scholar] [CrossRef] [PubMed]
- Rivero, S.; Ceballos-Chavez, M.; Bhattacharya, S.S.; Reyes, J.C. HMG20A is required for SNAI1-mediated epithelial to mesenchymal transition. Oncogene 2015, 34, 5264–5276. [Google Scholar] [CrossRef] [PubMed]
- Mellado-Gil, J.M.; Fuente-Martin, E.; Lorenzo, P.I.; Cobo-Vuilleumier, N.; Lopez-Noriega, L.; Martin-Montalvo, A.; Gomez, I.G.H.; Ceballos-Chavez, M.; Gomez-Jaramillo, L.; Campos-Caro, A.; et al. The type 2 diabetes-associated HMG20A gene is mandatory for islet β cell functional maturity. Cell Death Dis. 2018, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Kooner, J.S.; Saleheen, D.; Sim, X.; Sehmi, J.; Zhang, W.; Frossard, P.; Been, L.F.; Chia, K.S.; Dimas, A.S.; Hassanali, N.; et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat. Genet. 2011, 43, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Sim, X.; Ong, R.T.; Suo, C.; Tay, W.T.; Liu, J.; Ng, D.P.; Boehnke, M.; Chia, K.S.; Wong, T.Y.; Seielstad, M.; et al. Transferability of type 2 diabetes implicated loci in multi-ethnic cohorts from Southeast Asia. PLoS Genet. 2011, 7, e1001363. [Google Scholar] [CrossRef]
- Perry, J.R.; Voight, B.F.; Yengo, L.; Amin, N.; Dupuis, J.; Ganser, M.; Grallert, H.; Navarro, P.; Li, M.; Qi, L.; et al. Stratifying type 2 diabetes cases by BMI identifies genetic risk variants in LAMA1 and enrichment for risk variants in lean compared to obese cases. PLoS Genet. 2012, 8, e1002741. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Lopez-Noriega, L.; Jimenez-Moreno, C.; Herranz, A.; Lorenzo, P.I.; Cobo-Vuilleumier, N.; Tamayo, A.; Gonzalez-Guerrero, C.; Hofsteede, J.; Lebreton, F.; et al. Transient PAX8 expression in islets during pregnancy correlates with β-cell survival, revealing a novel candidate gene in gestational diabetes mellitus. Diabetes 2019, 68, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Cobo-Vuilleumier, N.; Lorenzo, P.I.; Rodriguez, N.G.; Herrera Gomez, I.G.; Fuente-Martin, E.; Lopez-Noriega, L.; Mellado-Gil, J.M.; Romero-Zerbo, S.Y.; Baquie, M.; Lachaud, C.C.; et al. LRH-1 agonism favours an immune-islet dialogue which protects against diabetes mellitus. Nat. Commun. 2018, 9, 1488. [Google Scholar] [CrossRef]
- Reimann, F.; Habib, A.M.; Tolhurst, G.; Parker, H.E.; Rogers, G.J.; Gribble, F.M. Glucose sensing in L cells: A primary cell study. Cell Metab. 2008, 8, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. Sensing of glucose in the brain. Handb. Exp. Pharmacol. 2012, 277–294. [Google Scholar] [CrossRef]
- Morton, G.J.; Muta, K.; Kaiyala, K.J.; Rojas, J.M.; Scarlett, J.M.; Matsen, M.E.; Nelson, J.T.; Acharya, N.K.; Piccinini, F.; Stefanovski, D.; et al. Evidence that the sympathetic nervous system elicits rapid, coordinated, and reciprocal adjustments of insulin secretion and insulin sensitivity during cold exposure. Diabetes 2017, 66, 823–834. [Google Scholar] [CrossRef]
- Levin, B.E.; Routh, V.H.; Kang, L.; Sanders, N.M.; Dunn-Meynell, A.A. Neuronal glucosensing: What do we know after 50 years? Diabetes 2004, 53, 2521–2528. [Google Scholar] [CrossRef]
- Grmek, M. Claude Bernard: Cahier de Notes (1850-60); Gallimard: Paris, France, 1965. [Google Scholar]
- Hoebel, B.G. Hypothalamic lesions by electrocauterization: Disinhibition of feeding and self-stimulation. Science 1965, 149, 452–453. [Google Scholar] [CrossRef]
- Bray, G.A. Autonomic and endocrine factors in the regulation of food intake. Brain Res. Bull. 1985, 14, 505–510. [Google Scholar] [CrossRef]
- King, B.M. The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight. Physiol. Behav. 2006, 87, 221–244. [Google Scholar] [CrossRef]
- Garfield, A.S.; Shah, B.P.; Madara, J.C.; Burke, L.K.; Patterson, C.M.; Flak, J.; Neve, R.L.; Evans, M.L.; Lowell, B.B.; Myers, M.G., Jr.; et al. A parabrachial-hypothalamic cholecystokinin neurocircuit controls counterregulatory responses to hypoglycemia. Cell Metab. 2014, 20, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Scarlett, J.M.; Rojas, J.M.; Matsen, M.E.; Kaiyala, K.J.; Stefanovski, D.; Bergman, R.N.; Nguyen, H.T.; Dorfman, M.D.; Lantier, L.; Wasserman, D.H.; et al. Central injection of fibroblast growth factor 1 induces sustained remission of diabetic hyperglycemia in rodents. Nat. Med. 2016, 22, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Morton, G.J.; Matsen, M.E.; Bracy, D.P.; Meek, T.H.; Nguyen, H.T.; Stefanovski, D.; Bergman, R.N.; Wasserman, D.H.; Schwartz, M.W. FGF19 action in the brain induces insulin-independent glucose lowering. J. Clin. Investig. 2013, 123, 4799–4808. [Google Scholar] [CrossRef]
- Marcelin, G.; Jo, Y.H.; Li, X.; Schwartz, G.J.; Zhang, Y.; Dun, N.J.; Lyu, R.M.; Blouet, C.; Chang, J.K.; Chua, S., Jr. Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism. Mol. Metab. 2014, 3, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.K.; Kohli, R.; Gutierrez-Aguilar, R.; Gaitonde, S.G.; Woods, S.C.; Seeley, R.J. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology 2013, 154, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.K.; Gutierrez-Juarez, R.; Pocai, A.; Rossetti, L. Regulation of blood glucose by hypothalamic pyruvate metabolism. Science 2005, 309, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Guillod-Maximin, E.; Lorsignol, A.; Alquier, T.; Penicaud, L. Acute intracarotid glucose injection towards the brain induces specific c-fos activation in hypothalamic nuclei: Involvement of astrocytes in cerebral glucose-sensing in rats. J. Neuroendocrinol. 2004, 16, 464–471. [Google Scholar] [CrossRef]
- Meek, T.H.; Nelson, J.T.; Matsen, M.E.; Dorfman, M.D.; Guyenet, S.J.; Damian, V.; Allison, M.B.; Scarlett, J.M.; Nguyen, H.T.; Thaler, J.P.; et al. Functional identification of a neurocircuit regulating blood glucose. Proc. Natl. Acad. Sci. USA 2016, 113, E2073–E2082. [Google Scholar] [CrossRef]
- Cai, D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol. Metab. 2013, 24, 40–47. [Google Scholar] [CrossRef]
- Li, J.; Tang, Y.; Cai, D. IKKβ/NF-κB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat. Cell Biol. 2012, 14, 999–1012. [Google Scholar] [CrossRef]
- Woods, S.C.; Lotter, E.C.; McKay, L.D.; Porte, D., Jr. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 1979, 282, 503–505. [Google Scholar] [CrossRef]
- Rossetti, L.; Shulman, G.I.; Zawalich, W.; DeFronzo, R.A. Effect of chronic hyperglycemia on in vivo insulin secretion in partially pancreatectomized rats. J. Clin. Investig. 1987, 80, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Mevorach, M.; Giacca, A.; Aharon, Y.; Hawkins, M.; Shamoon, H.; Rossetti, L. Regulation of endogenous glucose production by glucose per se is impaired in type 2 diabetes mellitus. J. Clin. Investig. 1998, 102, 744–753. [Google Scholar] [CrossRef]
- Williams, K.W.; Elmquist, J.K. From neuroanatomy to behavior: Central integration of peripheral signals regulating feeding behavior. Nat. Neurosci. 2012, 15, 1350–1355. [Google Scholar] [CrossRef]
- Jordan, S.D.; Konner, A.C.; Bruning, J.C. Sensing the fuels: Glucose and lipid signaling in the CNS controlling energy homeostasis. Cell. Mol. Life Sci. 2010, 67, 3255–3273. [Google Scholar] [CrossRef]
- Oomura, Y.; Kimura, K.; Ooyama, H.; Maeno, T.; Iki, M.; Kuniyoshi, M. Reciprocal activities of the ventromedial and lateral hypothalamic areas of cats. Science 1964, 143, 484–485. [Google Scholar] [CrossRef] [PubMed]
- Routh, V.H.; Hao, L.; Santiago, A.M.; Sheng, Z.; Zhou, C. Hypothalamic glucose sensing: Making ends meet. Front. Syst. Neurosci. 2014, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Kow, L.M.; Funabashi, T.; Mobbs, C.V. Hypothalamic glucose sensor: Similarities to and differences from pancreatic β-cell mechanisms. Diabetes 1999, 48, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Routh, V.H.; Kuzhikandathil, E.V.; Gaspers, L.D.; Levin, B.E. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes 2004, 53, 549–559. [Google Scholar] [CrossRef]
- Moriyama, R.; Tsukamura, H.; Kinoshita, M.; Okazaki, H.; Kato, Y.; Maeda, K. In vitro increase in intracellular calcium concentrations induced by low or high extracellular glucose levels in ependymocytes and serotonergic neurons of the rat lower brainstem. Endocrinology 2004, 145, 2507–2515. [Google Scholar] [CrossRef]
- Fioramonti, X.; Lorsignol, A.; Taupignon, A.; Penicaud, L. A new ATP-sensitive K+ channel-independent mechanism is involved in glucose-excited neurons of mouse arcuate nucleus. Diabetes 2004, 53, 2767–2775. [Google Scholar] [CrossRef]
- Song, Z.; Routh, V.H. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 2005, 54, 15–22. [Google Scholar] [CrossRef]
- O’Malley, D.; Reimann, F.; Simpson, A.K.; Gribble, F.M. Sodium-coupled glucose cotransporters contribute to hypothalamic glucose sensing. Diabetes 2006, 55, 3381–3386. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.A.; Reimann, F.; Burdakov, D. Dissociation between sensing and metabolism of glucose in sugar sensing neurones. J. Physiol. 2009, 587, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zhou, L.; Terwilliger, R.; Newton, S.S.; de Araujo, I.E. Sweet taste signaling functions as a hypothalamic glucose sensor. Front. Integr. Neurosci. 2009, 3, 12. [Google Scholar] [CrossRef]
- Toda, C.; Kim, J.D.; Impellizzeri, D.; Cuzzocrea, S.; Liu, Z.W.; Diano, S. UCP2 regulates mitochondrial fission and ventromedial nucleus control of glucose responsiveness. Cell 2016, 164, 872–883. [Google Scholar] [CrossRef]
- Silver, I.A.; Erecinska, M. Glucose-induced intracellular ion changes in sugar-sensitive hypothalamic neurons. J. Neurophysiol. 1998, 79, 1733–1745. [Google Scholar] [CrossRef]
- Fioramonti, X.; Marsollier, N.; Song, Z.; Fakira, K.A.; Patel, R.M.; Brown, S.; Duparc, T.; Pica-Mendez, A.; Sanders, N.M.; Knauf, C.; et al. Ventromedial hypothalamic nitric oxide production is necessary for hypoglycemia detection and counterregulation. Diabetes 2010, 59, 519–528. [Google Scholar] [CrossRef]
- Murphy, B.A.; Fakira, K.A.; Song, Z.; Beuve, A.; Routh, V.H. AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am. J. Physiol. Cell Physiol. 2009, 297, C750–C758. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, X.; Hentges, S.T.; Dunn-Meynell, A.A.; Levin, B.E.; Wang, W.; Routh, V.H. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 2004, 53, 1959–1965. [Google Scholar] [CrossRef]
- Romero-Zerbo, S.Y.; Bermudez-Silva, F.J. Cannabinoids, eating behaviour, and energy homeostasis. Drug Test. Anal. 2014, 6, 52–58. [Google Scholar] [CrossRef]
- Bermudez-Silva, F.J.; Cardinal, P.; Cota, D. The role of the endocannabinoid system in the neuroendocrine regulation of energy balance. J. Psychopharmacol. 2012, 26, 114–124. [Google Scholar] [CrossRef]
- Kohno, D.; Yada, T. Arcuate NPY neurons sense and integrate peripheral metabolic signals to control feeding. Neuropeptides 2012, 46, 315–319. [Google Scholar] [CrossRef]
- Coppari, R.; Ichinose, M.; Lee, C.E.; Pullen, A.E.; Kenny, C.D.; McGovern, R.A.; Tang, V.; Liu, S.M.; Ludwig, T.; Chua, S.C., Jr.; et al. The hypothalamic arcuate nucleus: A key site for mediating leptin’s effects on glucose homeostasis and locomotor activity. Cell Metab. 2005, 1, 63–72. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, C.; Uchida, A.; Chuang, J.C.; Walker, A.; Liu, T.; Osborne-Lawrence, S.; Mason, B.L.; Mosher, C.; Berglund, E.D.; et al. Arcuate AgRP neurons mediate orexigenic and glucoregulatory actions of ghrelin. Mol. Metab. 2014, 3, 64–72. [Google Scholar] [CrossRef]
- Muroya, S.; Yada, T.; Shioda, S.; Takigawa, M. Glucose-sensitive neurons in the rat arcuate nucleus contain neuropeptide Y. Neurosci. Lett. 1999, 264, 113–116. [Google Scholar] [CrossRef]
- Liu, T.; Kong, D.; Shah, B.P.; Ye, C.; Koda, S.; Saunders, A.; Ding, J.B.; Yang, Z.; Sabatini, B.L.; Lowell, B.B. Fasting activation of AgRP neurons requires NMDA receptors and involves spinogenesis and increased excitatory tone. Neuron 2012, 73, 511–522. [Google Scholar] [CrossRef]
- Yang, Y.; Atasoy, D.; Su, H.H.; Sternson, S.M. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 2011, 146, 992–1003. [Google Scholar] [CrossRef]
- Luquet, S.; Perez, F.A.; Hnasko, T.S.; Palmiter, R.D. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science 2005, 310, 683–685. [Google Scholar] [CrossRef]
- Gropp, E.; Shanabrough, M.; Borok, E.; Xu, A.W.; Janoschek, R.; Buch, T.; Plum, L.; Balthasar, N.; Hampel, B.; Waisman, A.; et al. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat. Neurosci. 2005, 8, 1289–1291. [Google Scholar] [CrossRef]
- Atasoy, D.; Betley, J.N.; Su, H.H.; Sternson, S.M. Deconstruction of a neural circuit for hunger. Nature 2012, 488, 172–177. [Google Scholar] [CrossRef]
- Ibrahim, N.; Bosch, M.A.; Smart, J.L.; Qiu, J.; Rubinstein, M.; Ronnekleiv, O.K.; Low, M.J.; Kelly, M.J. Hypothalamic proopiomelanocortin neurons are glucose responsive and express KATP channels. Endocrinology 2003, 144, 1331–1340. [Google Scholar] [CrossRef]
- Zhan, C.; Zhou, J.; Feng, Q.; Zhang, J.E.; Lin, S.; Bao, J.; Wu, P.; Luo, M. Acute and long-term suppression of feeding behavior by POMC neurons in the brainstem and hypothalamus, respectively. J. Neurosci. 2013, 33, 3624–3632. [Google Scholar] [CrossRef] [PubMed]
- Konner, A.C.; Janoschek, R.; Plum, L.; Jordan, S.D.; Rother, E.; Ma, X.; Xu, C.; Enriori, P.; Hampel, B.; Barsh, G.S.; et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007, 5, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.W.; Elias, C.F.; Fukuda, M.; Williams, K.W.; Berglund, E.D.; Holland, W.L.; Cho, Y.R.; Chuang, J.C.; Xu, Y.; Choi, M.; et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010, 11, 286–297. [Google Scholar] [CrossRef]
- Steculorum, S.M.; Ruud, J.; Karakasilioti, I.; Backes, H.; Engstrom Ruud, L.; Timper, K.; Hess, M.E.; Tsaousidou, E.; Mauer, J.; Vogt, M.C.; et al. AgRP neurons control systemic insulin sensitivity via myostatin expression in brown adipose tissue. Cell 2016, 165, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Krashes, M.J.; Koda, S.; Ye, C.; Rogan, S.C.; Adams, A.C.; Cusher, D.S.; Maratos-Flier, E.; Roth, B.L.; Lowell, B.B. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J. Clin. Investig. 2011, 121, 1424–1428. [Google Scholar] [CrossRef]
- Joly-Amado, A.; Denis, R.G.; Castel, J.; Lacombe, A.; Cansell, C.; Rouch, C.; Kassis, N.; Dairou, J.; Cani, P.D.; Ventura-Clapier, R.; et al. Hypothalamic AgRP-neurons control peripheral substrate utilization and nutrient partitioning. EMBO J. 2012, 31, 4276–4288. [Google Scholar] [CrossRef] [PubMed]
- Sainsbury, A.; Rohner-Jeanrenaud, F.; Cusin, I.; Zakrzewska, K.E.; Halban, P.A.; Gaillard, R.C.; Jeanrenaud, B. Chronic central neuropeptide Y infusion in normal rats: Status of the hypothalamo-pituitary-adrenal axis, and vagal mediation of hyperinsulinaemia. Diabetologia 1997, 40, 1269–1277. [Google Scholar] [CrossRef]
- Levin, B.E.; Magnan, C.; Dunn-Meynell, A.; Le Foll, C. Metabolic sensing and the brain: Who, what, where, and how? Endocrinology 2011, 152, 2552–2557. [Google Scholar] [CrossRef]
- Cortes-Campos, C.; Elizondo, R.; Carril, C.; Martinez, F.; Boric, K.; Nualart, F.; Garcia-Robles, M.A. MCT2 expression and lactate influx in anorexigenic and orexigenic neurons of the arcuate nucleus. PLoS ONE 2013, 8, e62532. [Google Scholar] [CrossRef]
- Garcia-Marin, V.; Garcia-Lopez, P.; Freire, M. Cajal’s contributions to glia research. Trends Neurosci. 2007, 30, 479–487. [Google Scholar] [CrossRef]
- Pellerin, L. How astrocytes feed hungry neurons. Mol. Neurobiol. 2005, 32, 59–72. [Google Scholar] [CrossRef]
- Yang, L.; Qi, Y.; Yang, Y. Astrocytes control food intake by inhibiting AGRP neuron activity via adenosine A1 receptors. Cell Rep. 2015, 11, 798–807. [Google Scholar] [CrossRef]
- Elizondo-Vega, R.; Cortes-Campos, C.; Barahona, M.J.; Oyarce, K.A.; Carril, C.A.; Garcia-Robles, M.A. The role of tanycytes in hypothalamic glucosensing. J. Cell Mol. Med. 2015, 19, 1471–1482. [Google Scholar] [CrossRef]
- Rodriguez, E.M.; Blazquez, J.L.; Pastor, F.E.; Pelaez, B.; Pena, P.; Peruzzo, B.; Amat, P. Hypothalamic tanycytes: A key component of brain-endocrine interaction. Int. Rev. Cytol. 2005, 247, 89–164. [Google Scholar] [CrossRef]
- Machler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; von Faber-Castell, A.; Kaelin, V.; Zuend, M.; San Martin, A.; Romero-Gomez, I.; et al. In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef]
- Wender, R.; Brown, A.M.; Fern, R.; Swanson, R.A.; Farrell, K.; Ransom, B.R. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J. Neurosci. 2000, 20, 6804–6810. [Google Scholar] [CrossRef]
- Scarlett, J.M.; Muta, K.; Brown, J.M.; Rojas, J.M.; Matsen, M.E.; Acharya, N.K.; Secher, A.; Ingvorsen, C.; Jorgensen, R.; Hoeg-Jensen, T.; et al. Peripheral mechanisms mediating the sustained antidiabetic action of FGF1 in the brain. Diabetes 2019, 68, 654–664. [Google Scholar] [CrossRef]
- Garcia, M.; Millan, C.; Balmaceda-Aguilera, C.; Castro, T.; Pastor, P.; Montecinos, H.; Reinicke, K.; Zuniga, F.; Vera, J.C.; Onate, S.A.; et al. Hypothalamic ependymal-glial cells express the glucose transporter GLUT2, a protein involved in glucose sensing. J. Neurochem. 2003, 86, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Frayling, C.; Britton, R.; Dale, N. ATP-mediated glucosensing by hypothalamic tanycytes. J. Physiol. 2011, 589, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Saez, P.J.; Cortes-Campos, C.; Elizondo, R.J.; Shoji, K.F.; Contreras-Duarte, S.; Figueroa, V.; Velarde, V.; Jiang, J.X.; Nualart, F.; et al. Glucose increases intracellular free Ca2+ in tanycytes via ATP released through connexin 43 hemichannels. Glia 2012, 60, 53–68. [Google Scholar] [CrossRef]
- Benford, H.; Bolborea, M.; Pollatzek, E.; Lossow, K.; Hermans-Borgmeyer, I.; Liu, B.; Meyerhof, W.; Kasparov, S.; Dale, N. A sweet taste receptor-dependent mechanism of glucosensing in hypothalamic tanycytes. Glia 2017, 65, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Meda, P.; Schuit, F. Glucose-stimulated insulin secretion: The hierarchy of its multiple cellular and subcellular mechanisms. Diabetologia 2013, 56, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Barahona, M.J.; Llanos, P.; Recabal, A.; Escobar-Acuna, K.; Elizondo-Vega, R.; Salgado, M.; Ordenes, P.; Uribe, E.; Sepulveda, F.J.; Araneda, R.C.; et al. Glial hypothalamic inhibition of GLUT2 expression alters satiety, impacting eating behavior. Glia 2018, 66, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Marty, N.; Dallaporta, M.; Foretz, M.; Emery, M.; Tarussio, D.; Bady, I.; Binnert, C.; Beermann, F.; Thorens, B. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J. Clin. Investig. 2005, 115, 3545–3553. [Google Scholar] [CrossRef]
- Thorens, B.; Guillam, M.T.; Beermann, F.; Burcelin, R.; Jaquet, M. Transgenic reexpression of GLUT1 or GLUT2 in pancreatic β cells rescues GLUT2-null mice from early death and restores normal glucose-stimulated insulin secretion. J. Biol. Chem. 2000, 275, 23751–23758. [Google Scholar] [CrossRef] [PubMed]
- Chari, M.; Yang, C.S.; Lam, C.K.; Lee, K.; Mighiu, P.; Kokorovic, A.; Cheung, G.W.; Lai, T.Y.; Wang, P.Y.; Lam, T.K. Glucose transporter-1 in the hypothalamic glial cells mediates glucose sensing to regulate glucose production in vivo. Diabetes 2011, 60, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Bolborea, M.; Dale, N. Hypothalamic tanycytes: Potential roles in the control of feeding and energy balance. Trends Neurosci. 2013, 36, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.; Diez, A.; Araque, A. Astrocytes in endocannabinoid signalling. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130599. [Google Scholar] [CrossRef]
- Bosier, B.; Bellocchio, L.; Metna-Laurent, M.; Soria-Gomez, E.; Matias, I.; Hebert-Chatelain, E.; Cannich, A.; Maitre, M.; Leste-Lasserre, T.; Cardinal, P.; et al. Astroglial CB1 cannabinoid receptors regulate leptin signaling in mouse brain astrocytes. Mol. Metab. 2013, 2, 393–404. [Google Scholar] [CrossRef]
- Perea, G.; Araque, A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science 2007, 317, 1083–1086. [Google Scholar] [CrossRef]
- Covelo, A.; Araque, A. Lateral regulation of synaptic transmission by astrocytes. Neuroscience 2016, 323, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Fuente-Martin, E.; Garcia-Caceres, C.; Argente-Arizon, P.; Diaz, F.; Granado, M.; Freire-Regatillo, A.; Castro-Gonzalez, D.; Ceballos, M.L.; Frago, L.M.; Dickson, S.L.; et al. Ghrelin regulates glucose and glutamate transporters in hypothalamic astrocytes. Sci. Rep. 2016, 6, 23673. [Google Scholar] [CrossRef]
- Fuente-Martin, E.; Garcia-Caceres, C.; Granado, M.; de Ceballos, M.L.; Sanchez-Garrido, M.A.; Sarman, B.; Liu, Z.W.; Dietrich, M.O.; Tena-Sempere, M.; Argente-Arizon, P.; et al. Leptin regulates glutamate and glucose transporters in hypothalamic astrocytes. J. Clin. Investig. 2012, 122, 3900–3913. [Google Scholar] [CrossRef]
- Fuente-Martin, E.; Garcia-Caceres, C.; Morselli, E.; Clegg, D.J.; Chowen, J.A.; Finan, B.; Brinton, R.D.; Tschop, M.H. Estrogen, astrocytes and the neuroendocrine control of metabolism. Rev. Endocr. Metab. Disord. 2013, 14, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Caceres, C.; Fuente-Martin, E.; Argente, J.; Chowen, J.A. Emerging role of glial cells in the control of body weight. Mol. Metab. 2012, 1, 37–46. [Google Scholar] [CrossRef]
- Garcia-Caceres, C.; Fuente-Martin, E.; Diaz, F.; Granado, M.; Argente-Arizon, P.; Frago, L.M.; Freire-Regatillo, A.; Barrios, V.; Argente, J.; Chowen, J.A. The opposing effects of ghrelin on hypothalamic and systemic inflammatory processes are modulated by its acylation status and food intake in male rats. Endocrinology 2014, 155, 2868–2880. [Google Scholar] [CrossRef]
- Sohl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef]
- Harris, A.L. Connexin channel permeability to cytoplasmic molecules. Prog. Biophys. Mol. Biol. 2007, 94, 120–143. [Google Scholar] [CrossRef]
- Allard, C.; Carneiro, L.; Grall, S.; Cline, B.H.; Fioramonti, X.; Chretien, C.; Baba-Aissa, F.; Giaume, C.; Penicaud, L.; Leloup, C. Hypothalamic astroglial connexins are required for brain glucose sensing-induced insulin secretion. J. Cereb. Blood Flow Metab. 2014, 34, 339–346. [Google Scholar] [CrossRef]
- Koulakoff, A.; Mei, X.; Orellana, J.A.; Saez, J.C.; Giaume, C. Glial connexin expression and function in the context of Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1818, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Barros, L.F. Metabolic signaling by lactate in the brain. Trends Neurosci. 2013, 36, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Allard, C.; Carneiro, L.; Collins, S.C.; Chretien, C.; Grall, S.; Penicaud, L.; Leloup, C. Alteration of hypothalamic glucose and lactate sensing in 48h hyperglycemic rats. Neurosci. Lett. 2013, 534, 75–79. [Google Scholar] [CrossRef]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and inflammatory adaptation of reactive astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef]
- Dello Russo, C.; Gavrilyuk, V.; Weinberg, G.; Almeida, A.; Bolanos, J.P.; Palmer, J.; Pelligrino, D.; Galea, E.; Feinstein, D.L. Peroxisome proliferator-activated receptor γ thiazolidinedione agonists increase glucose metabolism in astrocytes. J. Biol. Chem. 2003, 278, 5828–5836. [Google Scholar] [CrossRef]
- Fernandez, M.O.; Hsueh, K.; Park, H.T.; Sauceda, C.; Hwang, V.; Kumar, D.; Kim, S.; Rickert, E.; Mahata, S.; Webster, N.J.G. Astrocyte-specific deletion of peroxisome-proliferator activated receptor-γ impairs glucose metabolism and estrous cycling in female mice. J. Endocr. Soc. 2017, 1, 1332–1350. [Google Scholar] [CrossRef]
- Ferris, H.A.; Perry, R.J.; Moreira, G.V.; Shulman, G.I.; Horton, J.D.; Kahn, C.R. Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole-body metabolism. Proc.Natl. Acad. Sci. USA 2017, 114, 1189–1194. [Google Scholar] [CrossRef]
- Fuente-Martin, E.; Garcia-Caceres, C.; Diaz, F.; Argente-Arizon, P.; Granado, M.; Barrios, V.; Argente, J.; Chowen, J.A. Hypothalamic inflammation without astrogliosis in response to high sucrose intake is modulated by neonatal nutrition in male rats. Endocrinology 2013, 154, 2318–2330. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Caceres, C.; Fuente-Martin, E.; Burgos-Ramos, E.; Granado, M.; Frago, L.M.; Barrios, V.; Horvath, T.; Argente, J.; Chowen, J.A. Differential acute and chronic effects of leptin on hypothalamic astrocyte morphology and synaptic protein levels. Endocrinology 2011, 152, 1809–1818. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.J.; Bruning, J.C.; Lannon, S.; Kahn, C.R. Insulin signaling in the central nervous system is critical for the normal sympathoadrenal response to hypoglycemia. Diabetes 2005, 54, 1447–1451. [Google Scholar] [CrossRef][Green Version]
- Diggs-Andrews, K.A.; Zhang, X.; Song, Z.; Daphna-Iken, D.; Routh, V.H.; Fisher, S.J. Brain insulin action regulates hypothalamic glucose sensing and the counterregulatory response to hypoglycemia. Diabetes 2010, 59, 2271–2280. [Google Scholar] [CrossRef]
- Diano, S.; Kalra, S.P.; Horvath, T.L. Leptin receptor immunoreactivity is associated with the Golgi apparatus of hypothalamic neurons and glial cells. J. Neuroendocrinol. 1998, 10, 647–650. [Google Scholar] [CrossRef]
- Garcia-Segura, L.M.; Chowen, J.A.; Duenas, M.; Parducz, A.; Naftolin, F. Gonadal steroids and astroglial plasticity. Cell Mol. Neurobiol. 1996, 16, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.A.; Smith, R.G.; Diano, S.; Tschop, M.; Pronchuk, N.; Grove, K.L.; Strasburger, C.J.; Bidlingmaier, M.; Esterman, M.; Heiman, M.L.; et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003, 37, 649–661. [Google Scholar] [CrossRef]
- Briggs, D.I.; Andrews, Z.B. A recent update on the role of ghrelin in glucose homeostasis. Curr. Diabetes Rev. 2011, 7, 201–207. [Google Scholar] [CrossRef]
- Marina, N.; Turovsky, E.; Christie, I.N.; Hosford, P.S.; Hadjihambi, A.; Korsak, A.; Ang, R.; Mastitskaya, S.; Sheikhbahaei, S.; Theparambil, S.M.; et al. Brain metabolic sensing and metabolic signaling at the level of an astrocyte. Glia 2018, 66, 1185–1199. [Google Scholar] [CrossRef] [PubMed]
- Kamohara, S.; Burcelin, R.; Halaas, J.L.; Friedman, J.M.; Charron, M.J. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature 1997, 389, 374–377. [Google Scholar] [CrossRef]
- Minokoshi, Y.; Haque, M.S.; Shimazu, T. Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes 1999, 48, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Pelleymounter, M.A.; Cullen, M.J.; Baker, M.B.; Hecht, R.; Winters, D.; Boone, T.; Collins, F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995, 269, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdan, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Aponte, Y.; Atasoy, D.; Sternson, S.M. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat. Neurosci. 2011, 14, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Suyama, S.; Koch, M.; Jin, S.; Argente-Arizon, P.; Argente, J.; Liu, Z.W.; Zimmer, M.R.; Jeong, J.K.; Szigeti-Buck, K.; et al. Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat. Neurosci. 2014, 17, 908–910. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Woods, S.C.; Porte, D., Jr. Effect of cerebral intraventricular insulin on pancreatic insulin secretion in the dog. Diabetes 1975, 24, 910–914. [Google Scholar] [CrossRef]
- Cai, W.; Xue, C.; Sakaguchi, M.; Konishi, M.; Shirazian, A.; Ferris, H.A.; Li, M.; Yu, R.; Kleinridders, A.; Pothos, E.N.; et al. Insulin regulates astrocyte gliotransmission and modulates behavior. J. Clin. Investig. 2018, 128, 2914–2926. [Google Scholar] [CrossRef]
- Garcia-Caceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.X.; et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Hernandez-Garzon, E.; Perez-Domper, P.; Perez-Alvarez, A.; Mederos, S.; Matsui, T.; Santi, A.; Trueba-Saiz, A.; Garcia-Guerra, L.; Pose-Utrilla, J.; et al. Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes 2017, 66, 64–74. [Google Scholar] [CrossRef]
- Hernandez-Garzon, E.; Fernandez, A.M.; Perez-Alvarez, A.; Genis, L.; Bascunana, P.; Fernandez de la Rosa, R.; Delgado, M.; Angel Pozo, M.; Moreno, E.; McCormick, P.J.; et al. The insulin-like growth factor I receptor regulates glucose transport by astrocytes. Glia 2016, 64, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Milanski, M.; Coope, A.; Torsoni, A.S.; Ropelle, E.; Carvalho, D.P.; Carvalheira, J.B.; Velloso, L.A. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology 2011, 152, 1314–1326. [Google Scholar] [CrossRef]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Rom, S.; Zuluaga-Ramirez, V.; Gajghate, S.; Seliga, A.; Winfield, M.; Heldt, N.A.; Kolpakov, M.A.; Bashkirova, Y.V.; Sabri, A.K.; Persidsky, Y. Hyperglycemia-driven neuroinflammation compromises BBB leading to memory loss in both diabetes mellitus (DM) type 1 and type 2 mouse models. Mol. Neurobiol. 2019, 56, 1883–1896. [Google Scholar] [CrossRef]
- Calegari, V.C.; Torsoni, A.S.; Vanzela, E.C.; Araujo, E.P.; Morari, J.; Zoppi, C.C.; Sbragia, L.; Boschero, A.C.; Velloso, L.A. Inflammation of the hypothalamus leads to defective pancreatic islet function. J. Biol. Chem. 2011, 286, 12870–12880. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Schur, E.A.; Melhorn, S.J.; Oh, S.K.; Lacy, J.M.; Berkseth, K.E.; Guyenet, S.J.; Sonnen, J.A.; Tyagi, V.; Rosalynn, M.; De Leon, B.; et al. Radiologic evidence that hypothalamic gliosis is associated with obesity and insulin resistance in humans. Obesity 2015, 23, 2142–2148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Reichel, J.M.; Han, C.; Zuniga-Hertz, J.P.; Cai, D. Astrocytic process plasticity and IKKβ/NF-κB in central control of blood glucose, blood pressure, and body weight. Cell Metab. 2017, 25, 1091–1102. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, L.; Du, H.; Sun, Q.; Wang, X.; Cong, L.; Liu, X.; Yin, L.; Li, S.; Du, Y. Insulin attenuates β-amyloid-associated insulin/Akt/EAAT signaling perturbations in human astrocytes. Cell Mol. Neurobiol. 2016, 36, 851–864. [Google Scholar] [CrossRef]
- Magistretti, P.J. Neuron-glia metabolic coupling and plasticity. Exp. Physiol. 2011, 96, 407–410. [Google Scholar] [CrossRef]
- Ferreira, I.L.; Resende, R.; Ferreiro, E.; Rego, A.C.; Pereira, C.F. Multiple defects in energy metabolism in Alzheimer’s disease. Curr. Drug Targets 2010, 11, 1193–1206. [Google Scholar] [CrossRef]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef]
- Lu, F.P.; Lin, K.P.; Kuo, H.K. Diabetes and the risk of multi-system aging phenotypes: A systematic review and meta-analysis. PLoS ONE 2009, 4, e4144. [Google Scholar] [CrossRef]
- Beydoun, M.A.; Beydoun, H.A.; Wang, Y. Obesity and central obesity as risk factors for incident dementia and its subtypes: A systematic review and meta-analysis. Obes. Rev. 2008, 9, 204–218. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKβ/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef]
- Mitchell, J.P.; Carmody, R.J. NF-κB and the transcriptional control of inflammation. Int. Rev. Cell Mol. Biol. 2018, 335, 41–84. [Google Scholar] [CrossRef]
- Kim, D.; Nam, H.J.; Lee, W.; Yim, H.Y.; Ahn, J.Y.; Park, S.W.; Shin, H.R.; Yu, R.; Won, K.J.; Bae, J.S.; et al. PKCα-LSD1-NF-κB-signaling cascade is crucial for epigenetic control of the inflammatory response. Mol. Cell. 2018, 69, 398–411. [Google Scholar] [CrossRef]
- Fuente-Martin, E.; Mellado-Gil, J.M.; Cobo-Vuilleumier, N.; Romero-Zerbo, S.Y.; Bermudez-Silva, F.J.; Reyes, J.C.; Gauthier, B.R. HMG20A, bridging brain and islet in insulin expression and glucose homeostasis. Endocrinol. Diabetes Nutr. 2018, 65, 22. [Google Scholar]
- Mellado-Gil, J.M.; Fuente-Martin, E.; Lorenzo, P.I.; Cobo-Vuilleumier, N.; Romero-Zerbo, S.Y.; Garcia Fuentes, E.; Reyes, J.C.; Hmadcha, A.; Soria, B.; Martin Bermudo, F.; et al. A metabolic and neuronal network connected by HMG20A. manuscript in preparation 2019.
- Dolai, S.; Xie, L.; Zhu, D.; Liang, T.; Qin, T.; Xie, H.; Kang, Y.; Chapman, E.R.; Gaisano, H.Y. Synaptotagmin-7 functions to replenish insulin granules for exocytosis in human islet β-cells. Diabetes 2016, 65, 1962–1976. [Google Scholar] [CrossRef]
- Turecek, J.; Regehr, W.G. Synaptotagmin 7 mediates both facilitation and asynchronous release at granule cell synapses. J. Neurosci. 2018, 38, 3240–3251. [Google Scholar] [CrossRef]
- Wynder, C.; Hakimi, M.A.; Epstein, J.A.; Shilatifard, A.; Shiekhattar, R. Recruitment of MLL by HMG-domain protein iBRAF promotes neural differentiation. Nat. Cell Biol. 2005, 7, 1113–1117. [Google Scholar] [CrossRef]
- Gauthier, B.R.; Lorenzo, P.I.; Fuente-Martin, E.; Cobo-Vuilleumier, N.; Mellado-Gil, J.M.; Bermudez-Silva, F.J.; Rojo Martinez, G.; Reyes, J.C. LSD1 inhibitors for use in the treatment of type 2 Diabetes. Patent application EP19382051, 2019. [Google Scholar]
- Lorenzo, P.I.; Fuente-Martin, E.; Mellado-Gil, J.M.; Cobo-Vuilleumier, N.; Reyes, J.C.; Gauthier, B.R. Modulation of HMG20A signaling as possible treatment for T2DM. Endocrinol. Diabetes Nutr. 2019, 66, 100. [Google Scholar]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuente-Martín, E.; Mellado-Gil, J.M.; Cobo-Vuilleumier, N.; Martín-Montalvo, A.; Romero-Zerbo, S.Y.; Diaz Contreras, I.; Hmadcha, A.; Soria, B.; Martin Bermudo, F.; Reyes, J.C.; et al. Dissecting the Brain/Islet Axis in Metabesity. Genes 2019, 10, 350. https://doi.org/10.3390/genes10050350
Fuente-Martín E, Mellado-Gil JM, Cobo-Vuilleumier N, Martín-Montalvo A, Romero-Zerbo SY, Diaz Contreras I, Hmadcha A, Soria B, Martin Bermudo F, Reyes JC, et al. Dissecting the Brain/Islet Axis in Metabesity. Genes. 2019; 10(5):350. https://doi.org/10.3390/genes10050350
Chicago/Turabian StyleFuente-Martín, Esther, Jose M. Mellado-Gil, Nadia Cobo-Vuilleumier, Alejandro Martín-Montalvo, Silvana Y. Romero-Zerbo, Irene Diaz Contreras, Abdelkrim Hmadcha, Bernat Soria, Francisco Martin Bermudo, Jose C. Reyes, and et al. 2019. "Dissecting the Brain/Islet Axis in Metabesity" Genes 10, no. 5: 350. https://doi.org/10.3390/genes10050350
APA StyleFuente-Martín, E., Mellado-Gil, J. M., Cobo-Vuilleumier, N., Martín-Montalvo, A., Romero-Zerbo, S. Y., Diaz Contreras, I., Hmadcha, A., Soria, B., Martin Bermudo, F., Reyes, J. C., Bermúdez-Silva, F. J., Lorenzo, P. I., & Gauthier, B. R. (2019). Dissecting the Brain/Islet Axis in Metabesity. Genes, 10(5), 350. https://doi.org/10.3390/genes10050350