Congenital Leptin Deficiency and Leptin Gene Missense Mutation Found in Two Colombian Sisters with Severe Obesity

 , , ,
, , ,
Abstract
{kind=link}
{kind=link}
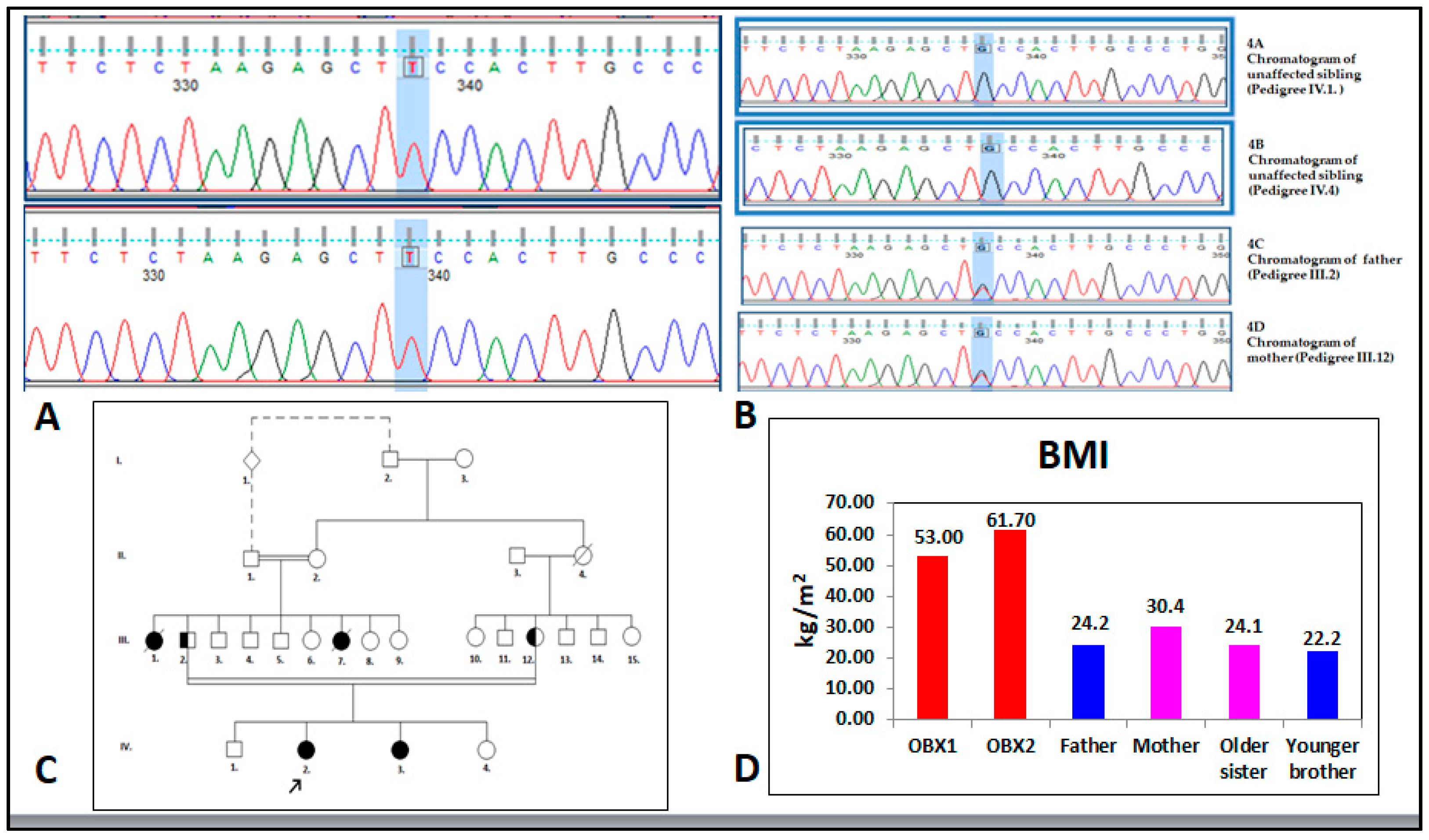{kind=link}
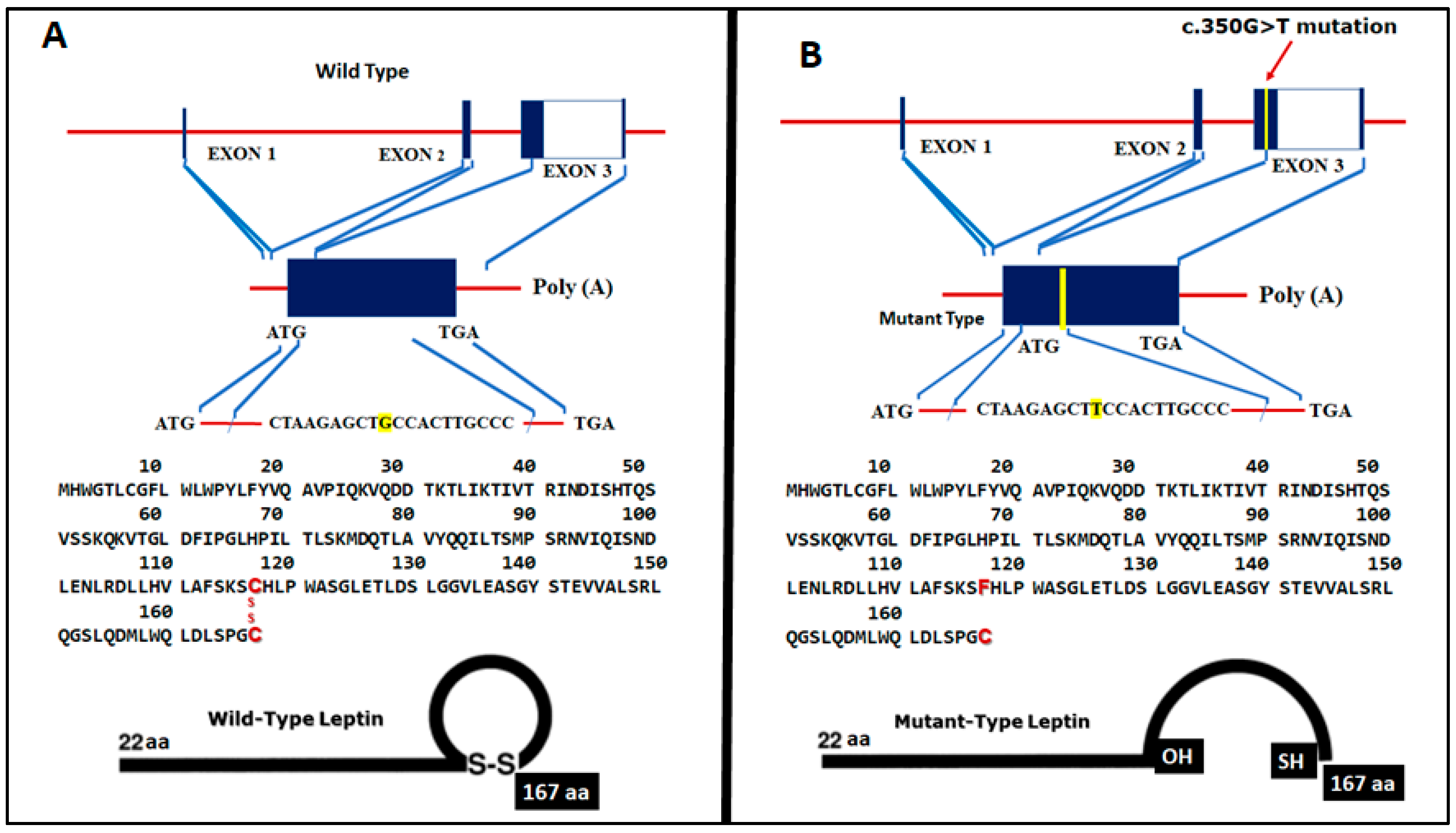{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethics Statement, Consent, and Permissions
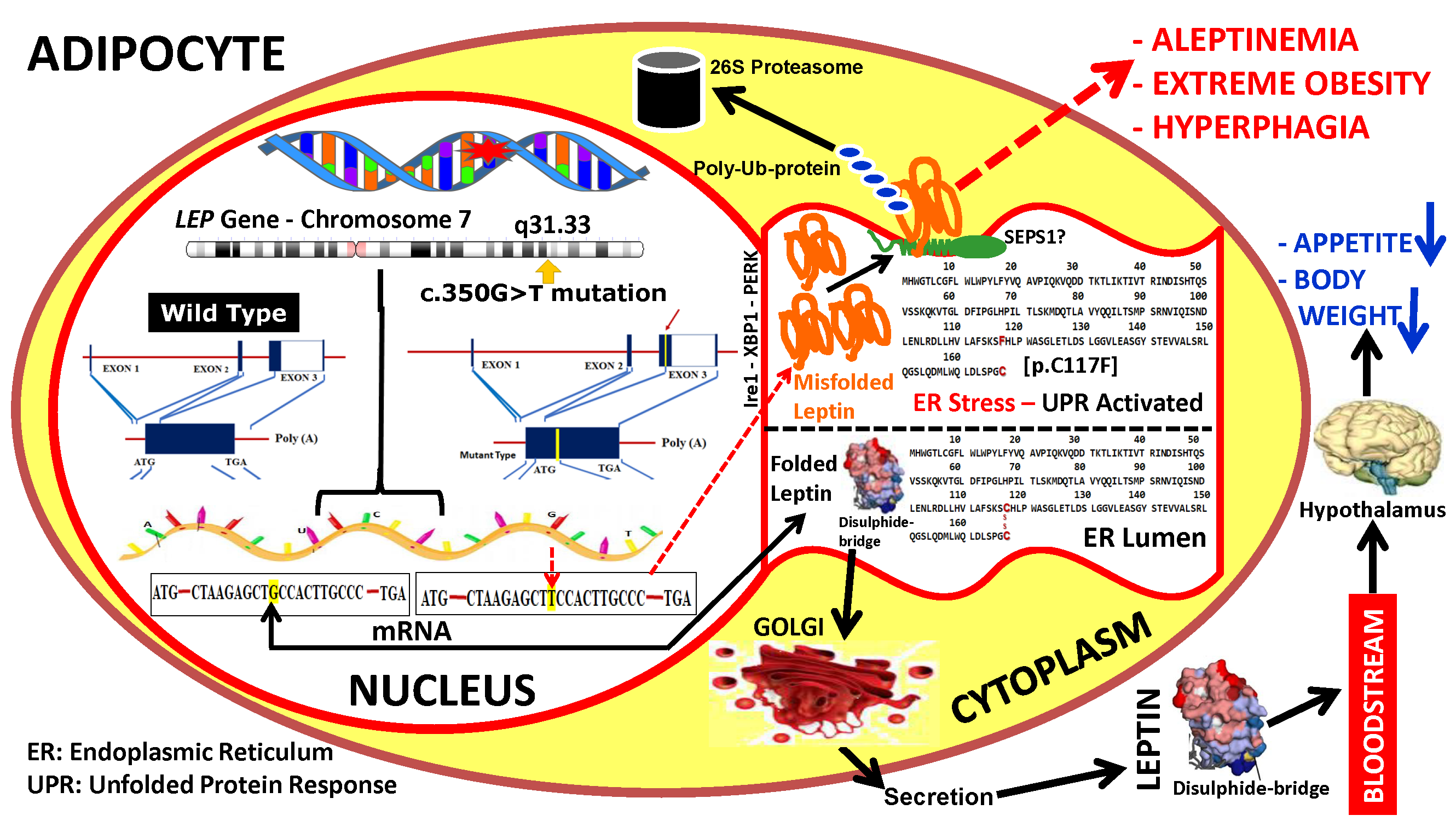
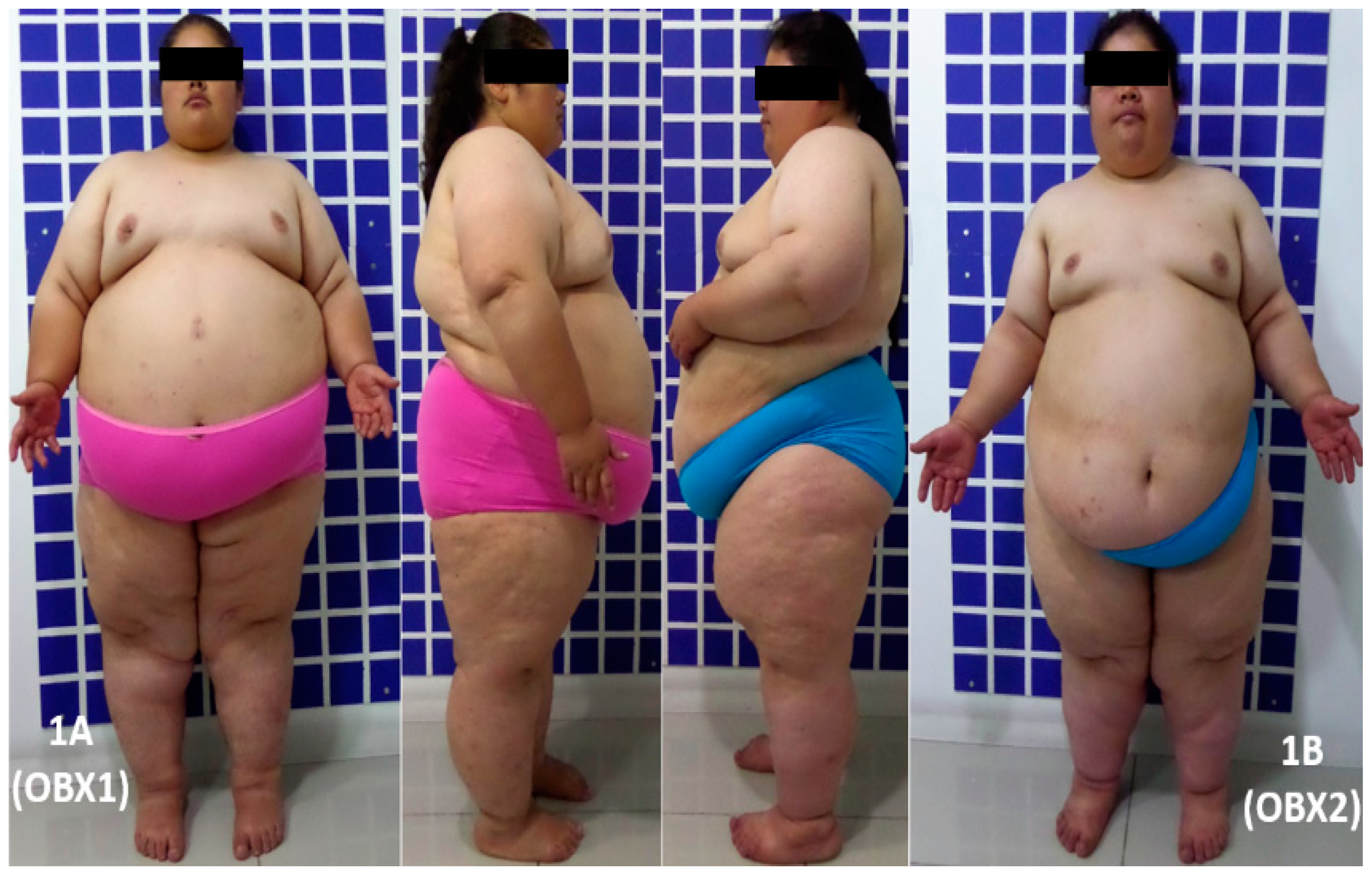
2.2. Subjects
2.3. Sequencing of Leptin Genomic DNA
2.4. Bioinformatics Analysis to Identify Point Mutations, Small Deletions, Insertions and Alterations
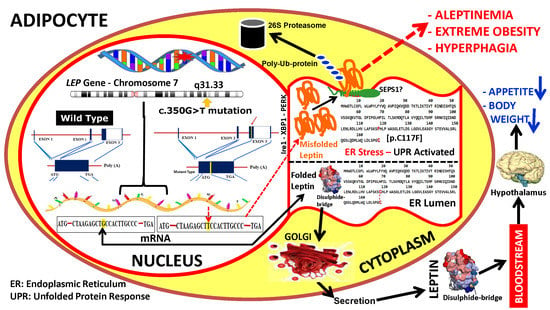
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Knight, J.A. Diseases and disorders associated with excess body weight. Ann. Clin. Lab. Sci. 2011, 41, 107–121. [Google Scholar]
- Styne, D.M.; Arslanian, S.A.; Connor, E.L.; Farooqi, I.S.; Murad, M.H.; Silverstein, J.H.; Yanovski, J.A. Pediatric Obesity-Assessment, Treatment, and Prevention: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2017, 102, 709–757. [Google Scholar] [CrossRef] [PubMed]
- Butte, N.F.; Comuzzie, A.G.; Cole, S.A.; Mehta, N.R.; Cai, G.; Tejero, M.; Bastarrachea, R.; Smith, E.O. Quantitative genetic analysis of the metabolic syndrome in Hispanic children. Pediatr. Res. 2005, 58, 1243–1248. [Google Scholar] [CrossRef][Green Version]
- O’Rahilly, S.; Farooqi, I.S. Human obesity as a heritable disorder of the central control of energy balance. Int. J. Obes. (2005) 2008, 32 (Suppl. 7), S55–S61. [Google Scholar] [CrossRef] [PubMed]
- Bastarrachea, R.A.; Gallegos-Cabriales, E.C.; Nava-González, E.J.; Haack, K.; Voruganti, V.S.; Charlesworth, J.; Laviada-Molina, H.A.; Veloz-Garza, R.A.; Cardenas-Villarreal, V.M.; Valdovinos-Chavez, S.B.; et al. Integrating genomic analysis with the genetic basis of gene expression: Preliminary evidence of the identification of causal genes for cardiovascular and metabolic traits related to nutrition in Mexicans. Adv. Nutr. 2012, 3, 596S–604S. [Google Scholar] [CrossRef]
- Bastarrachea, R.A.; Cole, S.A.; Comuzzie, A.G. Genomics of body weight regulation: Unraveling the molecular mechanisms predisposing to obesity. Med. Clin. (Barc.) 2004, 123, 104–117. [Google Scholar] [CrossRef]
- Hinney, A.; Vogel, C.I.; Hebebrand, J. From monogenic to polygenic obesity: Recent advances. Eur. Child Adolesc. Psychiatry 2010, 19, 297–310. [Google Scholar] [CrossRef]
- Nordang, G.B.; Busk, Ø.L.; Tveten, K.; Hanevik, H.I.; Fell, A.K.; Hjelmesæth, J.; Holla, Ø.L.; Hertel, J.K. Next-generation sequencing of the monogenic obesity genes LEP, LEPR, MC4R, PCSK1 and POMC in a Norwegian cohort of patients with morbid obesity and normal weight controls. Mol. Genet. Metab. 2017, 121, 51–56. [Google Scholar] [CrossRef]
- Saeed, S.; Bonnefond, A.; Manzoor, J.; Shabir, F.; Ayesha, H.; Philippe, J.; Durand, E.; Crouch, H.; Sand, O.; Ali, M.; et al. Genetic variants in LEP, LEPR, and MC4R explain 30% of severe obesity in children from a consanguineous population. Obesity (Silver Spring) 2015, 23, 1687–1695. [Google Scholar] [CrossRef]
- Chung, W.K. An overview of mongenic and syndromic obesities in humans. Pediatr. Blood Cancer 2012, 58, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Huvenne, H.; Dubern, B.; Clement, K.; Poitou, C. Rare Genetic Forms of Obesity: Clinical Approach and Current Treatments in 2016. Obes. Facts 2016, 9, 158–173. [Google Scholar] [CrossRef]
- Saeed, S.; Arslan, M.; Froguel, P. Genetics of Obesity in Consanguineous Populations: Toward Precision Medicine and the Discovery of Novel Obesity Genes. Obesity (Silver Spring) 2018, 26, 474–484. [Google Scholar] [CrossRef]
- Mantzoros, C.S.; Magkos, F.; Brinkoetter, M.; Sienkiewicz, E.; Dardeno, T.A.; Kim, S.Y.; Hamnvik, O.P.; Koniaris, A. Leptin in human physiology and pathophysiology. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E567–E584. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J. 20 years of leptin: Leptin at 20: An overview. J. Endocrinol. 2014, 223, T1–T8. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.J.; Tsai, P.J.; Cheng, C.Y.; Chou, C.K.; Jheng, H.F.; Chuang, Y.C.; Yang, C.N.; Lin, Y.T.; Hsu, C.W.; Cheng, I.H.; et al. ENU mutagenesis identifies mice with morbid obesity and severe hyperinsulinemia caused by a novel mutation in leptin. PLoS ONE 2010, 5, e15333. [Google Scholar] [CrossRef]
- Farooqi, I.S. The severely obese patient—A genetic work-up. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 172–177, quiz following 7. [Google Scholar] [CrossRef]
- Comuzzie, A.G.; Hixson, J.E.; Almasy, L.; Mitchell, B.D.; Mahaney, M.C.; Dyer, T.D.; Stern, M.P.; MacCluer, J.W.; Blangero, J. A major quantitative trait locus determining serum leptin levels and fat mass is located on human chromosome 2. Nat. Genet. 1997, 15, 273–276. [Google Scholar] [CrossRef]
- Dayal, D.; Seetharaman, K.; Panigrahi, I.; Muthuvel, B.; Agarwal, A. Severe Early Onset Obesity due to a Novel Missense Mutation in Exon 3 of the Leptin Gene in an Infant from Northwest India. J. Clin. Res. Pediatr. Endocrinol. 2018, 10, 274–278. [Google Scholar] [CrossRef]
- Montague, C.T.; Farooqi, I.S.; Whitehead, J.P.; Soos, M.A.; Rau, H.; Wareham, N.J.; Sewter, C.P.; Digby, J.E.; Mohammed, S.N.; Hurst, J.A.; et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997, 387, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Strobel, A.; Issad, T.; Camoin, L.; Ozata, M.; Strosberg, A.D. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat. Genet. 1998, 18, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Chekhranova, M.K.; Karpova, S.K.; Iatsyshina, S.B.; Pankov Iu, A. A new mutation c.422C>G (p.S141C) in homo- and heterozygous forms of the human leptin gene. Bioorg. Khim. 2008, 34, 854–856. [Google Scholar]
- Mazen, I.; El-Gammal, M.; Abdel-Hamid, M.; Amr, K. A novel homozygous missense mutation of the leptin gene (N103K) in an obese Egyptian patient. Mol. Genet. Metab. 2009, 97, 305–308. [Google Scholar] [CrossRef]
- Fischer-Posovszky, P.; von Schnurbein, J.; Moepps, B.; Lahr, G.; Strauss, G.; Barth, T.F.; Kassubek, J.; Muhleder, H.; Möller, P.; Debatin, K.M.; et al. A new missense mutation in the leptin gene causes mild obesity and hypogonadism without affecting T cell responsiveness. J. Clin. Endocrinol. Metab. 2010, 95, 2836–2840. [Google Scholar] [CrossRef]
- Fatima, W.; Shahid, A.; Imran, M.; Manzoor, J.; Hasnain, S.; Rana, S.; Mahmood, S. Leptin deficiency and leptin gene mutations in obese children from Pakistan. Int. J. Pediatr. Obes. 2011, 6, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Kumar, A.; Dubey, S.; Saxena, R.; Peters, A.N.; Singhal, A. A novel mutation of the leptin gene in an Indian patient. Clin. Genet. 2014, 86, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hong, N.; Liu, X.; Wu, B.; Tang, S.; Yang, J.; Hu, C.; Jia, W. A novel mutation in leptin gene is associated with severe obesity in Chinese individuals. BioMed Res. Int. 2014, 2014, 912052. [Google Scholar] [CrossRef]
- Wabitsch, M.; Funcke, J.B.; von Schnurbein, J.; Denzer, F.; Lahr, G.; Mazen, I.; El-Gammal, M.; Denzer, C.; Moss, A.; Debatin, K.M.; et al. Severe Early-Onset Obesity Due to Bioinactive Leptin Caused by a p.N103K Mutation in the Leptin Gene. J. Clin. Endocrinol. Metab. 2015, 100, 3227–3230. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Deng, Y.M.; Spirason, N.; Iannello, P.; Jelley, L.; Lau, H.; Barr, I.G. A simplified Sanger sequencing method for full genome sequencing of multiple subtypes of human influenza A viruses. J. Clin. Virol. 2015, 68, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Aswini, R.; Jayapalan, S. Modified Ferriman-Gallwey Score in Hirsutism and its Association with Metabolic Syndrome. Int. J. Trichol. 2017, 9, 7–13. [Google Scholar]
- Addo, O.Y.; Sarafoglou, K.; Miller, B.S. Effect of Adjusting for Tanner Stage Age on Prevalence of Short and Tall Stature of Youths in the United States. J. Pediatr. 2018, 201, 93.e4–99.e4. [Google Scholar] [CrossRef] [PubMed]
- Whiffin, N.; Minikel, E.; Walsh, R.; O’Donnell-Luria, A.H.; Karczewski, K.; Ing, A.Y.; Barton, P.J.; Funke, B.; Cook, S.A.; MacArthur, D.; et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 2017, 19, 1151–1158. [Google Scholar] [CrossRef]
- Denver, R.J.; Bonett, R.M.; Boorse, G.C. Evolution of leptin structure and function. Neuroendocrinology 2011, 94, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Nava-Gonzalez, E.J.; Gallegos-Cabriales, E.C.; Leal-Berumen, I.; Bastarrachea, R.A. Mini-Review: The Contribution of Intermediate Phenotypes to GxE Effects on Disorders of Body Composition in the New OMICS Era. Int. J. Environ. Res. Public Health 2017, 14, 1079. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.W.; Mao, P. Recent advances in obesity: Genetics and beyond. ISRN Endocrinol. 2012, 2012, 536905. [Google Scholar] [CrossRef] [PubMed]
- Chesi, A.; Grant, S.F.A. The Genetics of Pediatric Obesity. Trends Endocrinol. Metab. 2015, 26, 711–721. [Google Scholar] [CrossRef]
- Serra-Juhé, C.; Martos-Moreno, G.Á.; de Pieri, F.B.; Flores, R.; González, J.R.; Rodríguez-Santiago, B.; Argente, J.; Pérez-Jurado, L.A. Novel genes involved in severe early-onset obesity revealed by rare copy number and sequence variants. PLoS Genet. 2017, 13, e1006657. [Google Scholar] [CrossRef]
- Khan, S.A.; Muhammad, N.; Khan, M.A.; Kamal, A.; Rehman, Z.U.; Khan, S. Genetics of human Bardet-Biedl syndrome, an updates. Clin. Genet. 2016, 90, 3–15. [Google Scholar] [CrossRef]
- Abdilla, Y.; Andria Barbara, M.; Calleja-Agius, J. Prader-Willi Syndrome: Background and Management. Neonatal Netw. 2017, 36, 134–141. [Google Scholar] [CrossRef]
- Zammit, M.; Caruana, E.; Cassar, D.; Calleja-Agius, J. Beckwith-Wiedemann Syndrome Review: A Guide for the Neonatal Nurse. Neonatal Netw. 2017, 36, 129–133. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Haglund, E.; Nguyen, L.; Schafer, N.P.; Lammert, H.; Jennings, P.A.; Onuchic, J.N. Uncovering the molecular mechanisms behind disease-associated leptin variants. J. Biol. Chem. 2018, 293, 12919–12933. [Google Scholar] [CrossRef]
- Hainerova, I.A.; Lebl, J. Treatment options for children with monogenic forms of obesity. World Rev. Nutr. Diet. 2013, 106, 105–112. [Google Scholar] [PubMed]
- Paz-Filho, G.; Wong, M.L.; Licinio, J. Ten years of leptin replacement therapy. Obes Rev. 2011, 12, e315–e323. [Google Scholar] [CrossRef]
- Oral, E.A.; Chan, J.L. Rationale for leptin-replacement therapy for severe lipodystrophy. Endocr. Pract. 2010, 16, 324–333. [Google Scholar] [CrossRef]
- Paz-Filho, G.; Mastronardi, C.A.; Licinio, J. Leptin treatment: Facts and expectations. Metabolism 2015, 64, 146–156. [Google Scholar] [CrossRef]
- Meehan, C.A.; Cochran, E.; Kassai, A.; Brown, R.J.; Gorden, P. Metreleptin for injection to treat the complications of leptin deficiency in patients with congenital or acquired generalized lipodystrophy. Expert Rev. Clin. Pharmacol. 2016, 9, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Sepulveda, C.; Ardagna, Y.; Dutour, O. Paleoepidemiology of vertebral degenerative disease in a Pre-Columbian Muisca series from Colombia. Am. J. Phys. Anthropol. 2008, 135, 416–430. [Google Scholar] [CrossRef]
- Casas-Vargas, A.; Romero, L.M.; Usaquén, W.; Zea, S.; Silva, M.; Briceño, I.; Gómez, A.; Rodríguez, J.V. Mitochondrial DNA diversity in prehispanic bone remains on the eastern Colombian Andes. Biomedica 2017, 37, 548–560. [Google Scholar] [CrossRef]
- Liascovich, R.; Rittler, M.; Castilla, E.E. Consanguinity in South America: Demographic aspects. Hum. Hered. 2001, 51, 27–34. [Google Scholar] [CrossRef]
- De Castro, M.; Restrepo, C.M. Genetics and genomic medicine in Colombia. Mol. Genet. Genom. Med. 2015, 3, 84–91. [Google Scholar] [CrossRef]
- Boute, N.; Zilberfarb, V.; Camoin, L.; Bonnafous, S.; Le Marchand-Brustel, Y.; Issad, T. The formation of an intrachain disulfide bond in the leptin protein is necessary for efficient leptin secretion. Biochimie 2004, 86, 351–356. [Google Scholar] [CrossRef]
- Zhang, F.; Basinski, M.B.; Beals, J.M.; Briggs, S.L.; Churgay, L.M.; Clawson, D.K.; DiMarchi, R.D.; Furman, T.C.; Hale, J.E.; Hsiung, H.M.; et al. Crystal structure of the obese protein leptin-E100. Nature 1997, 387, 206–209. [Google Scholar] [CrossRef]
- Wittrup, K.D. Disulfide bond formation and eukaryotic secretory productivity. Curr. Opin. Biotechnol. 1995, 6, 203–208. [Google Scholar] [CrossRef]
- Sawyer, J.T.; Lukaczyk, T.; Yilla, M. Dithiothreitol treatment induces heterotypic aggregation of newly synthesized secretory proteins in HepG2 cells. J. Biol. Chem. 1994, 269, 22440–22445. [Google Scholar]
- Farooqi, I.S.; O’Rahilly, S. New advances in the genetics of early onset obesity. Int. J. Obes. (2005) 2005, 29, 1149–1152. [Google Scholar] [CrossRef][Green Version]
- Ozata, M.; Ozdemir, I.C.; Licinio, J. Human leptin deficiency caused by a missense mutation: Multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J. Clin. Endocrinol. Metab. 1999, 84, 3686–3695. [Google Scholar]
- Kopera, D.; Wehr, E.; Obermayer-Pietsch, B. Endocrinology of hirsutism. Int. J. Trichol. 2010, 2, 30–35. [Google Scholar] [CrossRef]
- Elghblawi, E. Idiopathic hirsutism: Excessive bodily and facial hair in women. Br. J. Nurs. 2008, 17, 192–197. [Google Scholar] [CrossRef]
- Serafini, P.; Lobo, R.A. Increased 5 α-reductase activity in idiopathic hirsutism. Fertil. Steril. 1985, 43, 74–78. [Google Scholar] [CrossRef]
- Sawaya, M.E.; Shalita, A.R. Androgen receptor polymorphisms (CAG repeat lengths) in androgenetic alopecia, hirsutism, and acne. J. Cutan. Med. Surg. 1998, 3, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Talaei, A.; Adgi, Z.; Mohamadi Kelishadi, M. Idiopathic hirsutism and insulin resistance. Int. J. Endocrinol. 2013, 2013, 593197. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yupanqui-Lozno, H.; Bastarrachea, R.A.; Yupanqui-Velazco, M.E.; Alvarez-Jaramillo, M.; Medina-Méndez, E.; Giraldo-Peña, A.P.; Arias-Serrano, A.; Torres-Forero, C.; Garcia-Ordoñez, A.M.; Mastronardi, C.A.; et al. Congenital Leptin Deficiency and Leptin Gene Missense Mutation Found in Two Colombian Sisters with Severe Obesity. Genes 2019, 10, 342. https://doi.org/10.3390/genes10050342
Yupanqui-Lozno H, Bastarrachea RA, Yupanqui-Velazco ME, Alvarez-Jaramillo M, Medina-Méndez E, Giraldo-Peña AP, Arias-Serrano A, Torres-Forero C, Garcia-Ordoñez AM, Mastronardi CA, et al. Congenital Leptin Deficiency and Leptin Gene Missense Mutation Found in Two Colombian Sisters with Severe Obesity. Genes. 2019; 10(5):342. https://doi.org/10.3390/genes10050342
Chicago/Turabian StyleYupanqui-Lozno, Hernan, Raul A. Bastarrachea, Maria E. Yupanqui-Velazco, Monica Alvarez-Jaramillo, Esteban Medina-Méndez, Aida P. Giraldo-Peña, Alexandra Arias-Serrano, Carolina Torres-Forero, Angelica M. Garcia-Ordoñez, Claudio A. Mastronardi, and et al. 2019. "Congenital Leptin Deficiency and Leptin Gene Missense Mutation Found in Two Colombian Sisters with Severe Obesity" Genes 10, no. 5: 342. https://doi.org/10.3390/genes10050342
APA StyleYupanqui-Lozno, H., Bastarrachea, R. A., Yupanqui-Velazco, M. E., Alvarez-Jaramillo, M., Medina-Méndez, E., Giraldo-Peña, A. P., Arias-Serrano, A., Torres-Forero, C., Garcia-Ordoñez, A. M., Mastronardi, C. A., Restrepo, C. M., Rodriguez-Ayala, E., Nava-Gonzalez, E. J., Arcos-Burgos, M., Kent, J. W., Jr., Cole, S. A., Licinio, J., & Celis-Regalado, L. G. (2019). Congenital Leptin Deficiency and Leptin Gene Missense Mutation Found in Two Colombian Sisters with Severe Obesity. Genes, 10(5), 342. https://doi.org/10.3390/genes10050342