Metagenomics-Guided Survey, Isolation, and Characterization of Uranium Resistant Microbiota from the Savannah River Site, USA


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Sample Collection
2.2. Establishment of Diffusion Chambers/ Microbial Traps and Isolation of U-Resistant Microbiota
2.3. Microbiome Analysis
2.4. Identification of the Isolated Strains
2.5. Evaluation of Uranium Resistance of the Isolated Strains
2.6. Evaluation of Uranium Depletion by the Isolated Microbiota Using Soil Microcosms
2.7. Genome Sequencing and Bioinformatic Characterization of Strain MT2
2.8. Comparative Genomics of Strain MT2
2.9. Nucleotide Sequence Accession Number
2.10. Metagenomic Sequence Accession Number
2.11. Whole Genome Sequence Submission
3. Results and Discussion
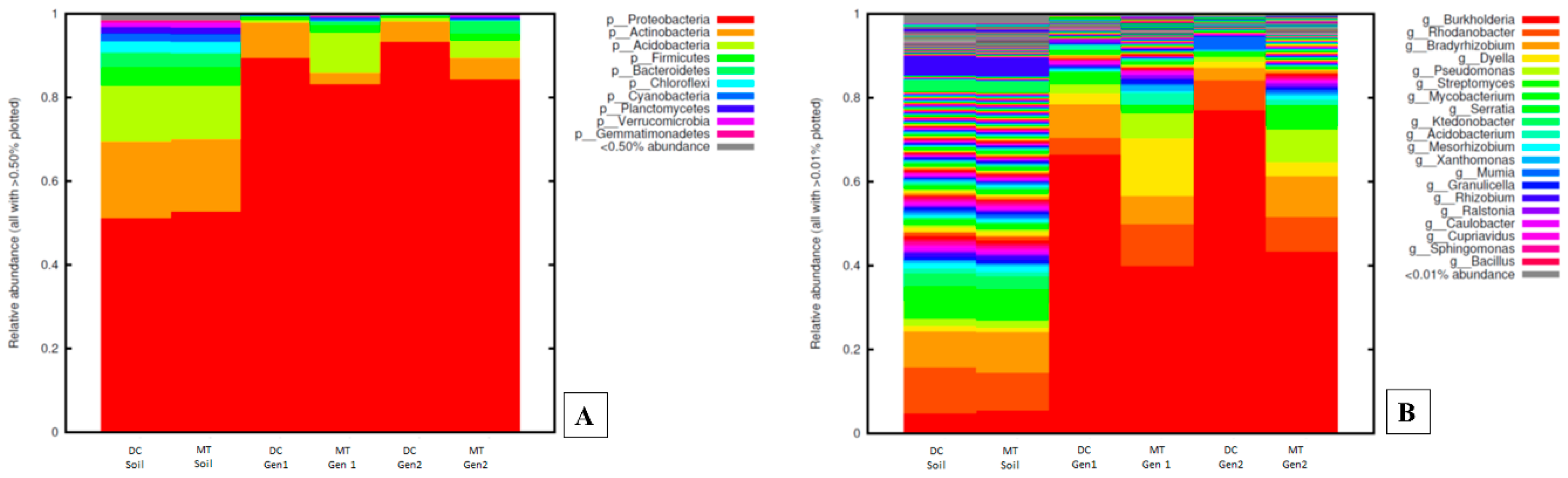
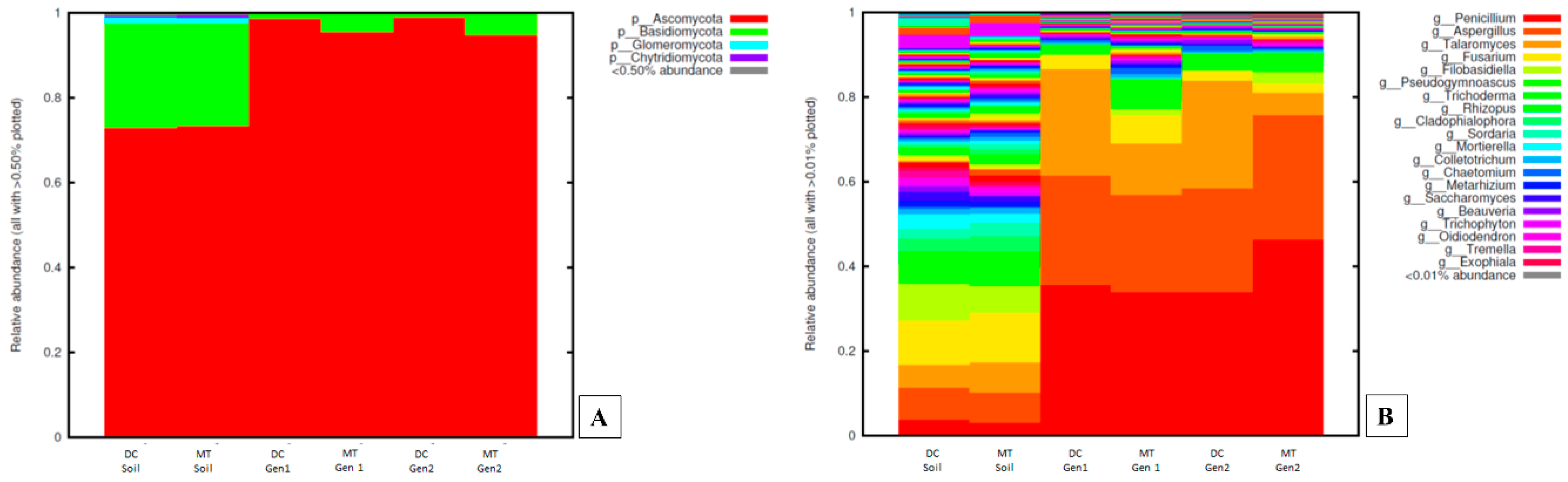
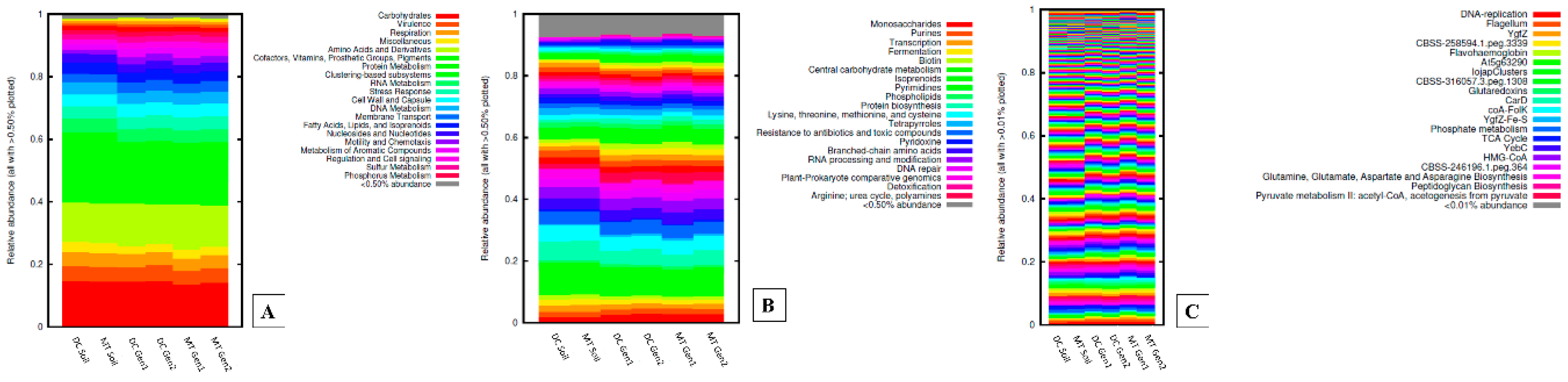
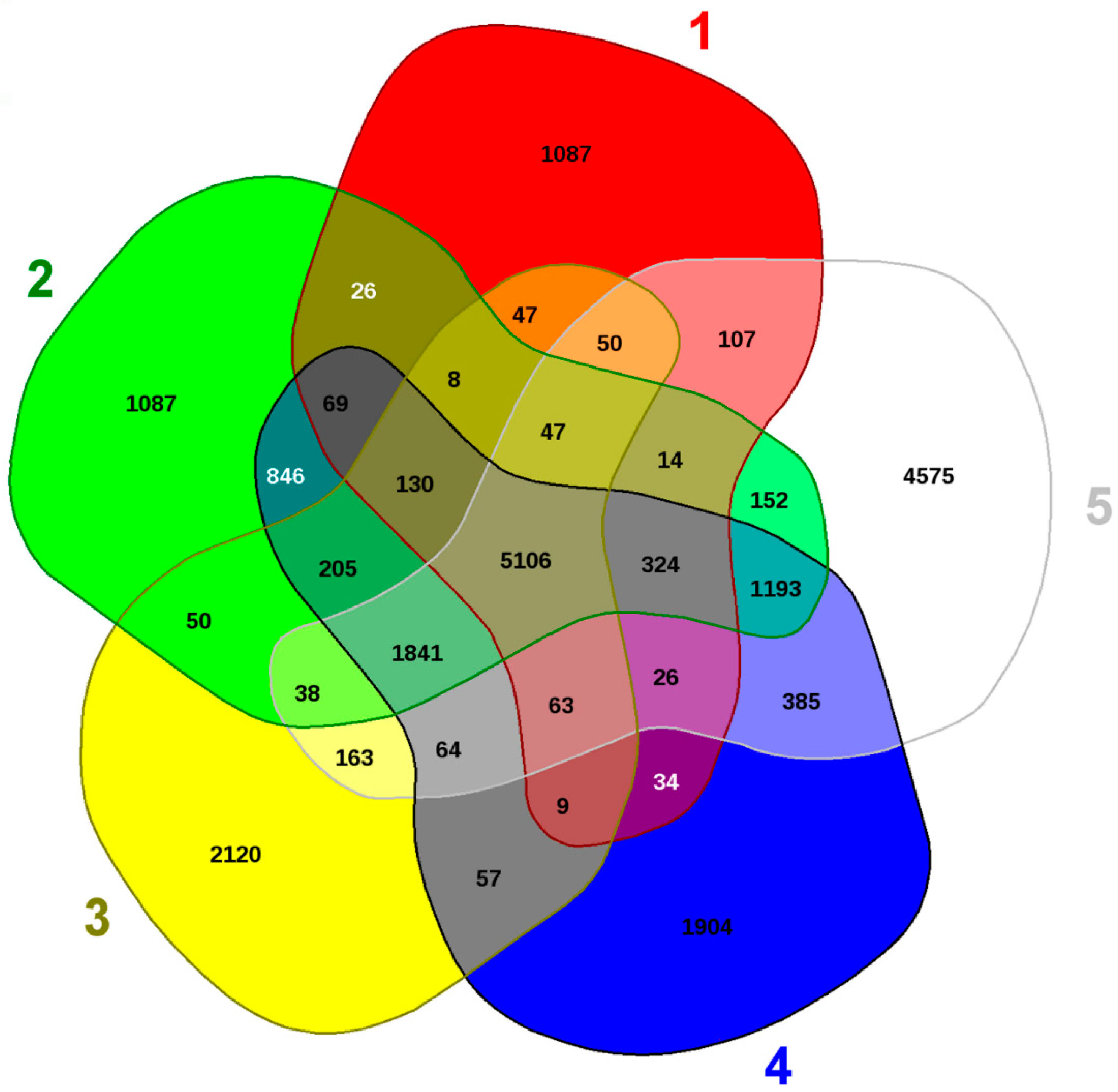
3.1. Changes in the Bacterial, Archaeal, and Fungal Assemblages between DC and MT Generations
3.2. Gene Functional Analysis in Soils Relative to DC and MT Chambers
3.3. Isolation of Bacteria and Fungi from DC/MT Chambers
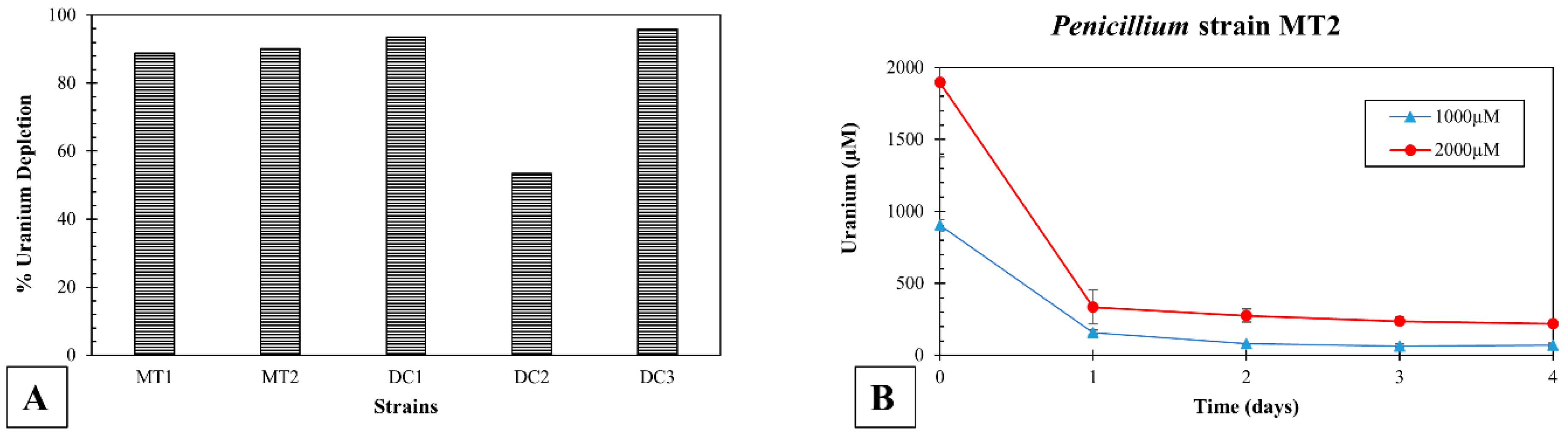
3.4. Screening for Uranium Resistance in the Isolated Strains
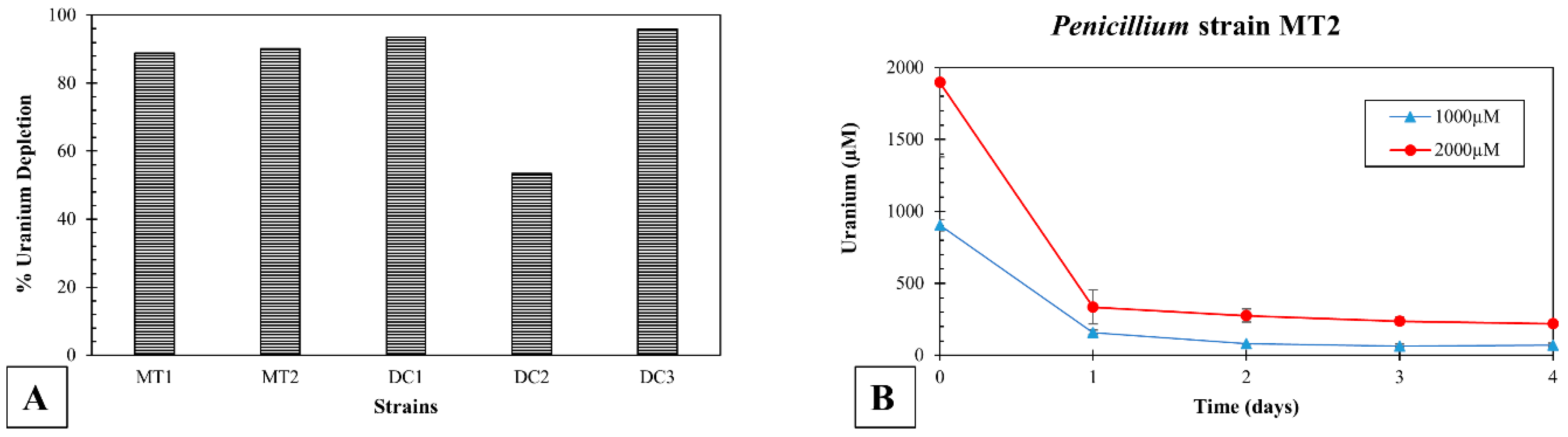
3.5. Uranium Depletion by the Isolated Microbiota in Soil Microcosms
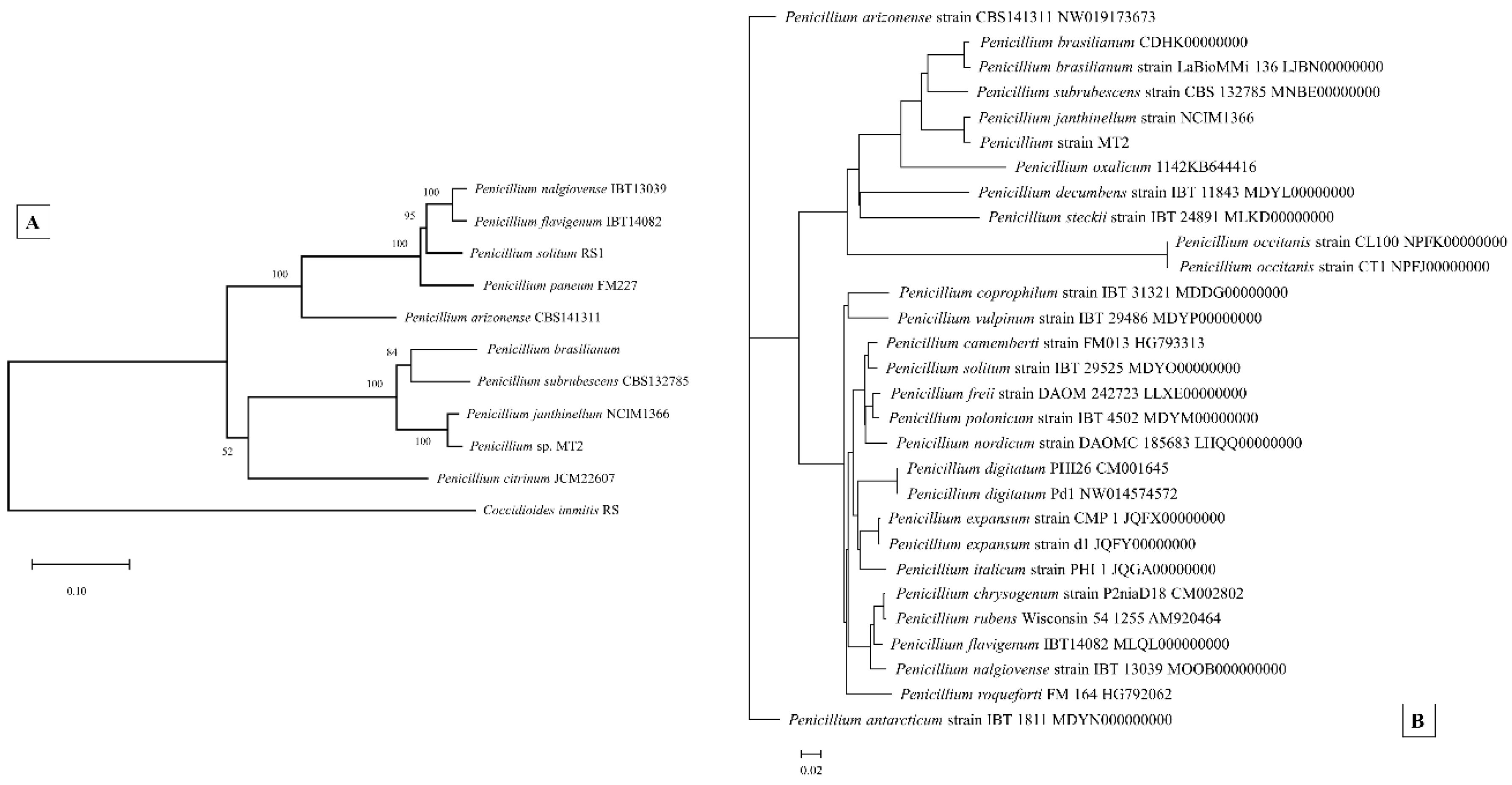
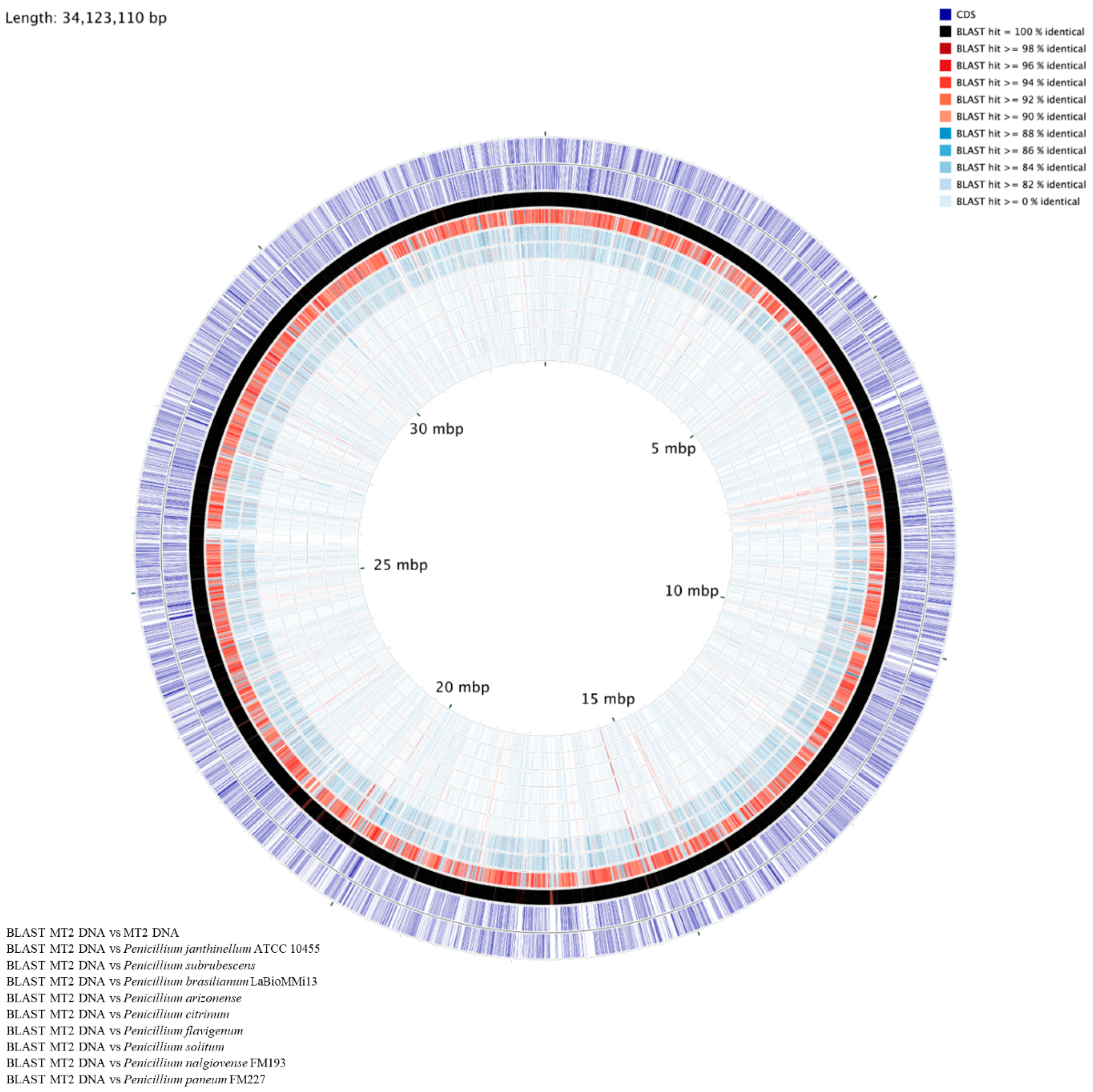
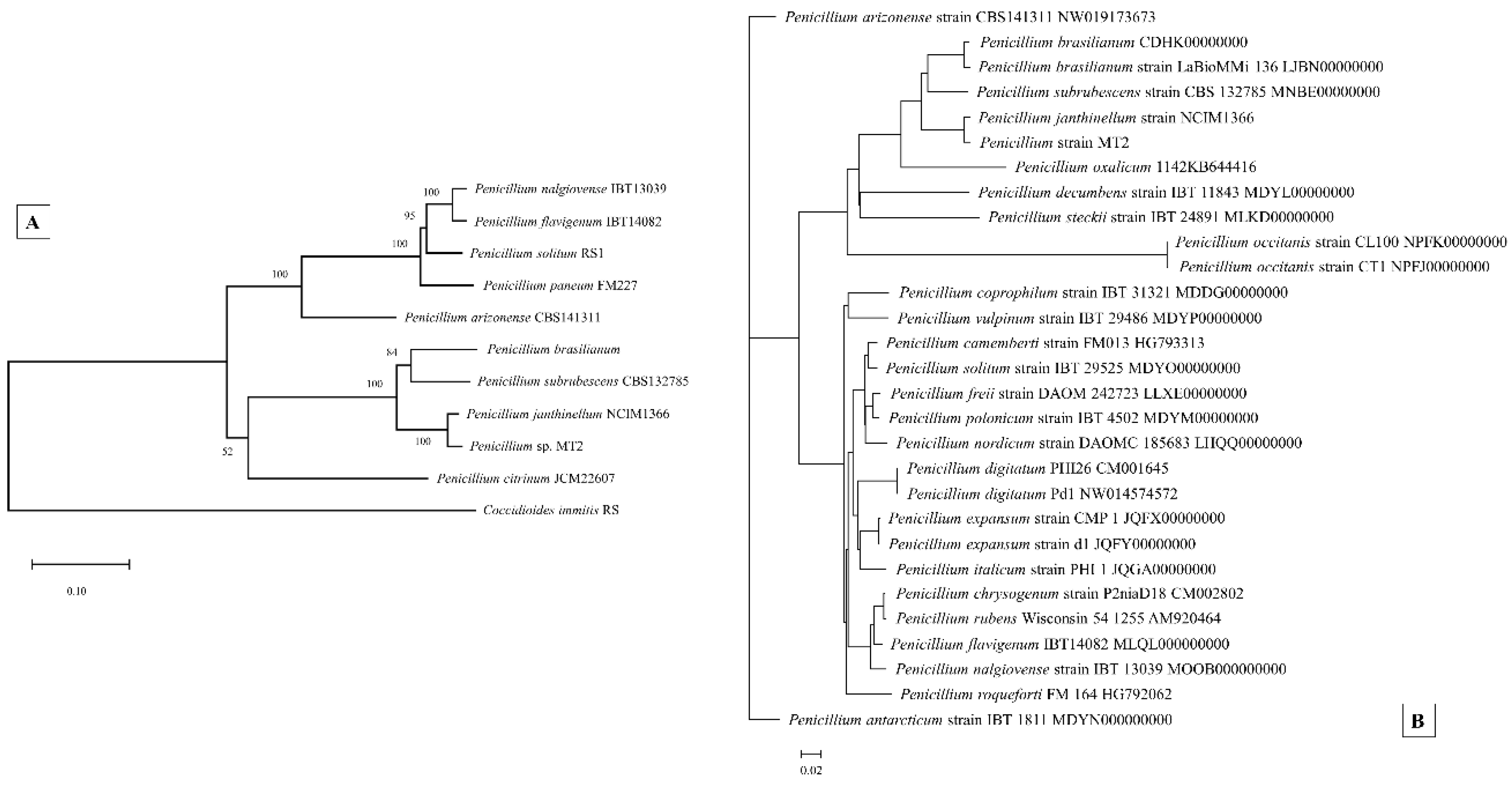
3.6. Genome-Centric Evaluation and Comparative Genomics of Strain MT2
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Flandroy, L.; Poutahidis, T.; Berg, G.; Clarke, G.; Dao, M.-C.; Decaestecker, E.; Furman, E.; Haahtela, T.; Massart, S.; Plovier, H.; et al. The impact of human activities and lifestyles on the interlinked microbiota and health of humans and of ecosystems. Sci. Total Environ. 2018, 627, 1018–1038. [Google Scholar] [CrossRef]
- Keller, M.; Zengler, K. Tapping into microbial diversity. Nat. Rev. Microbiol. 2004, 2, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.; Lewis, K.; Orjala, J.; Mo, S.; Ortenberg, R.; O’Connor, P.; Zhao, C.; Vouros, P.; Kaeberlein, T.; Epstein, S.S. Short peptide induces an “uncultivable” microorganism to grow in vitro. Appl. Environ. Microbiol. 2008, 74, 4889–4897. [Google Scholar] [CrossRef]
- Joseph, S.J.; Hugenholtz, P.; Sangwan, P.; Osborne, C.A.; Janssen, P.H. Laboratory cultivation of widespread and previously uncultured soil bacteria. Appl. Environ. Microbiol. 2003, 69, 7210–7215. [Google Scholar] [CrossRef]
- Rappé, M.S.; Giovannoni, S.J. The uncultured microbial majority. Annu. Rev. Microbiol. 2003, 57, 369–394. [Google Scholar] [CrossRef] [PubMed]
- Zengler, K.; Toledo, G.; Rappe, M.; Elkins, J.; Mathur, E.J.; Short, J.M.; Keller, M. Cultivating the uncultured. Proc. Natl. Acad. Sci. USA 2002, 99, 15681–15686. [Google Scholar] [CrossRef]
- Bollmann, A.; Palumbo, A.V.; Lewis, K.; Epstein, S.S. Isolation and physiology of bacteria from contaminated subsurface sediments. Appl. Environ. Microbiol. 2010, 76, 7413–7419. [Google Scholar] [CrossRef]
- Bruns, A.; Cypionka, H.; Overmann, J. Cyclic AMP and acyl homoserine lactones increase the cultivation efficiency of heterotrophic bacteria from the central Baltic Sea. Appl. Environ. Microbiol. 2002, 68, 3978–3987. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, H.; Hanada, S.; Sekiguchi, Y.; Tanaka, Y.; Kamagata, Y. Effect of gelling agent on colony formation in solid cultivation of microbial community in lake sediment. Environ. Microbiol. 2009, 11, 1827–1834. [Google Scholar] [CrossRef]
- Davis, K.E.R.; Joseph, S.J.; Janssen, P.H. Effects of growth medium, inoculum size, and incubation time on culturability and isolation of soil bacteria. Appl. Environ. Microbiol. 2005, 71, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Yamagishi, A.; Daimon, S.; Kawasaki, K.; Tamaki, H.; Kitagawa, W.; Abe, A.; Tanaka, M.; Sone, T.; Asano, K.; et al. Isolation of Previously Uncultured Slow-Growing Bacteria by Using a Simple Modification in the Preparation of Agar Media. Appl. Environ. Microbiol. 2018, 84, e00807-18. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Seo, C.; Ji, M.; Paik, M.-J.; Myung, S.-W.; Kim, J. Effective soil extraction method for cultivating previously uncultured soil bacteria. bioRxiv 2018, 322735. [Google Scholar] [CrossRef] [PubMed]
- Ringeisen, B.R.; Rincon, K.; Fitzgerald, L.A.; Fulmer, P.A.; Wu, P.K. Printing soil: a single-step, high-throughput method to isolate micro-organisms and near-neighbour microbial consortia from a complex environmental sample. Methods Ecol. Evolut. 2015, 6, 209–217. [Google Scholar] [CrossRef]
- Feng, G.; Xie, T.; Wang, X.; Bai, J.; Tang, L.; Zhao, H.; Wei, W.; Wang, M.; Zhao, Y. Metagenomic analysis of microbial community and function involved in cd-contaminated soil. BMC Microbiol. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Hemme, C.L.; Tu, Q.; Shi, Z.; Qin, Y.; Gao, W.; Deng, Y.; Nostrand, J.D.V.; Wu, L.; He, Z.; Chain, P.S.G.; et al. Comparative metagenomics reveals impact of contaminants on groundwater microbiomes. Front Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Liu, Y.-R.; Delgado-Baquerizo, M.; Bi, L.; Zhu, J.; He, J.-Z. Consistent responses of soil microbial taxonomic and functional attributes to mercury pollution across China. Microbiome 2018, 6. [Google Scholar] [CrossRef]
- Kumar, R.; Nongkhlaw, M.; Acharya, C.; Joshi, S.R. Uranium (U)-tolerant bacterial diversity from U ore deposit of Domiasiat in North-East India and its prospective utilisation in bioremediation. Microbes Environ. 2013, 28, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Misra, C.S.; Gupta, A.; Ballal, A.; Apte, S.K. Interaction of Uranium with Bacterial Cell Surfaces: Inferences from Phosphatase-Mediated Uranium Precipitation. Appl. Environ. Microbiol. 2016, 82, 4965–4974. [Google Scholar] [CrossRef] [PubMed]
- Gadd, G.M.; Fomina, M. Uranium and Fungi. Geomicrobiol. J. 2011, 28, 471–482. [Google Scholar] [CrossRef]
- Mumtaz, S.; Streten-Joyce, C.; Parry, D.L.; McGuinness, K.A.; Lu, P.; Gibb, K.S. Fungi outcompete bacteria under increased uranium concentration in culture media. J. Environ. Radioact. 2013, 120, 39–44. [Google Scholar] [CrossRef]
- Grijseels, S.; Nielsen, J.C.; Randelovic, M.; Nielsen, J.; Nielsen, K.F.; Workman, M.; Frisvad, J.C. Penicillium arizonense, a new, genome sequenced fungal species, reveals a high chemical diversity in secreted metabolites. Sci. Rep. 2016, 6, 35112. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.C.; Grijseels, S.; Prigent, S.; Ji, B.; Dainat, J.; Nielsen, K.F.; Frisvad, J.C.; Workman, M.; Nielsen, J. Global analysis of biosynthetic gene clusters reveals vast potential of secondary metabolite production in Penicillium species. Nat. Microbiol. 2017, 2, 17044. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, T.; Lewis, K.; Epstein, S.S. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 2002, 296, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Bollmann, A.; Lewis, K.; Epstein, S.S. Incubation of Environmental Samples in a Diffusion Chamber Increases the Diversity of Recovered Isolates. Appl. Environ. Microbiol. 2007, 73, 6386–6390. [Google Scholar] [CrossRef] [PubMed]
- Steinert, G.; Whitfield, S.; Taylor, M.W.; Thoms, C.; Schupp, P.J. Application of diffusion growth chambers for the cultivation of marine sponge-associated bacteria. Mar. Biotechnol. 2014, 16, 594–603. [Google Scholar] [CrossRef]
- Rajapaksha, R.M.C.P.; Tobor-Kapłon, M.A.; Bååth, E. Metal Toxicity Affects Fungal and Bacterial Activities in Soil Differently. Appl. Environ. Microbiol. 2004, 70, 2966–2973. [Google Scholar] [CrossRef]
- Oladipo, O.G.; Awotoye, O.O.; Olayinka, A.; Bezuidenhout, C.C.; Maboeta, M.S. Heavy metal tolerance traits of filamentous fungi isolated from gold and gemstone mining sites. Braz. J. Microbiol. 2018, 49, 29–37. [Google Scholar] [CrossRef]
- Maddela, N.R.; Masabanda, M.; Leiva-Mora, M. Novel diesel-oil-degrading bacteria and fungi from the Ecuadorian Amazon rainforest. Water Sci. Technol. 2015, 71, 1554–1561. [Google Scholar] [CrossRef]
- Wufuer, R.; Wei, Y.; Lin, Q.; Wang, H.; Song, W.; Liu, W.; Zhang, D.; Pan, X.; Gadd, G.M. Uranium Bioreduction and Biomineralization. Adv. Appl. Microbiol. 2017, 101, 137–168. [Google Scholar]
- Beazley, M.J.; Martinez, R.J.; Sobecky, P.A.; Webb, S.M.; Taillefert, M. Uranium Biomineralization as a Result of Bacterial Phosphatase Activity: Insights from Bacterial Isolates from a Contaminated Subsurface. Environ. Sci. Technol. 2007, 41, 5701–5707. [Google Scholar]
- Yung, M.C.; Jiao, Y. Biomineralization of uranium by PhoY phosphatase activity aids cell survival in Caulobacter crescentus. Appl. Environ. Microbiol. 2014, 80, 4795–4804. [Google Scholar] [CrossRef] [PubMed]
- Gallois, N.; Alpha-Bazin, B.; Ortet, P.; Barakat, M.; Piette, L.; Long, J.; Berthomieu, C.; Armengaud, J.; Chapon, V. Proteogenomic insights into uranium tolerance of a Chernobyl’s Microbacterium bacterial isolate. J. Proteom. 2018, 177, 148–157. [Google Scholar] [CrossRef]
- Chauhan, A.; Pathak, A.; Jaswal, R.; Edwards III, B.; Chappell, D.; Ball, C.; Garcia-Sillas, R.; Stothard, P.; Seaman, J. Physiological and Comparative Genomic Analysis of Arthrobacter sp. SRS-W-1-2016 Provides Insights on Niche Adaptation for Survival in Uraniferous Soils. Genes 2018, 9, 31. [Google Scholar] [CrossRef]
- Pathak, A.; Chauhan, A.; Stothard, P.; Green, S.; Maienschein-Cline, M.; Jaswal, R.; Seaman, J. Genome-centric evaluation of Burkholderia sp. strain SRS-W-2-2016 resistant to high concentrations of uranium and nickel isolated from the Savannah River Site (SRS), USA. Genom. Data 2017, 12, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Banfield, J.F. Resistance to, and Accumulation of, Uranium by Bacteria from a Uranium-Contaminated Site. Geomicrobiol. J. 2004, 21, 113–121. [Google Scholar] [CrossRef]
- Katsenovich, Y.; Carvajal, D.; Guduru, R.; Lagos, L.; Li, C.-Z. Assessment of the Resistance to Uranium (VI) Exposure by Arthrobacter sp. Isolated from Hanford Site Soil. Geomicrobiol. J. 2013, 30, 120–130. [Google Scholar] [CrossRef]
- Agarwal, M.; Pathak, A.; Rathore, R.S.; Prakash, O.; Singh, R.; Jaswal, R.; Seaman, J.; Chauhan, A. Proteogenomic Analysis of Burkholderia Species Strains 25 and 46 Isolated from Uraniferous Soils Reveals Multiple Mechanisms to Cope with Uranium Stress. Cells 2018, 7, 269. [Google Scholar] [CrossRef]
- Liang, Y.; Van Nostrand, J.D.; N’guessan, L.A.; Peacock, A.D.; Deng, Y.; Long, P.E.; Resch, C.T.; Wu, L.; He, Z.; Li, G.; et al. Microbial functional gene diversity with a shift of subsurface redox conditions during In Situ uranium reduction. Appl. Environ. Microbiol. 2012, 78, 2966–2972. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, B.; Chariton, A.A.; Harford, A.J.; Hose, G.C.; Greenfield, P.; Elbourne, L.D.H.; Oytam, Y.; Stephenson, S.; Midgley, D.J.; Paulsen, I.T. Effects of uranium concentration on microbial community structure and functional potential. Environ. Microbiol. 2017, 19, 3323–3341. [Google Scholar] [CrossRef]
- Yan, X.; Luo, X.; Zhao, M. Metagenomic analysis of microbial community in uranium-contaminated soil. Appl. Microbiol. Biotechnol. 2016, 100, 299–310. [Google Scholar] [CrossRef]
- Evans, A.G.; Bauer, L.R.; Haselow, J.S.; Hayes, D.W.; Martin, H.L.; McDowell, W.L.; Pickett, J.B. Uranium in the Savannah River Site Environment; Westinghouse Savannah River Co.: Aiken, SC, USA, 1992. [Google Scholar]
- United States Environmental Protection Agency. National Primary Drinking Water Regulations. Available online: https://www.epa.gov/ground-water-and-drinking-water/national-primary-drinking-water-regulations (accessed on 25 October 2018).
- Gavrish, E.; Bollmann, A.; Epstein, S.; Lewis, K. A trap for in situ cultivation of filamentous actinobacteria. J. Microbiol. Methods 2008, 72, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.G.Z.; Green, K.T.; Dutilh, B.E.; Edwards, R.A. SUPER-FOCUS: a tool for agile functional analysis of shotgun metagenomic data. Bioinformatics 2016, 32, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- LANE, D.J. 16S/23S rRNA sequencing. Nucleic Acid Tech. Bact. Syst. 1991, 115–175. [Google Scholar]
- Vainio, E.J.; Hantula, J. Direct analysis of wood-inhabiting fungi using denaturing gradient gel electrophoresis of amplified ribosomal DNA. Mycol. Res. 2000, 104, 927–936. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef]
- Blom, J.; Albaum, S.P.; Doppmeier, D.; Pühler, A.; Vorhölter, F.-J.; Zakrzewski, M.; Goesmann, A. EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinf. 2009, 10, 154. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Yoon, S.-H.; Ha, S.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef]
- Genome-to-Genome Distance Calculator. Available online: http://ggdc.dsmz.de/home.php (accessed on 18 December 2018).
- NCBI. Metagenomics-Guided Isolation of Uranium Resistant Microbiota Using Diffusion Chamber and or Microbial Trap Approaches. Available online: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA509237 (accessed on 16 January 2019).
- NCBI. Rhizomucor sp. MT2 MMC-2018, Whole Genome Shotgun Sequencing Project. Available online: https://www.ncbi.nlm.nih.gov/nuccore/PZKB00000000 (accessed on 21 January 2018).
- Akob, D.M.; Mills, H.J.; Kostka, J.E. Metabolically active microbial communities in uranium-contaminated subsurface sediments. FEMS Microbiol. Ecol. 2007, 59, 95–107. [Google Scholar] [CrossRef]
- Islam, E.; Dhal, P.K.; Kazy, S.K.; Sar, P. Molecular analysis of bacterial communities in uranium ores and surrounding soils from Banduhurang open cast uranium mine, India: A comparative study. J. Environ. Sci. Health A Tox Hazard Subst. Environ. Eng. 2011, 46, 271–280. [Google Scholar] [CrossRef]
- Mondani, L.; Benzerara, K.; Carrière, M.; Christen, R.; Mamindy-Pajany, Y.; Février, L.; Marmier, N.; Achouak, W.; Nardoux, P.; Berthomieu, C.; et al. Influence of Uranium on Bacterial Communities: A Comparison of Natural Uranium-Rich Soils with Controls. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Green, S.J.; Prakash, O.; Jasrotia, P.; Overholt, W.A.; Cardenas, E.; Hubbard, D.; Tiedje, J.M.; Watson, D.B.; Schadt, C.W.; Brooks, S.C.; et al. Denitrifying bacteria from the genus Rhodanobacter dominate bacterial communities in the highly contaminated subsurface of a nuclear legacy waste site. Appl. Environ. Microbiol. 2012, 78, 1039–1047. [Google Scholar] [CrossRef]
- Islam, E.; Sar, P. Culture-dependent and -independent molecular analysis of the bacterial community within uranium ore. J. Basic Microbiol. 2011, 51, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Koribanics, N.M.; Tuorto, S.J.; Lopez-Chiaffarelli, N.; McGuinness, L.R.; Häggblom, M.M.; Williams, K.H.; Long, P.E.; Kerkhof, L.J. Spatial Distribution of an Uranium-Respiring Betaproteobacterium at the Rifle, CO Field Research Site. PLoS ONE 2015, 10, e0123378. [Google Scholar] [CrossRef]
- Miller, C.S.; Handley, K.M.; Wrighton, K.C.; Frischkorn, K.R.; Thomas, B.C.; Banfield, J.F. Short-Read Assembly of Full-Length 16S Amplicons Reveals Bacterial Diversity in Subsurface Sediments. PLoS ONE 2013, 8, e56018. [Google Scholar] [CrossRef] [PubMed]
- Moreels, D.; Crosson, G.; Garafola, C.; Monteleone, D.; Taghavi, S.; Fitts, J.P.; van der Lelie, D. Microbial community dynamics in uranium contaminated subsurface sediments under biostimulated conditions with high nitrate and nickel pressure. Environ. Sci. Pollut. Res. Int. 2008, 15, 481–491. [Google Scholar] [CrossRef]
- North, N.N.; Dollhopf, S.L.; Petrie, L.; Istok, J.D.; Balkwill, D.L.; Kostka, J.E. Change in bacterial community structure during in situ biostimulation of subsurface sediment cocontaminated with uranium and nitrate. Appl. Environ. Microbiol. 2004, 70, 4911–4920. [Google Scholar] [CrossRef]
- Vishnivetskaya, T.A.; Brandt, C.C.; Madden, A.S.; Drake, M.M.; Kostka, J.E.; Akob, D.M.; Küsel, K.; Palumbo, A.V. Microbial community changes in response to ethanol or methanol amendments for U(VI) reduction. Appl. Environ. Microbiol. 2010, 76, 5728–5735. [Google Scholar] [CrossRef] [PubMed]
- Coenye, T.; Vandamme, P. Diversity and significance of Burkholderia species occupying diverse ecological niches. Environ. Microbiol. 2003, 5, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Ormeño-Orrillo, E.; Rogel, M.A.; Chueire, L.M.O.; Tiedje, J.M.; Martínez-Romero, E.; Hungria, M. Genome sequences of Burkholderia sp. strains CCGE1002 and H160, isolated from legume nodules in Mexico and Brazil. J. Bacteriol. 2012, 194, 6927. [Google Scholar] [CrossRef]
- Suárez-Moreno, Z.R.; Caballero-Mellado, J.; Coutinho, B.G.; Mendonça-Previato, L.; James, E.K.; Venturi, V. Common features of environmental and potentially beneficial plant-associated Burkholderia. Microb. Ecol. 2012, 63, 249–266. [Google Scholar] [CrossRef]
- Pérez-Pantoja, D.; Nikel, P.I.; Chavarría, M.; de Lorenzo, V. Endogenous stress caused by faulty oxidation reactions fosters evolution of 2,4-dinitrotoluene-degrading bacteria. PLoS Genet. 2013, 9, e1003764. [Google Scholar] [CrossRef]
- Oyetibo, G.O.; Ilori, M.O.; Obayori, O.S.; Amund, O.O. Biodegradation of petroleum hydrocarbons in the presence of nickel and cobalt. J. Basic Microbiol. 2013, 53, 917–927. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, Z.; Chai, L.; Wang, Y.; Liu, Y.; Xiao, R. Bioleaching remediation of heavy metal-contaminated soils using Burkholderia sp. Z-90. J. Hazard. Mater. 2016, 301, 145–152. [Google Scholar] [CrossRef]
- Holmes, D.E.; Giloteaux, L.; Chaurasia, A.K.; Williams, K.H.; Luef, B.; Wilkins, M.J.; Wrighton, K.C.; Thompson, C.A.; Comolli, L.R.; Lovley, D.R. Evidence of Geobacter-associated phage in a uranium-contaminated aquifer. ISME J. 2015, 9, 333–346. [Google Scholar] [CrossRef]
- Allison, S.D.; Martiny, J.B.H. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 2008, 105, 11512–11519. [Google Scholar] [CrossRef]
- Newsome, L.; Morris, K.; Lloyd, J.R. The biogeochemistry and bioremediation of uranium and other priority radionuclides. Chem. Geol. 2014, 363, 164–184. [Google Scholar] [CrossRef]
- Liang, X.; Hillier, S.; Pendlowski, H.; Gray, N.; Ceci, A.; Gadd, G.M. Uranium phosphate biomineralization by fungi. Environ. Microbiol. 2015, 17, 2064–2075. [Google Scholar] [CrossRef]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef]
- Santos, P.C.D.; Dean, D.R. A newly discovered role for iron-sulfur clusters. Proc. Natl. Acad. Sci. USA 2008, 105, 11589–11590. [Google Scholar] [CrossRef]
- Glukhova, L.B.; Frank, Y.A.; Danilova, E.V.; Avakyan, M.R.; Banks, D.; Tuovinen, O.H.; Karnachuk, O.V. Isolation, Characterization, and Metal Response of Novel, Acid-Tolerant Penicillium spp. from Extremely Metal-Rich Waters at a Mining Site in Transbaikal (Siberia, Russia). Microb. Ecol. 2018, 76, 911–924. [Google Scholar] [CrossRef]
- Figueras, M.J.; Beaz-Hidalgo, R.; Hossain, M.J.; Liles, M.R. Taxonomic Affiliation of New Genomes Should Be Verified Using Average Nucleotide Identity and Multilocus Phylogenetic Analysis. Genome Announc. 2014, 2, e00927-14. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaswal, R.; Pathak, A.; Edwards III, B.; Lewis III, R.; Seaman, J.C.; Stothard, P.; Krivushin, K.; Blom, J.; Rupp, O.; Chauhan, A. Metagenomics-Guided Survey, Isolation, and Characterization of Uranium Resistant Microbiota from the Savannah River Site, USA. Genes 2019, 10, 325. https://doi.org/10.3390/genes10050325
Jaswal R, Pathak A, Edwards III B, Lewis III R, Seaman JC, Stothard P, Krivushin K, Blom J, Rupp O, Chauhan A. Metagenomics-Guided Survey, Isolation, and Characterization of Uranium Resistant Microbiota from the Savannah River Site, USA. Genes. 2019; 10(5):325. https://doi.org/10.3390/genes10050325
Chicago/Turabian StyleJaswal, Rajneesh, Ashish Pathak, Bobby Edwards III, Robert Lewis III, John C. Seaman, Paul Stothard, Kirill Krivushin, Jochen Blom, Oliver Rupp, and Ashvini Chauhan. 2019. "Metagenomics-Guided Survey, Isolation, and Characterization of Uranium Resistant Microbiota from the Savannah River Site, USA" Genes 10, no. 5: 325. https://doi.org/10.3390/genes10050325
APA StyleJaswal, R., Pathak, A., Edwards III, B., Lewis III, R., Seaman, J. C., Stothard, P., Krivushin, K., Blom, J., Rupp, O., & Chauhan, A. (2019). Metagenomics-Guided Survey, Isolation, and Characterization of Uranium Resistant Microbiota from the Savannah River Site, USA. Genes, 10(5), 325. https://doi.org/10.3390/genes10050325