Systematic Identification and Evolution Analysis of Sox Genes in Coturnix japonica Based on Comparative Genomics

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Incubation Conditions
2.3. Retrieval of Genome Sequences
2.4. Phylogenetic Analysis
2.5. Microsynteny, Selection, and Functional Analysis
2.6. RNA Extraction, Q-PCR, and RNA-Seq Expression Analysis
3. Results and Discussion
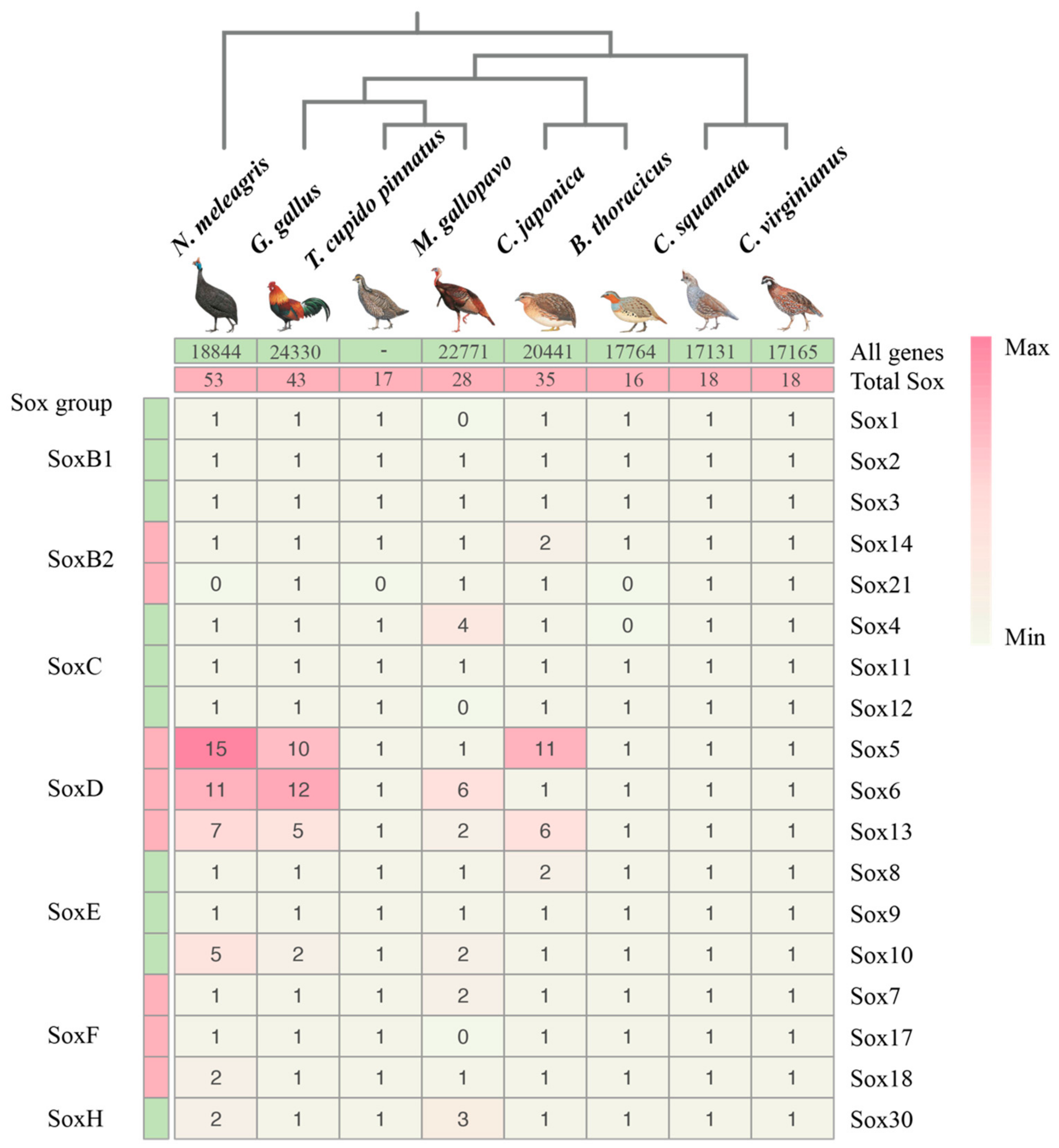
3.1. Genome-Wide Analysis of Galliformes Sox Genes
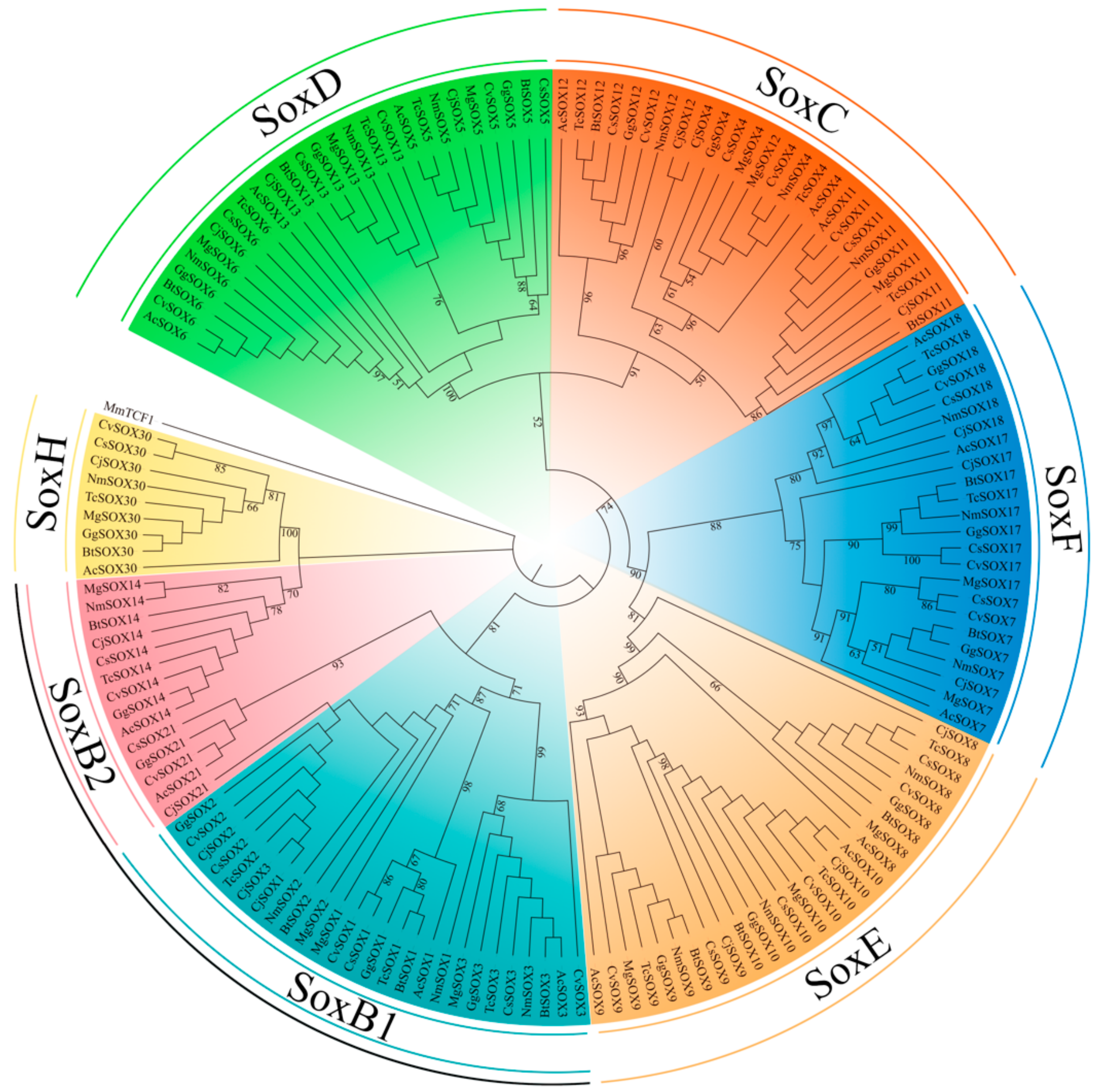
3.2. Phylogenetic and Classification of the Sox Gene Family
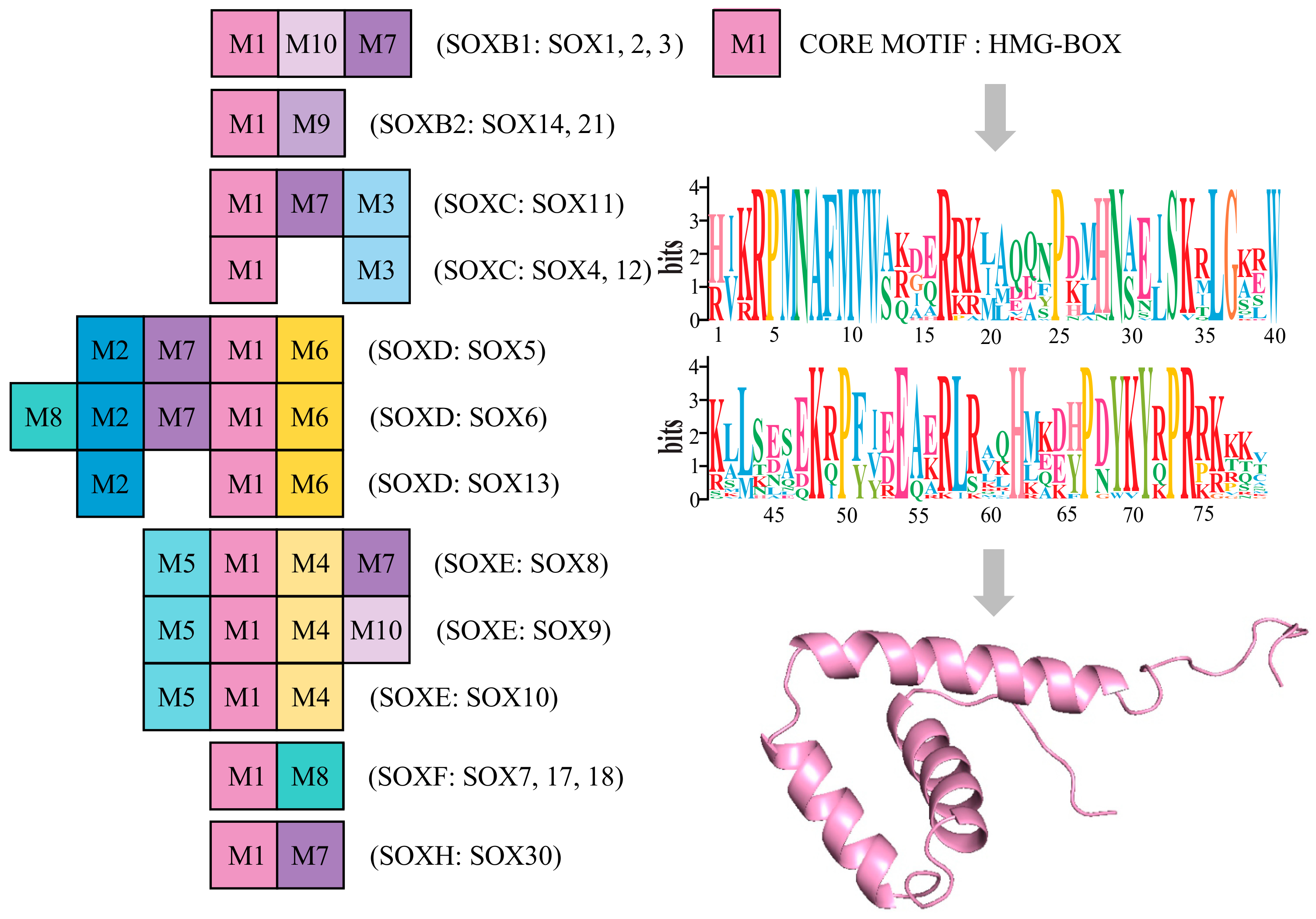
3.3. Conserved Motif Analysis of Sox Genes
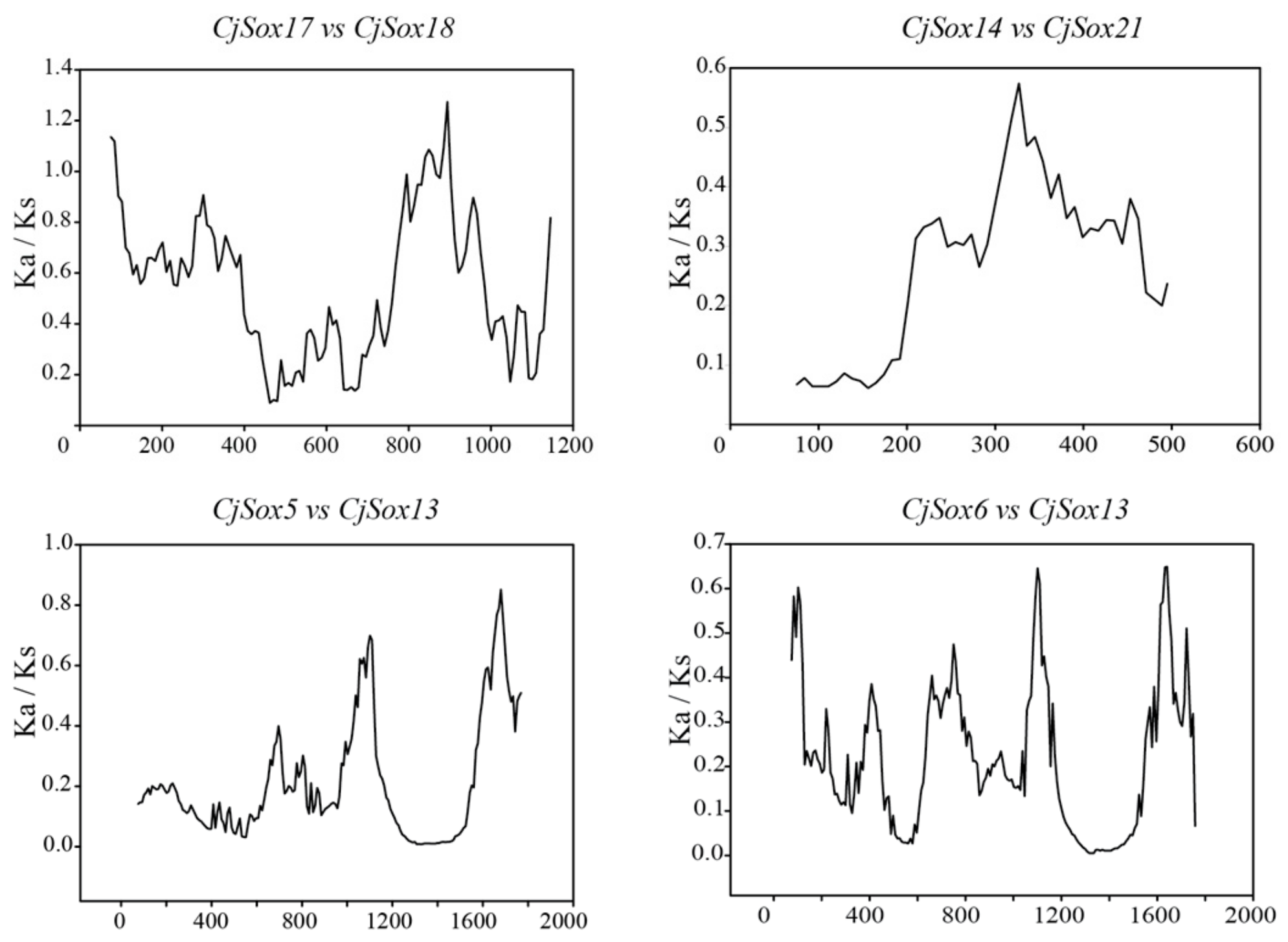
3.4. Gene Duplication of Sox Genes
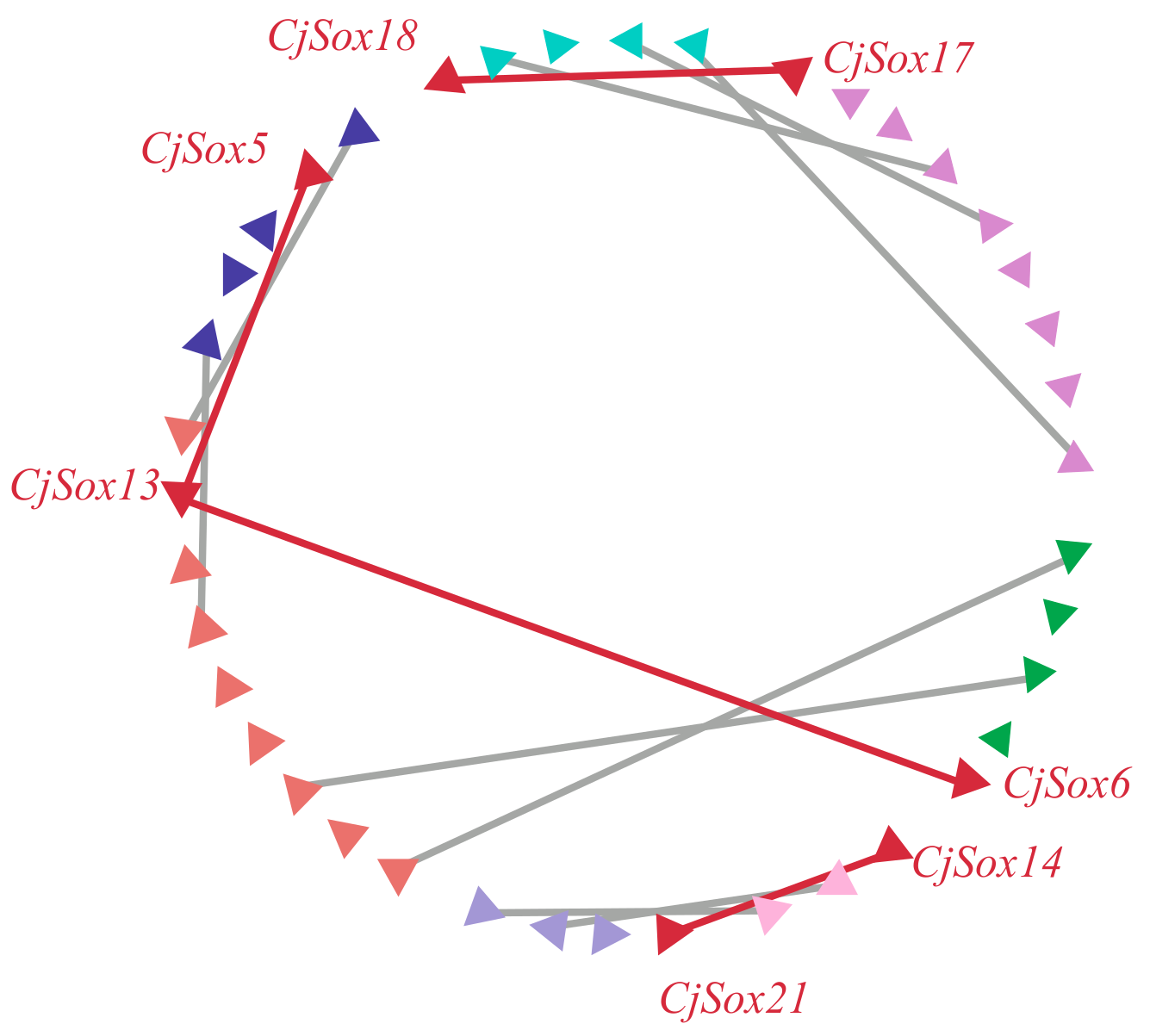
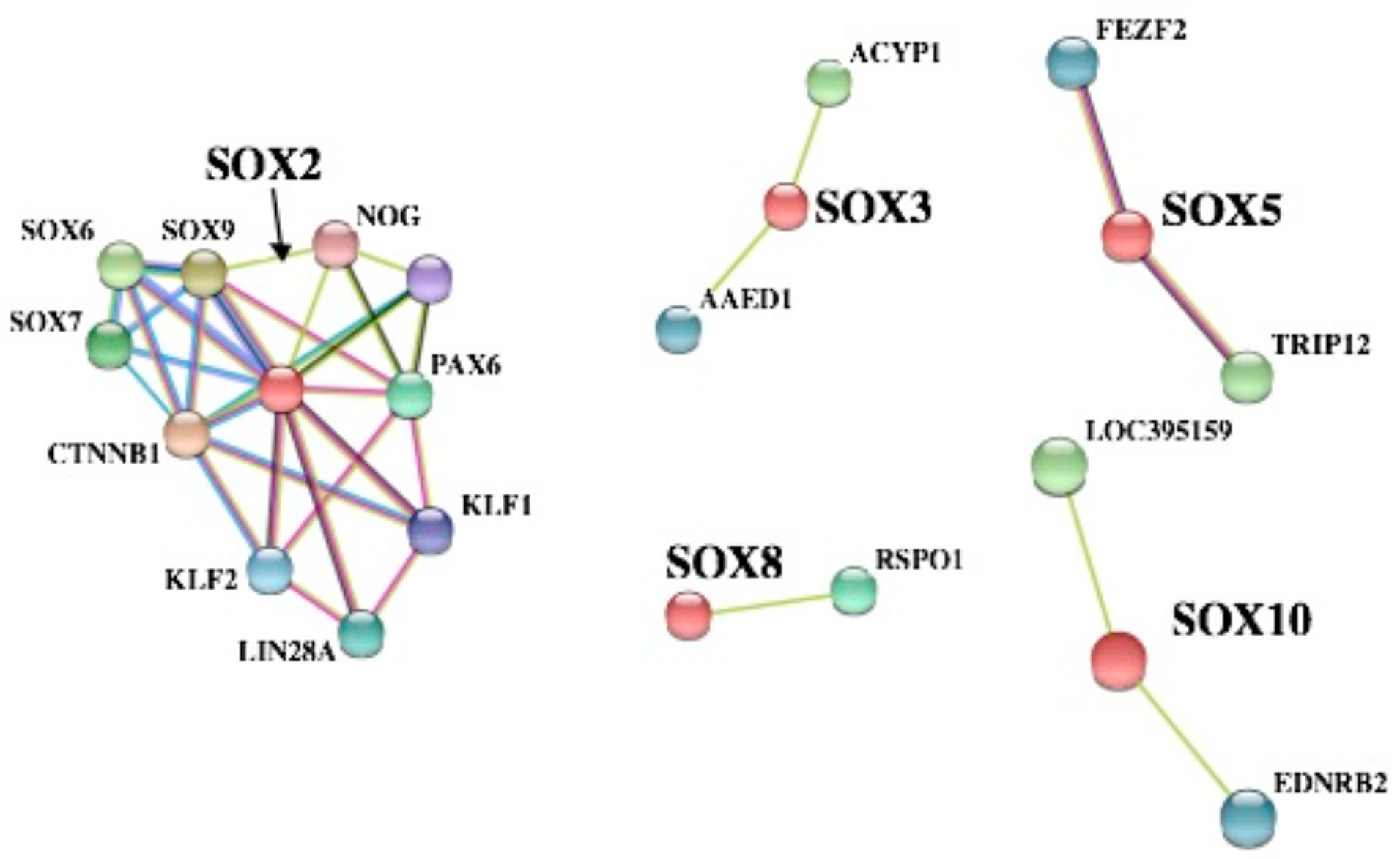
3.5. PPI Network Analyses of SOX Proteins and GO Enrichment Analysis
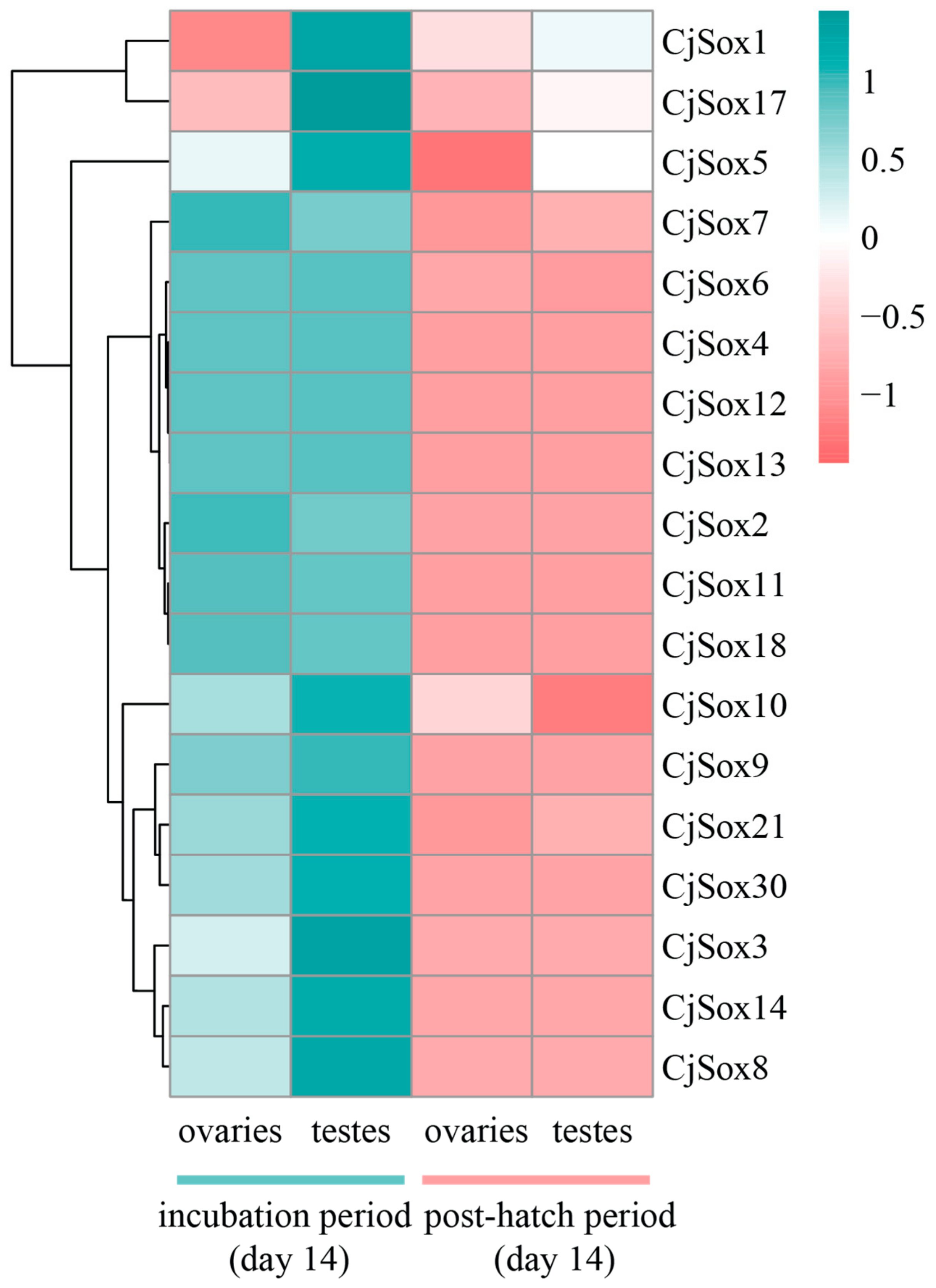
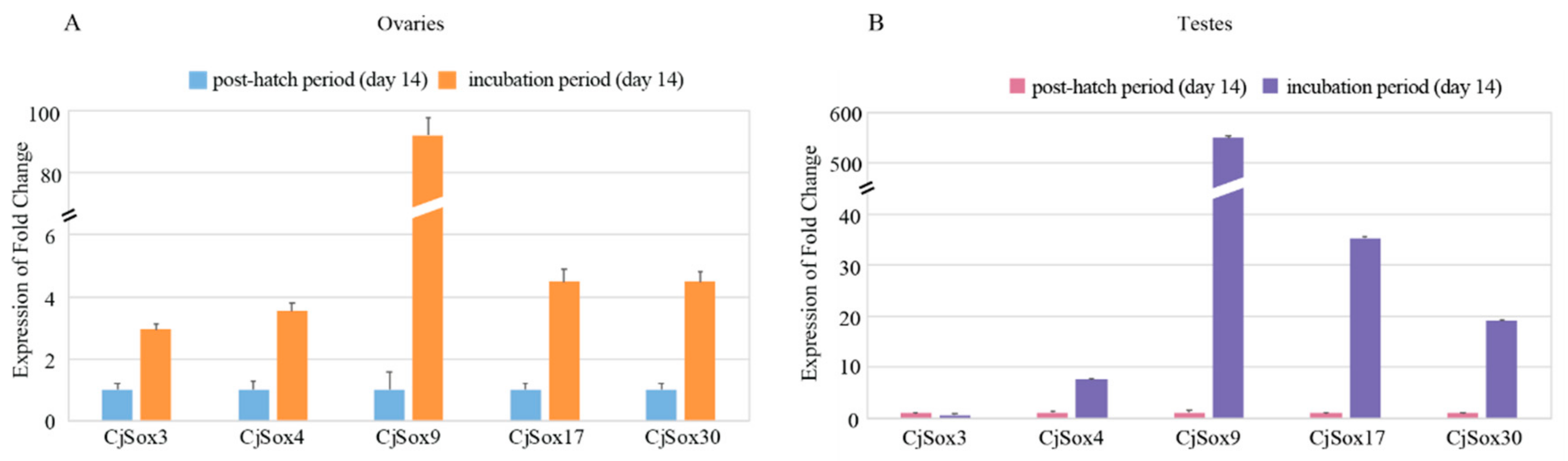
3.6. Gene Expression Analysis of C. Japonica Sox Genes in Gonads
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bowles, J.; Schepers, G.; Koopman, P. Phylogeny of the SOX family of developmental transcription factors based on sequence and structural indicators. Dev. Biol. 2000, 227, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Shinzato, C.; Iguchi, A.; Hayward, D.C.; Technau, U.; Ball, E.E.; Miller, D.J. Sox genes in the coral Acropora millepora: Divergent expression patterns reflect differences in developmental mechanisms within the Anthozoa. BMC Evol. Biol. 2008, 8, 311. [Google Scholar]
- Guth, S.; Wegner, M. Having it both ways: Sox protein function between conservation and innovation. Cell. Mol. Life Sci. 2008, 65, 3000–3018. [Google Scholar]
- Heenan, P.; Zondag, L.; Wilson, M.J. Evolution of the Sox gene family within the chordate phylum. Gene 2016, 575, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Kamachi, Y.; Kondoh, H. Sox proteins: Regulators of cell fate specification and differentiation. Development 2013, 140, 4129–4144. [Google Scholar] [CrossRef] [PubMed]
- Hokamp, K.; McLysaght, A.; Wolfe, K.H. The 2R hypothesis and the human genome sequence. J. Struct. Funct. Genomics 2003, 3, 95–110. [Google Scholar] [CrossRef]
- Sinclair, A.H.; Berta, P.; Palmer, M.S.; Hawkins, J.R.; Griffiths, B.L.; Smith, M.J.; Foster, J.W.; Frischauf, A.-M.; Lovell-Badge, R.; Goodfellow, P.N. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990, 346, 240. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shen, X.; Zhao, H.; Nagahama, Y. Genome-wide analysis of Sox genes in Medaka (Oryzias latipes) and their expression pattern in embryonic development. Cytogenet. Genome Res. 2011, 134, 283–294. [Google Scholar] [PubMed]
- Schepers, G.E.; Teasdale, R.D.; Koopman, P. Twenty pairs of sox: Extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev. Cell 2002, 3, 167–170. [Google Scholar] [CrossRef]
- Wei, L.; Cheng, D.; Li, D.; Meng, M.; Peng, L.; Tang, L.; Pan, M.; Xiang, Z.; Xia, Q.; Lu, C. Identification and characterization of Sox genes in the silkworm, Bombyx mori. Mol. Biol. Rep. 2011, 38, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, X.; Wang, M.; Zhang, W.; Pan, J.; Qin, Q.; Zhong, L.; Shao, J.; Sun, M.; Jiang, H.; et al. Genome-wide identification, phylogeny and expressional profile of the Sox gene family in channel catfish (Ictalurus punctatus). Comp. Biochem. Physiol. Part D Genomics Proteomics 2018, 28, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.; Queinnec, E.; Houliston, E.; Manuel, M. Expansion of the SOX gene family predated the emergence of the Bilateria. Mol. Phylogenet. Evol. 2006, 39, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Cremazy, F.; Berta, P.; Girard, F. Genome-wide analysis of Sox genes in Drosophila melanogaster. Mech. Dev. 2001, 109, 371–375. [Google Scholar] [CrossRef]
- Jiang, T.; Hou, C.-C.; She, Z.-Y.; Yang, W.-X. The SOX gene family: Function and regulation in testis determination and male fertility maintenance. Mol. Biol. Rep. 2013, 40, 2187–2194. [Google Scholar] [CrossRef]
- Ng, L.-J.; Wheatley, S.; Muscat, G.E.; Conway-Campbell, J.; Bowles, J.; Wright, E.; Bell, D.M.; Tam, P.P.; Cheah, K.S.; Koopman, P. SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev. Biol. 1997, 183, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Herpers, R.; van de Kamp, E.; Duckers, H.J.; Schulte-Merker, S. Redundant roles for sox7 and sox18 in arteriovenous specification in zebrafish. Circ. Res. 2008, 102, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, L.; Gowda, V.; Wang, M.; Li, X.; Kan, X. The mitochondrial genome of the Cinnamon Bittern, Ixobrychus cinnamomeus (Pelecaniformes: Ardeidae): Sequence, structure and phylogenetic analysis. Mol. Biol. Rep. 2012, 39, 8315–8326. [Google Scholar] [CrossRef]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, W.; Yang, J.; Jiang, L.; Zhang, Q.; Kan, X.; Yang, X. Comparative transcriptomics in three Passerida species provides insights into the evolution of avian mitochondrial complex I. Comp. Biochem. Physiol. Part D Genomics Proteomics 2018, 28, 27–36. [Google Scholar] [CrossRef]
- Ren, Q.; Qian, C.; Yuan, J.; Li, X.; Yang, J.; Wang, P.; Jiang, L.; Zhang, Q.; Wang, Y.; Kan, X. Complete mitochondrial genome of the Black-capped Bulbul, Pycnonotus melanicterus (Passeriformes: Pycnonotidae). Mitochondrial DNA Part A 2016, 27, 1378–1380. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, Q.; Yu, J.; Gowda, V.; Johnson, G.; Yang, J.; Kan, X.; Yang, X. miRNAome expression profiles in the gonads of adult Melopsittacus undulatus. PeerJ 2018, 6, e4615. [Google Scholar] [CrossRef]
- Jiang, L.; Chen, J.; Wang, P.; Ren, Q.; Yuan, J.; Qian, C.; Hua, X.; Guo, Z.; Zhang, L.; Yang, J. The Mitochondrial Genomes of Aquila fasciata and Buteo lagopus (Aves, Accipitriformes): Sequence, structure and phylogenetic analyses. PLoS ONE 2015, 10, e0136297. [Google Scholar] [CrossRef]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.; Faircloth, B.C.; Nabholz, B.; Howard, J.T. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef]
- Tiley, G.; Kimball, R.; Braun, E.; Burleigh, J. Comparison of the Chinese bamboo partridge and red Junglefowl genome sequences highlights the importance of demography in genome evolution. BMC Genomics 2018, 19, 336. [Google Scholar] [CrossRef]
- Wallis, J.W.; Aerts, J.; Groenen, M.A.; Crooijmans, R.P.; Layman, D.; Graves, T.A.; Scheer, D.E.; Kremitzki, C.; Fedele, M.J.; Mudd, N.K. A physical map of the chicken genome. Nature 2004, 432, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Consortium, I.C.G.S. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar]
- Dalloul, R.A.; Long, J.A.; Zimin, A.V.; Aslam, L.; Beal, K.; Blomberg, L.A.; Bouffard, P.; Burt, D.W.; Crasta, O.; Crooijmans, R.P. Multi-platform next-generation sequencing of the domestic turkey (Meleagris gallopavo): Genome assembly and analysis. PLoS Biol. 2010, 8, e1000475. [Google Scholar] [CrossRef]
- Kawahara-Miki, R.; Sano, S.; Nunome, M.; Shimmura, T.; Kuwayama, T.; Takahashi, S.; Kawashima, T.; Matsuda, Y.; Yoshimura, T.; Kono, T. Next-generation sequencing reveals genomic features in the Japanese quail. Genomics 2013, 101, 345–353. [Google Scholar] [CrossRef]
- Halley, Y.A.; Dowd, S.E.; Decker, J.E.; Seabury, P.M.; Bhattarai, E.; Johnson, C.D.; Rollins, D.; Tizard, I.R.; Brightsmith, D.J.; Peterson, M.J. A draft de novo genome assembly for the northern bobwhite (Colinus virginianus) reveals evidence for a rapid decline in effective population size beginning in the Late Pleistocene. PLoS ONE 2014, 9, e90240. [Google Scholar] [CrossRef]
- Oldeschulte, D.L.; Halley, Y.A.; Wilson, M.L.; Bhattarai, E.K.; Brashear, W.; Hill, J.; Metz, R.P.; Johnson, C.D.; Rollins, D.; Peterson, M.J. Annotated draft genome assemblies for the Northern Bobwhite (Colinus virginianus) and the Scaled Quail (Callipepla squamata) reveal disparate estimates of modern genome diversity and historic effective population size. G3: Genes, Genomes, Genetics 2017, 7, 3047–3058. [Google Scholar] [CrossRef]
- Pokorná, M.; Giovannotti, M.; Kratochvíl, L.; Kasai, F.; Trifonov, V.A.; O’Brien, P.C.; Caputo, V.; Olmo, E.; Ferguson-Smith, M.A.; Rens, W. Strong conservation of the bird Z chromosome in reptilian genomes is revealed by comparative painting despite 275 million years divergence. Chromosoma 2011, 120, 455–468. [Google Scholar] [CrossRef]
- Takada, S.; Ota, J.; Kansaku, N.; Yamashita, H.; Izumi, T.; Ishikawa, M.; Wada, T.; Kaneda, R.; Choi, Y.L.; Koinuma, K.; et al. Nucleotide sequence and embryonic expression of quail and duck Sox9 genes. Gen. Comp. Endocrinol. 2006, 145, 208–213. [Google Scholar] [CrossRef]
- Suzuki, T.; Sakai, D.; Osumi, N.; Wada, H.; Wakamatsu, Y. Sox genes regulate type 2 collagen expression in avian neural crest cells. Dev. Growth Differ. 2006, 48, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.F.; Schulenberg, T.S.; Iliff, M.J.; Roberson, D.; Fredericks, T.A.; Sullivan, B.L.; Wood, C.L. The eBird/Clements Checklist of Birds of the World: v2018. Available online: https://www.birdforum.net/showthread.php?t=365123 (accessed on 14 August 2018).
- McGowan, P.J.K.; Christie, G.M. Japanese Quail (Coturnix japonica). In Handbook of the Birds of the World Alive; Del Hoyo, J., Elliott, A., Sargatal, J., Christie, D.A., de Juana, E., Eds.; Lynx Edicions: Barcelona, Spain, 2018. [Google Scholar]
- Gecgel, U.; Yilmaz, I.; Gurcan, E.K.; Karasu, S.; Dulger, G.C. Comparison of fatty acid composition between female and male Japanese quail meats. J. Chem. 2015, 2015, 569746. [Google Scholar] [CrossRef]
- Poynter, G.; Huss, D.; Lansford, R. Japanese quail: An efficient animal model for the production of transgenic avians. Cold Spring Harb. Protoc. 2009, 4, pdb.emo112. [Google Scholar] [CrossRef]
- Minvielle, F. The future of Japanese quail for research and production. Worlds Poult. Sci. J. 2004, 60, 500–507. [Google Scholar] [CrossRef]
- Huss, D.; Poynter, G.; Lansford, R. Japanese quail (Coturnix japonica) as a laboratory animal model. Lab Anim. 2008, 37, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Huss, D.; Lansford, R. Fluorescent Quail: A Transgenic Model System for the Dynamic Study of Avian Development. In Avian and Reptilian Developmental Biology; Springer Humana Press: New York, NY, USA, 2017; pp. 125–147. [Google Scholar]
- Karcher, D.; Fleming-Waddell, J.; Applegate, T. Developmental changes in insulin-like growth factor (IGF)-I and-II mRNA abundance in extra-embryonic membranes and small intestine of avian embryos. Growth Horm. IGF Res. 2009, 19, 31–42. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Cao, Y.; Han, Y.; Meng, D.; Li, D.; Jin, Q.; Lin, Y.; Cai, Y. Structural, evolutionary, and functional analysis of the class III peroxidase gene family in Chinese Pear (Pyrus bretschneideri). Front. Plant Sci. 2016, 7, 1874. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Laudet, V.; Stehelin, D.; Clevers, H. Ancestry and diversity of the HMG box superfamily. Nucleic Acids Res. 1993, 21, 2493–2501. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2006, 23, 127–128. [Google Scholar] [CrossRef]
- Cao, Y.; Han, Y.; Jin, Q.; Lin, Y.; Cai, Y. Comparative genomic analysis of the GRF genes in Chinese pear (Pyrus bretschneideri Rehd), poplar (Populous), grape (Vitis vinifera), Arabidopsis and rice (Oryza sativa). Front. Plant Sci. 2016, 7, 1750. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Wu, J.; Vallenius, T.; Ovaska, K.; Westermarck, J.; Mäkelä, T.P.; Hautaniemi, S. Integrated network analysis platform for protein-protein interactions. Nat. Methods 2008, 6, 75. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Mandal, A.; Mishra, A.K.; Dulani, P.; Muthamilarasan, M.; Shweta, S.; Prasad, M. Identification, characterization, expression profiling, and virus-induced gene silencing of armadillo repeat-containing proteins in tomato suggest their involvement in tomato leaf curl New Delhi virus resistance. Funct. Integr. Genomics 2018, 18, 101–111. [Google Scholar] [CrossRef]
- Wei, L.; Yang, C.; Tao, W.; Wang, D. Genome-Wide Identification and Transcriptome-Based Expression Profiling of the Sox Gene Family in the Nile Tilapia (Oreochromis niloticus). Int. J. Mol. Sci. 2016, 17, 270. [Google Scholar] [CrossRef]
- Janssen, R.; Andersson, E.; Betnér, E.; Bijl, S.; Fowler, W.; Höök, L.; Leyhr, J.; Mannelqvist, A.; Panara, V.; Smith, K. Embryonic expression patterns and phylogenetic analysis of panarthropod sox genes: Insight into nervous system development, segmentation and gonadogenesis. BMC Evol. Biol. 2018, 18, 88. [Google Scholar] [CrossRef]
- Ohno, S. Gene duplication and the uniqueness of vertebrate genomes circa 1970–1999. Semin. Cell Dev. Biol. 1999, 10, 517–522. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Kerner, P.; Simionato, E.; Le Gouar, M.; Vervoort, M. Orthologs of key vertebrate neural genes are expressed during neurogenesis in the annelid Platynereis dumerilii. Evol. Dev. 2009, 11, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Pioro, H.L.; Stollewerk, A.J.D.g. The expression pattern of genes involved in early neurogenesis suggests distinct and conserved functions in the diplopod Glomeris marginata. Dev. Genes Evol. 2006, 216, 417–430. [Google Scholar] [CrossRef]
- Le Gouar, M.; Guillou, A.; Vervoort, M. Expression of a SoxB and a Wnt2/13 gene during the development of the mollusc Patella vulgata. Dev. Genes Evol. 2004, 214, 250–256. [Google Scholar] [CrossRef]
- Wegner, M.; Stolt, C.C. From stem cells to neurons and glia: A Soxist’s view of neural development. Trends Neurosci. 2005, 28, 583–588. [Google Scholar] [CrossRef]
- Pevny, L.H.; Sockanathan, S.; Placzek, M.; Lovell-Badge, R. A role for SOX1 in neural determination. Development 1998, 125, 1967–1978. [Google Scholar]
- Uchikawa, M.; Kamachi, Y.; Kondoh, H. Two distinct subgroups of Group B Sox genes for transcriptional activators and repressors: Their expression during embryonic organogenesis of the chicken. Mech. Dev. 1999, 84, 103–120. [Google Scholar] [CrossRef]
- Magie, C.R.; Pang, K.; Martindale, M.Q. Genomic inventory and expression of Sox and Fox genes in the cnidarian Nematostella vectensis. Dev. Genes Evol. 2005, 215, 618–630. [Google Scholar] [CrossRef]
- Kenny, N.J.; Shimeld, S.M. Additive multiple k-mer transcriptome of the keelworm Pomatoceros lamarckii (Annelida; Serpulidae) reveals annelid trochophore transcription factor cassette. Dev. Genes Evol. 2012, 222, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.; Quéinnec, E.; Chiori, R.; Le Guyader, H.; Manuel, M. Insights into the early evolution of SOX genes from expression analyses in a ctenophore. J. Exp. Zool. B Mol. Dev. Evol. 2008, 310, 650–667. [Google Scholar] [CrossRef]
- Štros, M.; Launholt, D.; Grasser, K.D. The HMG-box: A versatile protein domain occurring in a wide variety of DNA-binding proteins. Cell. Mol. Life Sci. 2007, 64, 2590–2606. [Google Scholar] [CrossRef]
- Soullier, S.; Jay, P.; Poulat, F.; Vanacker, J.-M.; Berta, P.; Laudet, V. Diversification pattern of the HMG and SOX family members during evolution. J. Mol. Evol. 1999, 48, 517–527. [Google Scholar] [CrossRef]
- Cao, Y.; Han, Y.; Li, D.; Lin, Y.; Cai, Y. MYB transcription factors in chinese pear (Pyrus bretschneideri Rehd.): Genome-wide identification, classification, and expression profiling during fruit development. Front. Plant Sci. 2016, 7, 577. [Google Scholar] [CrossRef]
- Sato, S.; Nakamura, Y.; Kaneko, T.; Asamizu, E.; Kato, T.; Nakao, M.; Sasamoto, S.; Watanabe, A.; Ono, A.; Kawashima, K. Genome structure of the legume, Lotus japonicus. DNA Res. 2008, 15, 227–239. [Google Scholar] [CrossRef]
- Lin, Y.; Cheng, Y.; Jin, J.; Jin, X.; Jiang, H.; Yan, H.; Cheng, B. Genome duplication and gene loss affect the evolution of heat shock transcription factor genes in legumes. PLoS ONE 2014, 9, e102825. [Google Scholar] [CrossRef] [PubMed]
- Voldoire, E.; Brunet, F.; Naville, M.; Volff, J.-N.; Galiana, D. Expansion by whole genome duplication and evolution of the sox gene family in teleost fish. PLoS ONE 2017, 12, e0180936. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, L.; Zhu, Y.; Li, Y.; Yan, H.; Xiang, Y. Comparative genomic analysis of the WRKY III gene family in populus, grape, arabidopsis and rice. Biol. Direct 2015, 10, 48. [Google Scholar] [CrossRef]
- Song, H.; Guo, Z.; Chen, T.; Sun, J.; Yang, G. Genome-wide identification of LRR-containing sequences and the response of these sequences to nematode infection in Arachis duranensis. BMC Plant Biol. 2018, 18, 279. [Google Scholar] [CrossRef]
- Consortium, M.G.S. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef]
- He, Y.; Zuo, Q.; Edwards, J.; Zhao, K.; Lei, J.; Cai, W.; Nie, Q.; Li, B.; Song, J. DNA Methylation and Regulatory Elements during Chicken Germline Stem Cell Differentiation. Stem Cell Rep. 2018, 10, 1793–1806. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.M.; Lim, W.; Jeong, W.; Lee, J.Y.; Kim, J.; Han, J.Y.; Bazer, F.W.; Song, G. Hormonal regulation of β-catenin during development of the avian oviduct and its expression in epithelial cell-derived ovarian carcinogenesis. Mol. Cell. Endocrinol. 2014, 382, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wei, J.; Yang, P.; Zhang, T.; Chen, Z.; He, F.; Wei, F.; Chen, H.; Hu, H.; Zhong, J. Genome-scale CRISPR-Cas9 knockout screening in gastrointestinal stromal tumor with Imatinib resistance. Mol. Cancer 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wu, J.; Cai, Y.; Luo, M.; Wang, B.; Gu, Y. AAED1 modulates proliferation and glycolysis in gastric cancer. Oncol. Rep. 2018, 40, 1156–1164. [Google Scholar] [CrossRef]
- Li, X.; Eishi, Y.; Bai, Y.; Sakai, H.; Akiyama, Y.; Tani, M.; Takizawa, T.; Koike, M.; Yuasa, Y. Expression of the SRY-related HMG box protein SOX2 in human gastric carcinoma. Int. J. Oncol. 2004, 24, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Nakazawa, M.; Kani, S.; Bae, Y.-K.; Shimizu, T.; Kageyama, R.; Hibi, M. Zinc finger genes Fezf1 and Fezf2 control neuronal differentiation by repressing Hes5 expression in the forebrain. Developmental 2010, 137, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Kajiro, M.; Tsuchiya, M.; Kawabe, Y.-I.; Furumai, R.; Iwasaki, N.; Hayashi, Y.; Katano, M.; Nakajima, Y.; Goto, N.; Watanabe, T. The E3 ubiquitin ligase activity of Trip12 is essential for mouse embryogenesis. PLoS ONE 2011, 6, e25871. [Google Scholar] [CrossRef] [PubMed]
- Barrionuevo, F.; Scherer, G. SOX E genes: SOX9 and SOX8 in mammalian testis development. Int. J. Biochem. Cell Biol. 2010, 42, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Arsenault, M.; Ng, E.T.; Longmuss, E.; Chau, T.C.-Y.; Hartwig, S.; Koopman, P. SOX4 regulates gonad morphogenesis and promotes male germ cell differentiation in mice. Dev. Biol. 2017, 423, 46–56. [Google Scholar] [CrossRef]
- Jeng, S.; Wu, G.; Yueh, W.; Kuo, S.; Dufour, S.; Chang, C. Gonadal development and expression of sex-specific genes during sex differentiation in the Japanese eel. Gen. Comp. Endocrinol. 2018, 257, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Irie, N.; Weinberger, L.; Tang, W.W.; Kobayashi, T.; Viukov, S.; Manor, Y.S.; Dietmann, S.; Hanna, J.H.; Surani, M.A. SOX17 is a critical specifier of human primordial germ cell fate. Cell 2015, 160, 253–268. [Google Scholar] [CrossRef]
- Han, F.; Dong, Y.; Liu, W.; Ma, X.; Shi, R.; Chen, H.; Cui, Z.; Ao, L.; Zhang, H.; Cao, J. Epigenetic regulation of sox30 is associated with testis development in mice. PLoS ONE 2014, 9, e97203. [Google Scholar] [CrossRef]
- Kamachi, Y.; Uchikawa, M.; Kondoh, H. Pairing SOX off: With partners in the regulation of embryonic development. Trends Genet. 2000, 16, 182–187. [Google Scholar] [CrossRef]
- Larney, C.; Bailey, T.L.; Koopman, P. Switching on sex: Transcriptional regulation of the testis-determining gene Sry. Development 2014, 141, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Sekido, R.; Lovell-Badge, R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 2008, 453, 930. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Liu, S.; Gao, X.; Jiang, Y.; Perera, D.; Wang, X.; Li, C.; Sun, L.; Zhang, J.; Kaltenboeck, L. Male-biased genes in catfish as revealed by RNA-Seq analysis of the testis transcriptome. PLoS ONE 2013, 8, e68452. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, S.M.; Hacker, A.; Harley, V.; Goodfellow, P.; Swain, A.; Lovell-Badge, R. Sox9 expression during gonadal development implies a conserved role for the gene in testis differentiation in mammals and birds. Nat. Genet. 1996, 14, 62. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Smith, M.J.; Sinclair, A.H. Gene expression during gonadogenesis in the chicken embryo. Gene 1999, 234, 395–402. [Google Scholar] [CrossRef]
- Nagai, K. Molecular evolution of Sry and Sox gene. Gene 2001, 270, 161–169. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence (5′–3′) | Primer Length (bp) |
|---|---|---|
| Sox4-F | CATCAAGCGACCGATGAACG | 20 |
| Sox4-R | CTTCTCGCTGTCCTTCAGCA | 20 |
| Sox3-F | CCGAGATCAAGACTCCGCAA | 20 |
| Sox3-R | GTTCTCCTGGGCCATCTTCC | 20 |
| Sox9-F | GGAGAACACCCGACCTCAAG | 20 |
| Sox9-R | CGTGGGGTTTGTTCTTGCTG | 20 |
| Sox17-F | TAAATCGTGGAAGGCGCTGT | 20 |
| Sox17-R | GCCGCTTCACCTGCTTCTTC | 20 |
| Sox30-F | CTAGGATTCACCGACCTGCC | 20 |
| Sox30-R | TGGTCGTGGCTGATAAACCC | 20 |
| β-action-F | TGTAACCCAACAAGTGTCTT | 20 |
| β-action-R | CCACATACTGGCACTTTACT | 20 |
| Species | C. japonica | G. gallus | C. virginianus | C. squamata | N. meleagris | B. thoracicus | M. gallopavo | |
|---|---|---|---|---|---|---|---|---|
| Singleton | Genome | 3158 | 2443 | 4562 | 4491 | 4259 | 6043 | 5847 |
| Sox | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Dispersed | Genome | 4526 | 4353 | 12332 | 12266 | 4197 | 11588 | 6459 |
| Sox | 13 | 12 | 13 | 14 | 9 | 13 | 9 | |
| Proximal | Genome | 2015 | 4018 | 183 | 132 | 2580 | 91 | 1214 |
| Sox | 0 | 1 | 1 | 0 | 1 | 0 | 0 | |
| Tandem | Genome | 28974 | 37843 | 67 | 104 | 31830 | 41 | 19539 |
| Sox | 4 | 3 | 0 | 0 | 5 | 0 | 4 | |
| WGD or segmental | Genome | 416 | 560 | 22 | 139 | 375 | 0 | 263 |
| Sox | 1 | 2 | 0 | 0 | 0 | 0 | 0 | |
| Total | Genome | 39089 | 45199 | 17166 | 17132 | 43241 | 17763 | 33322 |
| Sox | 18 | 18 | 14 | 14 | 15 | 13 | 13 | |
| Group | Gene | Intron Number | Length (aa) | Molecular Weight (KDa) | pI | GRAVY |
|---|---|---|---|---|---|---|
| B1 | Sox1 | 0 | 373 | 37.91857 | 9.70 | −0.505 |
| Sox2 | 0 | 404 | 44.21928 | 9.96 | −0.641 | |
| Sox3 | 0 | 316 | 34.02945 | 9.68 | −0.715 | |
| B2 | Sox14 | 0 | 240 | 26.66555 | 9.68 | −0.630 |
| Sox21 | 0 | 280 | 28.76797 | 9.74 | −0.218 | |
| C | Sox4 | 0 | 427 | 43.10559 | 7.10 | −0.658 |
| Sox11 | 0 | 396 | 43.18391 | 4.92 | −0.770 | |
| Sox12 | 0 | 285 | 31.26546 | 7.65 | −0.805 | |
| D | Sox5 | 21 | 773 | 85.07967 | 6.15 | −0.746 |
| Sox6 | 15 | 816 | 90.57472 | 6.66 | −0.817 | |
| Sox13 | 15 | 612 | 68.31525 | 6.13 | −0.810 | |
| E | Sox8 | 2 | 470 | 50.90391 | 6.37 | −0.820 |
| Sox9 | 2 | 495 | 55.03546 | 6.16 | −1.080 | |
| Sox10 | 3 | 565 | 60.96868 | 7.47 | −0.788 | |
| F | Sox17 | 1 | 387 | 40.38743 | 6.68 | −0.580 |
| Sox18 | 1 | 418 | 46.22470 | 6.40 | −0.791 | |
| Sox7 | 1 | 377 | 41.15915 | 6.62 | −0.759 | |
| H | Sox30 | 4 | 638 | 68.85069 | 6.07 | −0.490 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Bi, D.; Ding, H.; Wu, X.; Zhu, R.; Zeng, J.; Yang, X.; Kan, X. Systematic Identification and Evolution Analysis of Sox Genes in Coturnix japonica Based on Comparative Genomics. Genes 2019, 10, 314. https://doi.org/10.3390/genes10040314
Jiang L, Bi D, Ding H, Wu X, Zhu R, Zeng J, Yang X, Kan X. Systematic Identification and Evolution Analysis of Sox Genes in Coturnix japonica Based on Comparative Genomics. Genes. 2019; 10(4):314. https://doi.org/10.3390/genes10040314
Chicago/Turabian StyleJiang, Lan, De Bi, Hengwu Ding, Xuan Wu, Ran Zhu, Juhua Zeng, Xiaojun Yang, and Xianzhao Kan. 2019. "Systematic Identification and Evolution Analysis of Sox Genes in Coturnix japonica Based on Comparative Genomics" Genes 10, no. 4: 314. https://doi.org/10.3390/genes10040314
APA StyleJiang, L., Bi, D., Ding, H., Wu, X., Zhu, R., Zeng, J., Yang, X., & Kan, X. (2019). Systematic Identification and Evolution Analysis of Sox Genes in Coturnix japonica Based on Comparative Genomics. Genes, 10(4), 314. https://doi.org/10.3390/genes10040314