Correction of NR2E3 Associated Enhanced S-cone Syndrome Patient-specific iPSCs using CRISPR-Cas9

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient-Derived iPSCs
2.2. Cloning of CRISPR-Cas9 and HDR Donor Constructs
2.3. Screening of NR2E3-specific sgRNAs
2.4. Delivery of sgRNA-Cas9 and the HDR Contruct to NR2E3-Patient-Specific iPSCs
2.5. Retinal Differentiation
2.6. Reverse Transcription PCR
3. Results
3.1. Testing CRISPR-Cas9 Guide Cleavage in HEK293T Cells and iPSCs
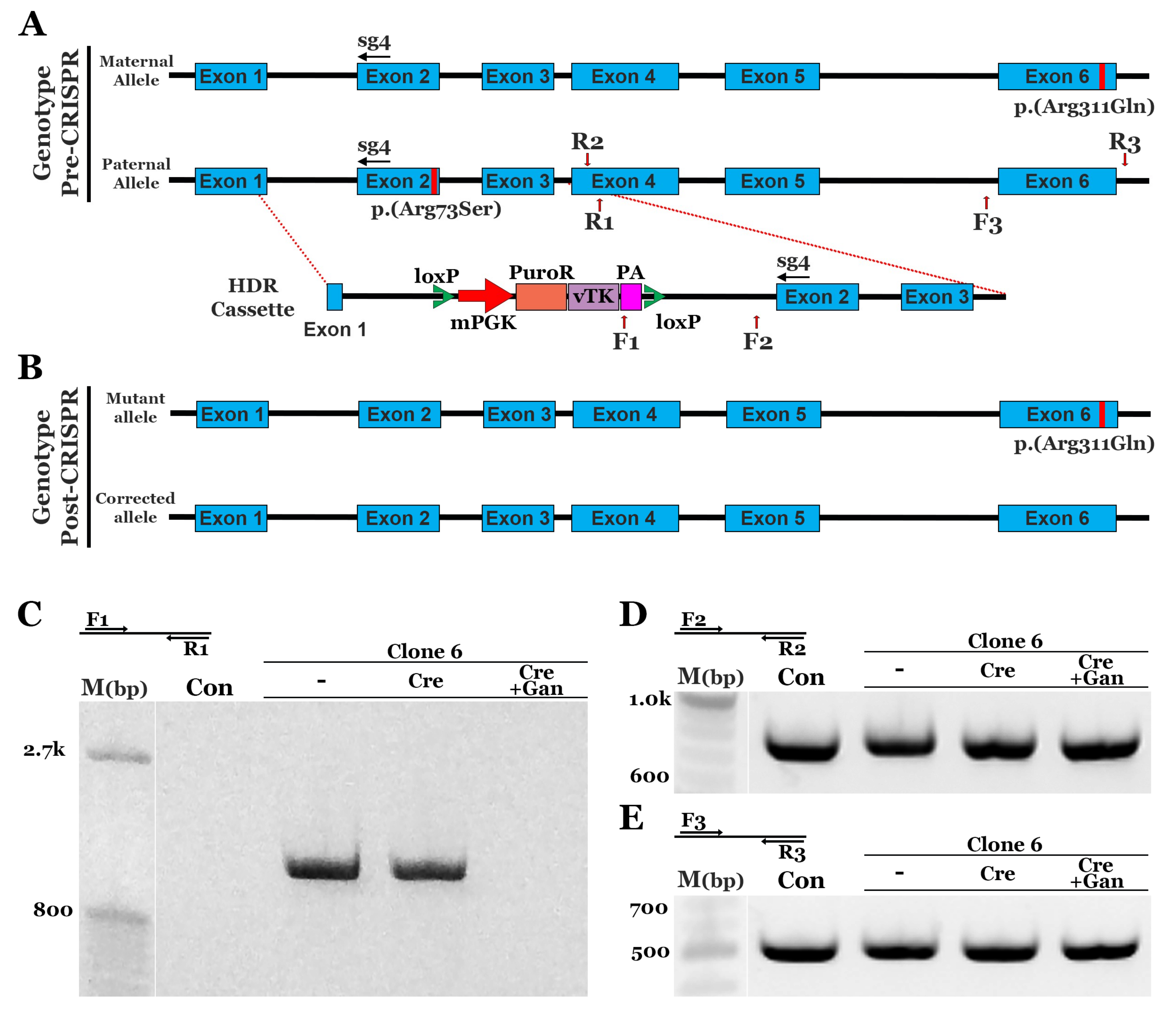
3.2. CRISPR Correction of the NR2E3 c.119-2A>C mutation in Patient-Specific iPSCs using Homology-Directed Repair (HDR)
3.3. CRISPR Correction of the NR2E3 p.(Arg73Ser) Mutation in Patient-Specific iPSCs using HDR
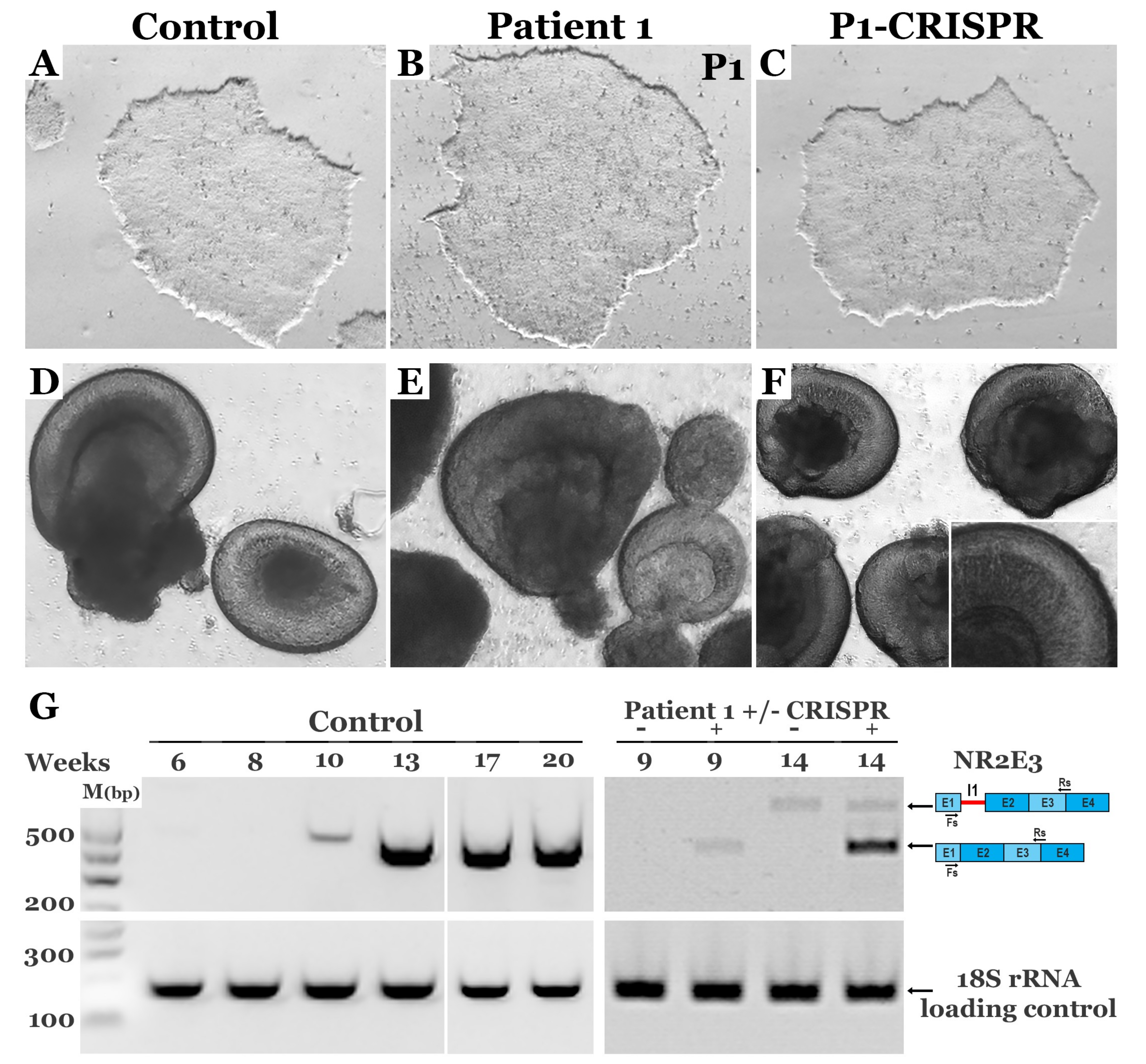
3.4. CRISPR-based Restoration of the NR2E3 Transcript in Patient-derived Retinal Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haider, N.B.; Jacobson, S.G.; Cideciyan, A.V.; Swiderski, R.; Streb, L.M.; Searby, C.; Beck, G.; Hockey, R.; Hanna, D.B.; Gorman, S.; et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet. 2000, 24, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.B.; Naggert, J.K.; Nishina, P.M. Excess cone cell proliferation due to lack of a functional NR2E3 causes retinal dysplasia and degeneration in rd7/rd7 mice. Hum. Mol. Genet. 2001, 10, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.B.; Demarco, P.; Nystuen, A.M.; Huang, X.; Smith, R.S.; McCall, M.A.; Naggert, J.K.; Nishina, P.M. The transcription factor Nr2e3 functions in retinal progenitors to suppress cone cell generation. Vis. Neurosci. 2006, 23, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.B.; Mollema, N.; Gaule, M.; Yuan, Y.; Sachs, A.J.; Nystuen, A.M.; Naggert, J.K.; Nishina, P.M. Nr2e3-directed transcriptional regulation of genes involved in photoreceptor development and cell-type specific phototransduction. Exp. Eye Res. 2009, 89, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Khan, N.W.; Roger, J.E.; Swaroop, A. Excess cones in the retinal degeneration rd7 mouse, caused by the loss of function of orphan nuclear receptor Nr2e3, originate from early-born photoreceptor precursors. Hum. Mol. Genet. 2011, 20, 4102–4115. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rattner, A.; Nathans, J. The rod photoreceptor-specific nuclear receptor Nr2e3 represses transcription of multiple cone-specific genes. J. Neurosci. 2005, 25, 118–129. [Google Scholar] [CrossRef]
- Marmor, M.F.; Jacobson, S.G.; Foerster, M.H.; Kellner, U.; Weleber, R.G. Diagnostic clinical findings of a new syndrome with night blindness, maculopathy, and enhanced S cone sensitivity. Am. J. Ophthalmol. 1990, 110, 124–134. [Google Scholar] [CrossRef]
- Fishman, G.A.; Jampol, L.M.; Goldberg, M.F. Diagnostic features of the Favre-Goldmann syndrome. Br. J. Ophthalmol 1976, 60, 345–353. [Google Scholar] [CrossRef]
- Garafalo, A.V.; Calzetti, G.; Cideciyan, A.V.; Roman, A.J.; Saxena, S.; Sumaroka, A.; Choi, W.; Wright, A.F.; Jacobson, S.G. Cone Vision Changes in the Enhanced S-Cone Syndrome Caused by NR2E3 Gene Mutations. Invest. Ophthalmol. Vis. Sci. 2018, 59, 3209–3219. [Google Scholar] [CrossRef]
- Yzer, S.; Barbazetto, I.; Allikmets, R.; van Schooneveld, M.J.; Bergen, A.; Tsang, S.H.; Jacobson, S.G.; Yannuzzi, L.A. Expanded clinical spectrum of enhanced S-cone syndrome. JAMA Ophthalmol 2013, 131, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Schorderet, D.F.; Escher, P. NR2E3 mutations in enhanced S-cone sensitivity syndrome (ESCS), Goldmann-Favre syndrome (GFS), clumped pigmentary retinal degeneration (CPRD), and retinitis pigmentosa (RP). Hum. Mutat. 2009, 30, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Escher, P.; Gouras, P.; Roduit, R.; Tiab, L.; Bolay, S.; Delarive, T.; Chen, S.; Tsai, C.-C.; Hayashi, M.; Zernant, J.; et al. Mutations in NR2E3 can cause dominant or recessive retinal degenerations in the same family. Hum. Mutat. 2009, 30, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef] [PubMed]
- Bernal, S.; Solans, T.; Gamundi, M.J.; Hernan, I.; de Jorge, L.; Carballo, M.; Navarro, R.; Tizzano, E.; Ayuso, C.; Baiget, M. Analysis of the involvement of the NR2E3 gene in autosomal recessive retinal dystrophies. Clin. Genet. 2008, 73, 360–366. [Google Scholar] [CrossRef]
- Burnight, E.R.; Giacalone, J.C.; Cooke, J.A.; Thompson, J.R.; Bohrer, L.R.; Chirco, K.R.; Drack, A.V.; Fingert, J.H.; Worthington, K.S.; Wiley, L.A.; et al. CRISPR-Cas9 genome engineering: Treating inherited retinal degeneration. Prog Retin Eye Res. 2018, 65, 28–49. [Google Scholar] [CrossRef] [PubMed]
- Giacalone, J.C.; Sharma, T.P.; Burnight, E.R.; Fingert, J.F.; Mullins, R.F.; Stone, E.M.; Tucker, B.A. CRISPR-Cas9-Based Genome Editing of Human Induced Pluripotent Stem Cells. Curr Protoc Stem Cell Biol 2018, 44, 5B.7.1–5B.7.22. [Google Scholar] [PubMed]
- Burnight, E.R.; Gupta, M.; Wiley, L.A.; Anfinson, K.R.; Tran, A.; Triboulet, R.; Hoffmann, J.M.; Klaahsen, D.L.; Andorf, J.L.; Jiao, C.; et al. Using CRISPR-Cas9 to Generate Gene-Corrected Autologous iPSCs for the Treatment of Inherited Retinal Degeneration. Mol. Ther. 2017, 25, 1999–2013. [Google Scholar] [CrossRef]
- Burnight, E.R.; Bohrer, L.R.; Giacalone, J.C.; Klaahsen, D.L.; Daggett, H.T.; East, J.S.; Madumba, R.A.; Worthington, K.S.; Mullins, R.F.; Stone, E.M.; et al. CRISPR-Cas9-Mediated Correction of the 1.02 kb Common Deletion in CLN3 in Induced Pluripotent Stem Cells from Patients with Batten Disease. The CRISPR Journal 2018, 1, 75–87. [Google Scholar] [CrossRef]
- Buskin, A.; Zhu, L.; Chichagova, V.; Basu, B.; Mozaffari-Jovin, S.; Dolan, D.; Droop, A.; Collin, J.; Bronstein, R.; Mehrotra, S.; et al. Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa. Nat. Commun 2018, 9, 4234. [Google Scholar] [CrossRef]
- Steyer, B.; Bu, Q.; Cory, E.; Jiang, K.; Duong, S.; Sinha, D.; Steltzer, S.; Gamm, D.; Chang, Q.; Saha, K. Scarless Genome Editing of Human Pluripotent Stem Cells via Transient Puromycin Selection. Stem Cell Reports 2018, 10, 642–654. [Google Scholar] [CrossRef]
- Deng, W.-L.; Gao, M.-L.; Lei, X.-L.; Lv, J.-N.; Zhao, H.; He, K.-W.; Xia, X.-X.; Li, L.-Y.; Chen, Y.-C.; Li, Y.-P.; et al. Gene Correction Reverses Ciliopathy and Photoreceptor Loss in iPSC-Derived Retinal Organoids from Retinitis Pigmentosa Patients. Stem Cell Reports 2018, 10, 1267–1281. [Google Scholar] [CrossRef]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Wiley, L.A.; Anfinson, K.R.; Cranston, C.M.; Kaalberg, E.E.; Collins, M.M.; Mullins, R.F.; Stone, E.M.; Tucker, B.A. Generation of Xeno-Free, cGMP-Compliant Patient-Specific iPSCs from Skin Biopsy. Curr Protoc Stem Cell Biol 2017, 42, 4A.12.1–4A.12.14. [Google Scholar] [PubMed]
- Wiley, L.A.; Burnight, E.R.; DeLuca, A.P.; Anfinson, K.R.; Cranston, C.M.; Kaalberg, E.E.; Penticoff, J.A.; Affatigato, L.M.; Mullins, R.F.; Stone, E.M.; et al. cGMP production of patient-specific iPSCs and photoreceptor precursor cells to treat retinal degenerative blindness. Sci Rep. 2016, 6, 30742. [Google Scholar] [CrossRef] [PubMed]
- Ohlemacher, S.K.; Iglesias, C.L.; Sridhar, A.; Gamm, D.M.; Meyer, J.S. Generation of highly enriched populations of optic vesicle-like retinal cells from human pluripotent stem cells. Curr Protoc Stem Cell Biol. 2015, 32, 1H.8.1–1H.8.20. [Google Scholar] [PubMed]
- Tucker, B.A.; Scheetz, T.E.; Mullins, R.F.; DeLuca, A.P.; Hoffmann, J.M.; Johnston, R.M.; Jacobson, S.G.; Sheffield, V.C.; Stone, E.M. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2011, 108, E569–E576. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.A.; Cranston, C.M.; Anfinson, K.A.; Shrestha, S.; Streb, L.M.; Leon, A.; Mullins, R.F.; Stone, E.M. Using patient-specific induced pluripotent stem cells to interrogate the pathogenicity of a novel retinal pigment epithelium-specific 65 kDa cryptic splice site mutation and confirm eligibility for enrollment into a clinical gene augmentation trial. Transl Res. 2015, 166, 740–749.e1. [Google Scholar] [CrossRef] [PubMed]
- Wiley, L.A.; Burnight, E.R.; Drack, A.V.; Banach, B.B.; Ochoa, D.; Cranston, C.M.; Madumba, R.A.; East, J.S.; Mullins, R.F.; Stone, E.M.; et al. Using Patient-Specific Induced Pluripotent Stem Cells and Wild-Type Mice to Develop a Gene Augmentation-Based Strategy to Treat CLN3-Associated Retinal Degeneration. Hum. Gene Ther. 2016, 27, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Llonch, S.; Carido, M.; Ader, M. Organoid technology for retinal repair. Dev. Biol. 2018, 433, 132–143. [Google Scholar] [CrossRef]
- Achberger, K.; Haderspeck, J.C.; Kleger, A.; Liebau, S. Stem cell-based retina models. Adv. Drug Deliv. Rev. 2018. [Google Scholar] [CrossRef]
- Tucker, B.A.; Park, I.-H.; Qi, S.D.; Klassen, H.J.; Jiang, C.; Yao, J.; Redenti, S.; Daley, G.Q.; Young, M.J. Transplantation of adult mouse iPS cell-derived photoreceptor precursors restores retinal structure and function in degenerative mice. PLoS ONE 2011, 6, e18992. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Onishi, A.; Ito, S.-I.; Nakamura, M.; Takahashi, M. Generation of three-dimensional retinal organoids expressing rhodopsin and S- and M-cone opsins from mouse stem cells. Biochem. Biophys. Res. Commun. 2018, 495, 2595–2601. [Google Scholar] [CrossRef] [PubMed]
- Riazifar, H.; Jia, Y.; Chen, J.; Lynch, G.; Huang, T. Chemically induced specification of retinal ganglion cells from human embryonic and induced pluripotent stem cells. Stem Cells Transl Med. 2014, 3, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.A.; Anfinson, K.R.; Mullins, R.F.; Stone, E.M.; Young, M.J. Use of a synthetic xeno-free culture substrate for induced pluripotent stem cell induction and retinal differentiation. Stem Cells Transl Med. 2013, 2, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.R.; Hartman, B.H.; Georgi, S.A.; Lan, M.S.; Reh, T.A. Transient inactivation of Notch signaling synchronizes differentiation of neural progenitor cells. Dev. Biol. 2007, 304, 479–498. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bohrer, L.R.; Wiley, L.A.; Burnight, E.R.; Cooke, J.A.; Giacalone, J.C.; Anfinson, K.R.; Andorf, J.L.; Mullins, R.F.; Stone, E.M.; Tucker, B.A. Correction of NR2E3 Associated Enhanced S-cone Syndrome Patient-specific iPSCs using CRISPR-Cas9. Genes 2019, 10, 278. https://doi.org/10.3390/genes10040278
Bohrer LR, Wiley LA, Burnight ER, Cooke JA, Giacalone JC, Anfinson KR, Andorf JL, Mullins RF, Stone EM, Tucker BA. Correction of NR2E3 Associated Enhanced S-cone Syndrome Patient-specific iPSCs using CRISPR-Cas9. Genes. 2019; 10(4):278. https://doi.org/10.3390/genes10040278
Chicago/Turabian StyleBohrer, Laura R., Luke A. Wiley, Erin R. Burnight, Jessica A. Cooke, Joseph C. Giacalone, Kristin R. Anfinson, Jeaneen L. Andorf, Robert F. Mullins, Edwin M. Stone, and Budd A. Tucker. 2019. "Correction of NR2E3 Associated Enhanced S-cone Syndrome Patient-specific iPSCs using CRISPR-Cas9" Genes 10, no. 4: 278. https://doi.org/10.3390/genes10040278
APA StyleBohrer, L. R., Wiley, L. A., Burnight, E. R., Cooke, J. A., Giacalone, J. C., Anfinson, K. R., Andorf, J. L., Mullins, R. F., Stone, E. M., & Tucker, B. A. (2019). Correction of NR2E3 Associated Enhanced S-cone Syndrome Patient-specific iPSCs using CRISPR-Cas9. Genes, 10(4), 278. https://doi.org/10.3390/genes10040278