XGBPRH: Prediction of Binding Hot Spots at Protein–RNA Interfaces Utilizing Extreme Gradient Boosting



Abstract
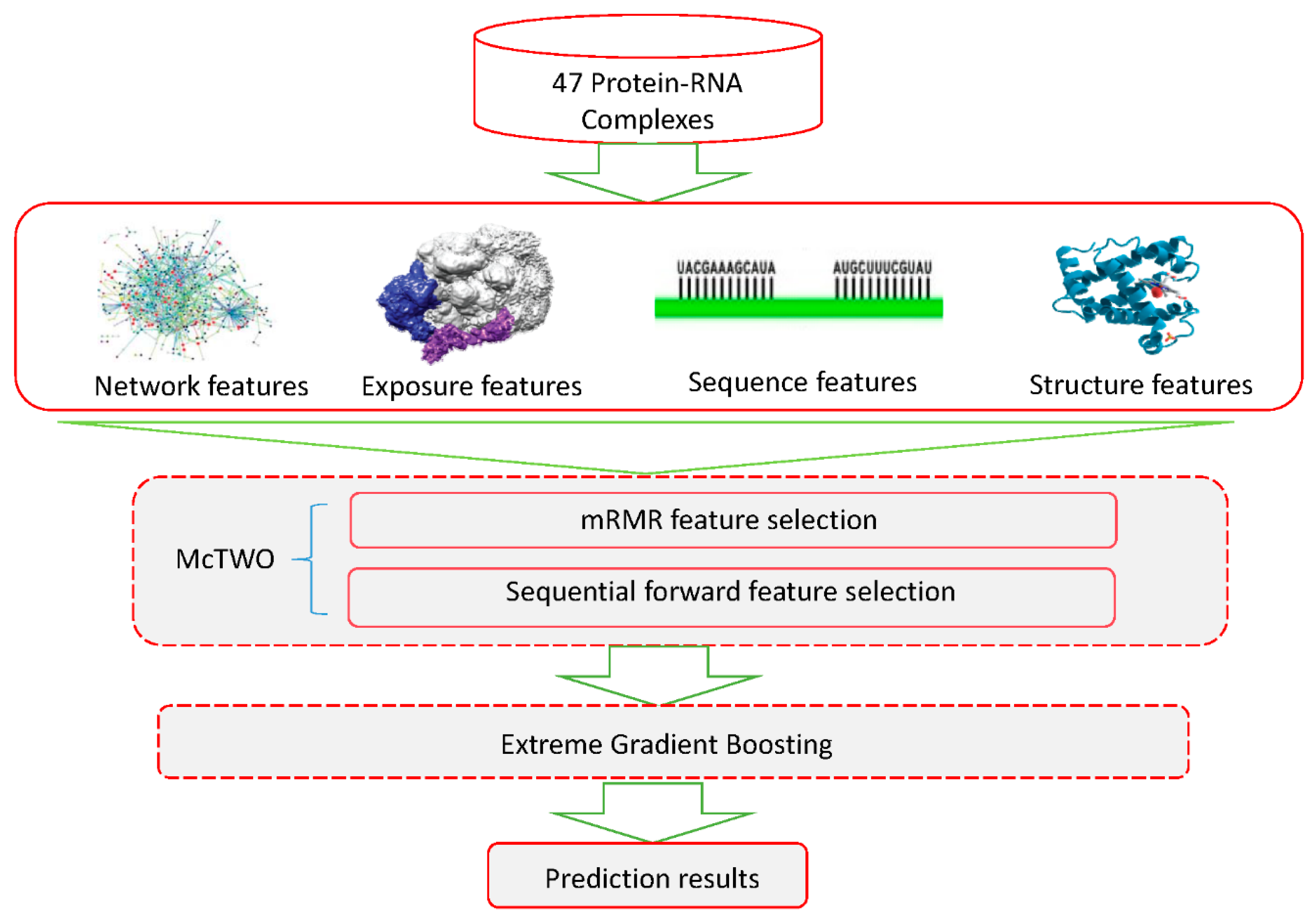
:1. Introduction
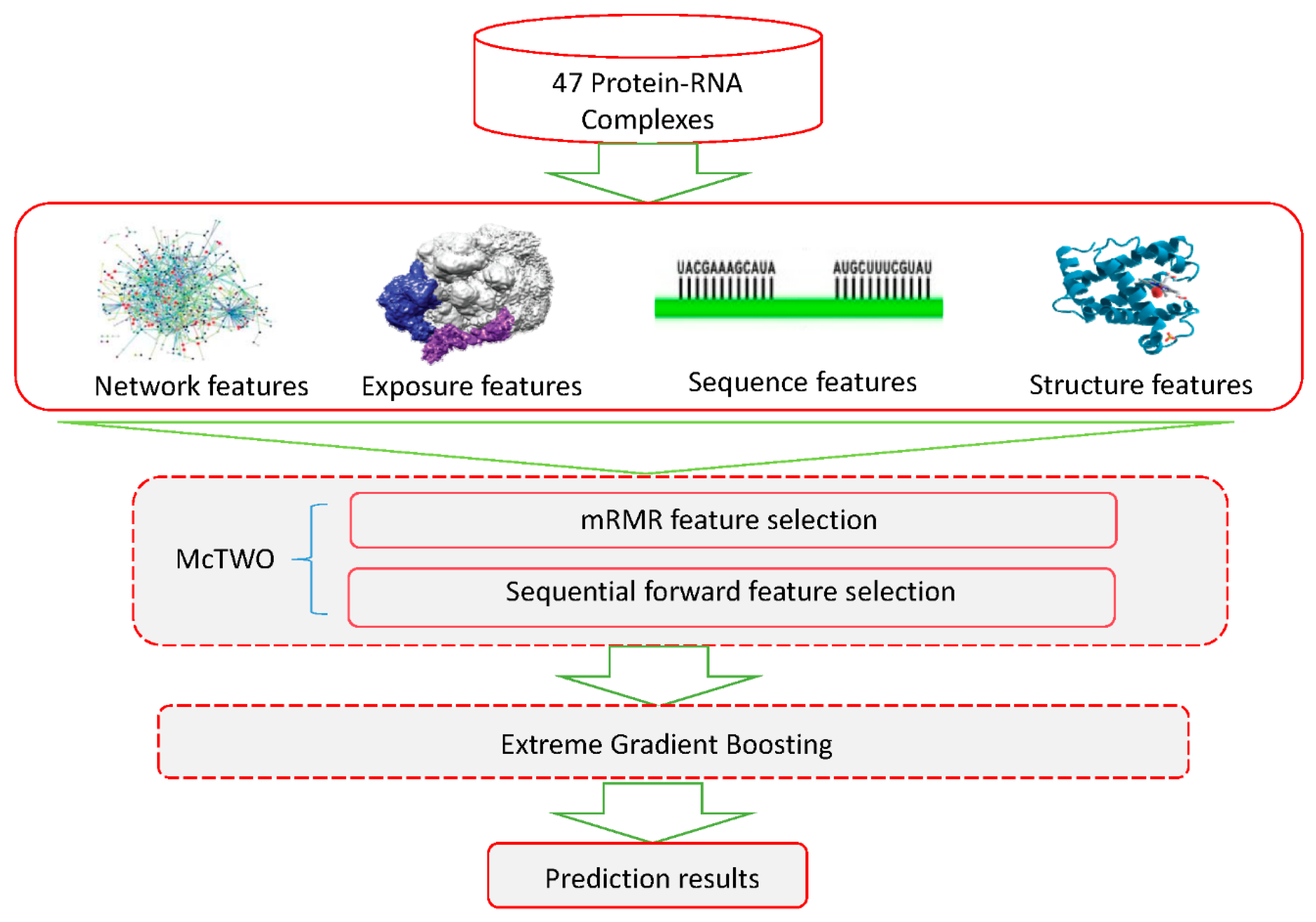
2. Materials and Methods
2.1. Datasets
2.2. Performance Evaluation
2.3. Feature Extraction
2.3.1. Features Based on Network
2.3.2. Features Based on Solvent Exposure
2.3.3. Features Based on 3D Structure
2.3.4. Features Based on Protein Structure
- Solvent accessible area (ASA). ASA represents the relatively accessible surface area, which can be calculated using the Naccess [36] program. These ASA features include values of all atoms (ASA_aaa), relative all atoms (ASA_raa), absolute total side (ASA_ats), and relative total side (ASA_rts). We also computed the ΔASA (the change in the solvent accessible surface area of the protein structure between bound and unbound states).
- Four-body statistical pseudo-potential (FBS2P). The FBS2P score, which is based on the Delaunay tessellation of proteins [39], can be written as the following formula.where i, j, p, and q are termed as the four amino acids in a Delaunay tetrahedron of the protein. represents the observed frequency of the residue component (ijpq) in a tetrahedron of type a over a set of protein structures, and represents the expected random frequency.
- Energy scores. We used ENDES [40] to calculate seven energy scores: residue energy (Enrich_re), side-chain energy (Enrich_se), conservation (Enrich_conserv), two combined scores (Enrich_com1 and Enrich_com2), relative solvent accessibility (Enrich_rsa), and interface propensity (Enrich_ip).
- Hydrogen bonds. The hydrogen bonds were calculated using HBPLUS [41].
- Helix and sheet. The features of α-helix and β-sheet secondary structure are represented with one-hot encoding [42].
2.3.5. Features Based on Protein Sequence
- Backbone flexibility. The protein is flexible and has a range of motion, especially when looking at intrinsically disordered proteins. The feature is calculated by DynaMine [43].
- Side-chain environment. The side-chain environment (pKa) represents an effective metric in determining the environmental characteristics of a protein. The value of pKa was acquired from Nelson and Cox, indicating a protein side-chain environmental factor, and has been utilized in previous research.
- Position-specific scoring matrices (PSSMs). The scoring matrices can be calculated by PSI-BLAST [44].
- Local structural entropy (LSE). LSE [45] is described as the degree of conformational heterogeneity in short protein sequences.
- Conservation score. We mainly used Jensen–Shannon divergence [46] to calculate the conservation score, which is calculated as follows:where is termed as the frequency of amino acid j at position i. The conservation score indicates the variability of residues at each position in the sequence. A value that is small at a position means that the residue is conserved.
- Physicochemical feature. The eight physicochemical features can be obtained from the AAindex database [47]. The eight features are as follows: propensities, average accessible surface area, hydrophobicity, atom-based hydrophobic moment, polarity, polarizability, flexibility parameter for no rigid neighbors, and hydrophilicity.
- Blocks substitution matrix. The substitution probabilities and their relative frequencies of amino acid can be counted by BLOSUM62 [53].
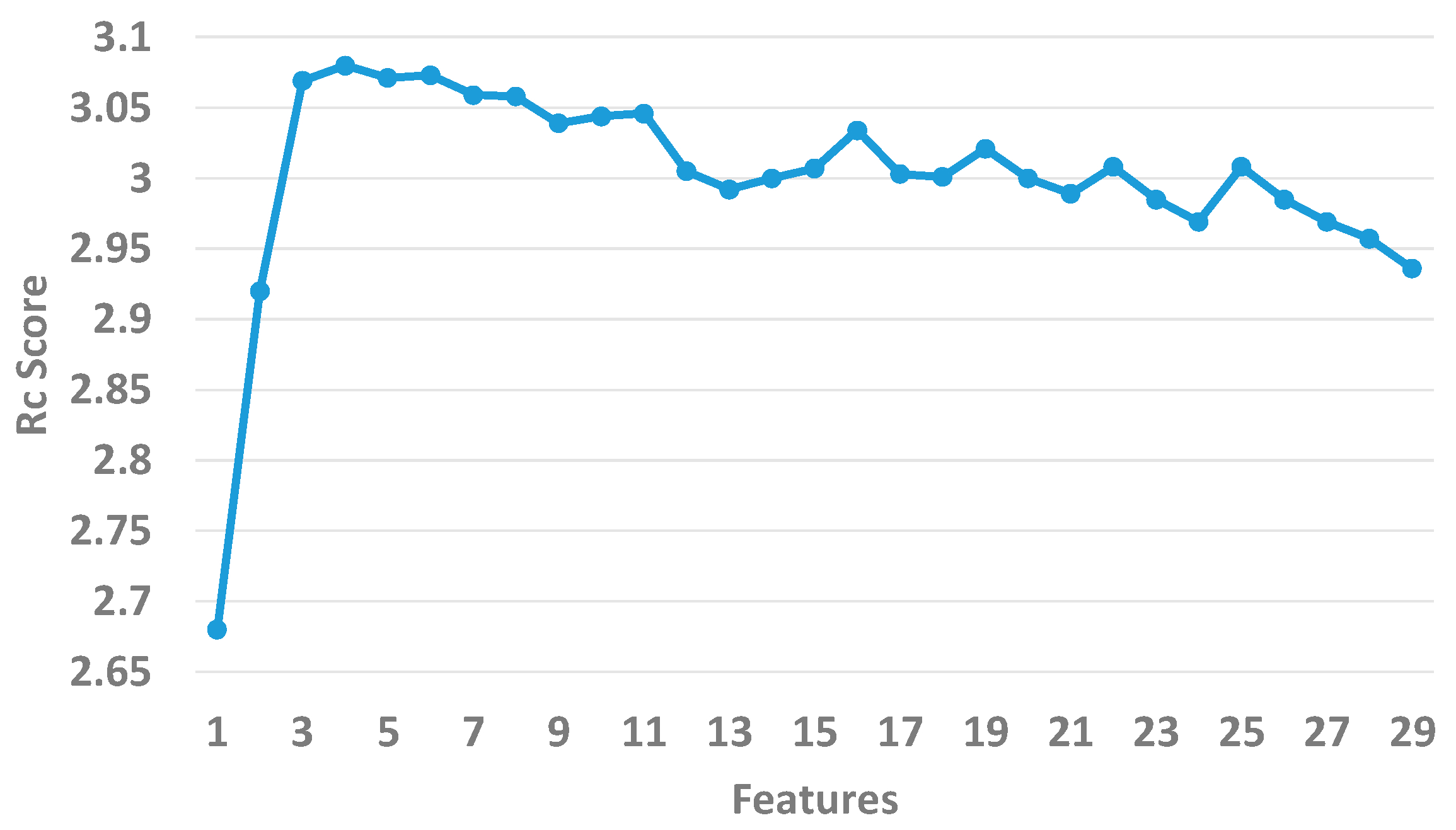
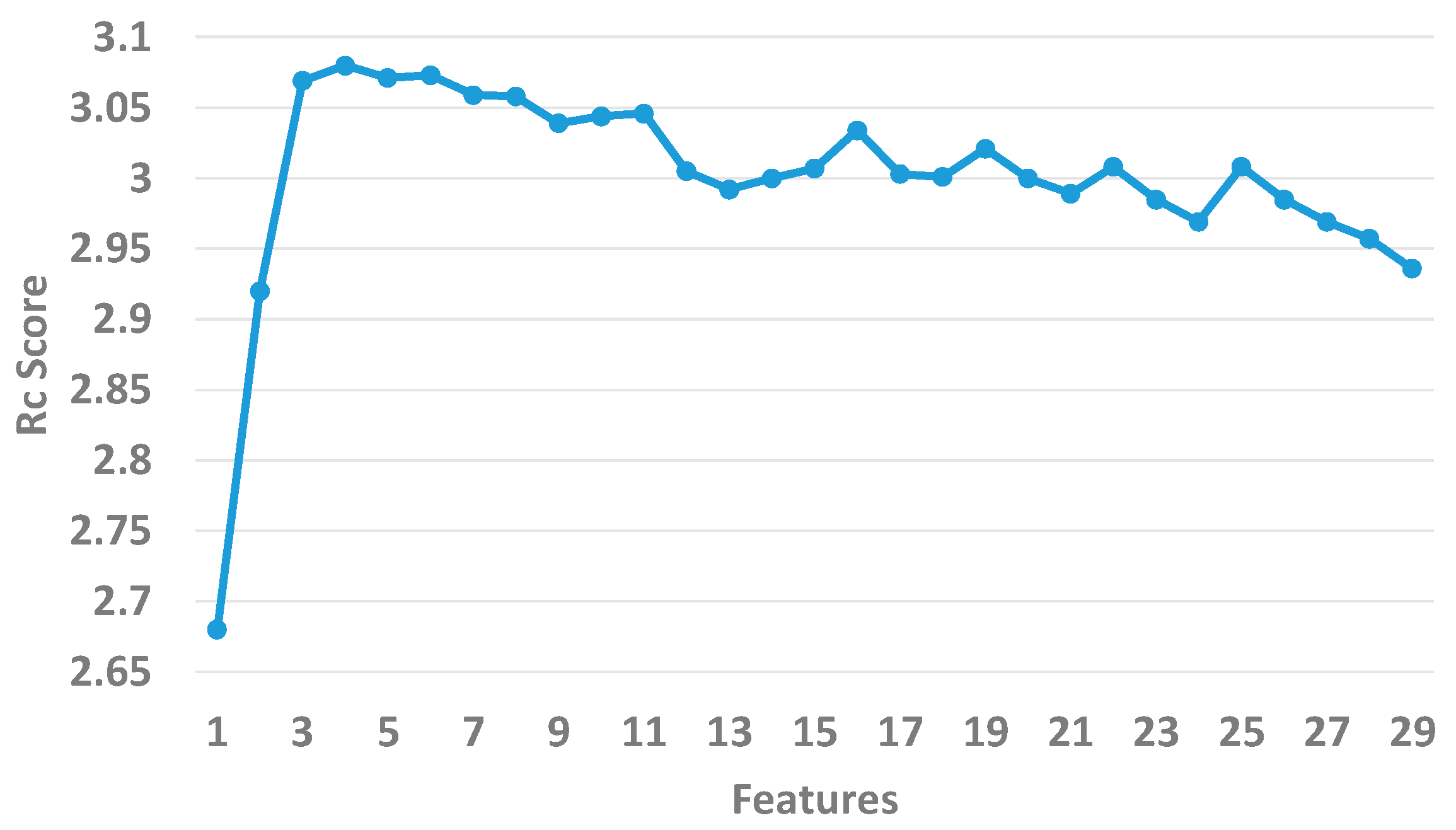
2.4. Feature Selection
2.5. Extreme Gradient Boosting Algorithm
2.6. The XGBPRH Approach
3. Results
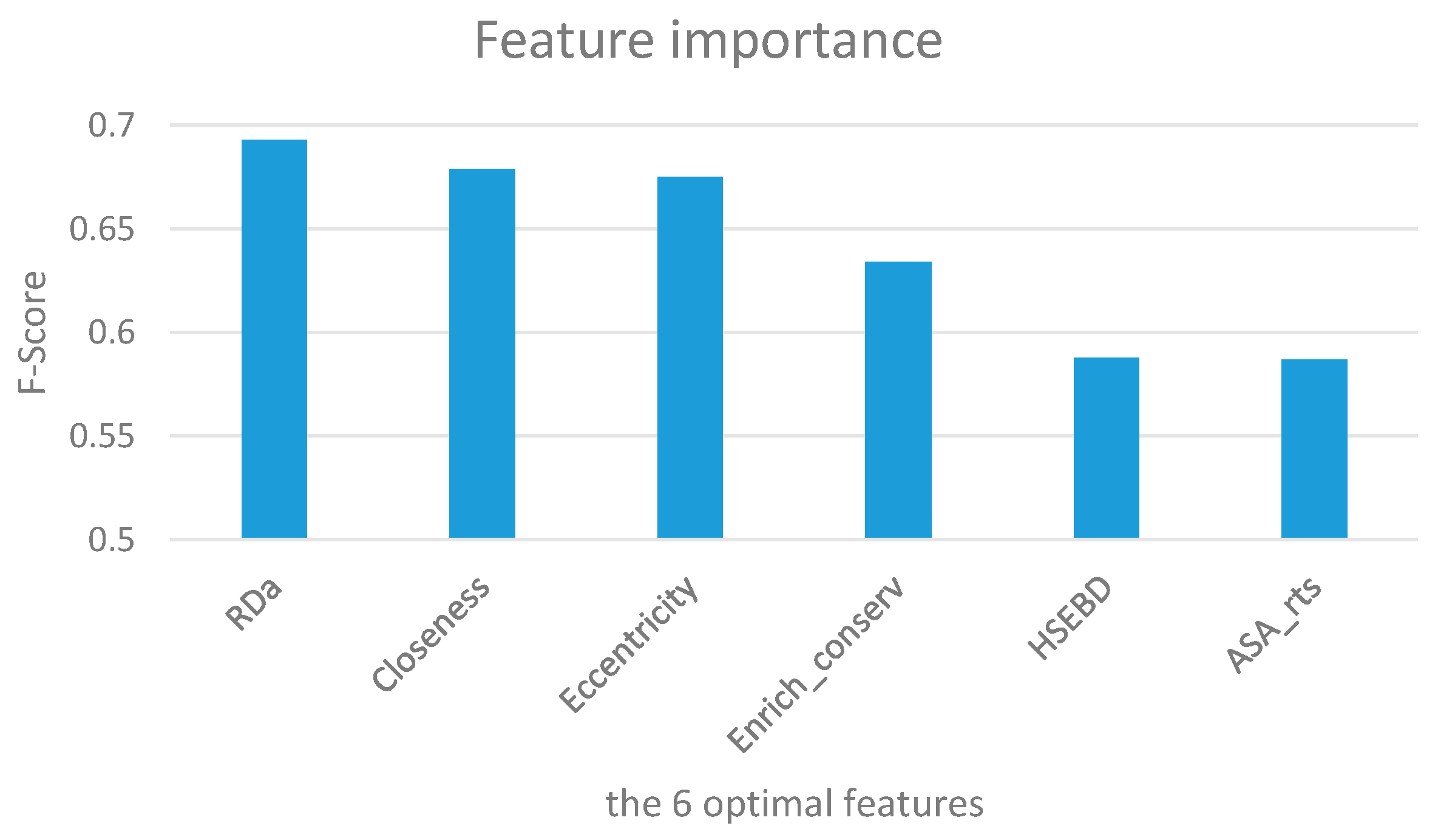
3.1. Assessment of Feature Importance
3.2. Comparison of Different Machine Learing Methods
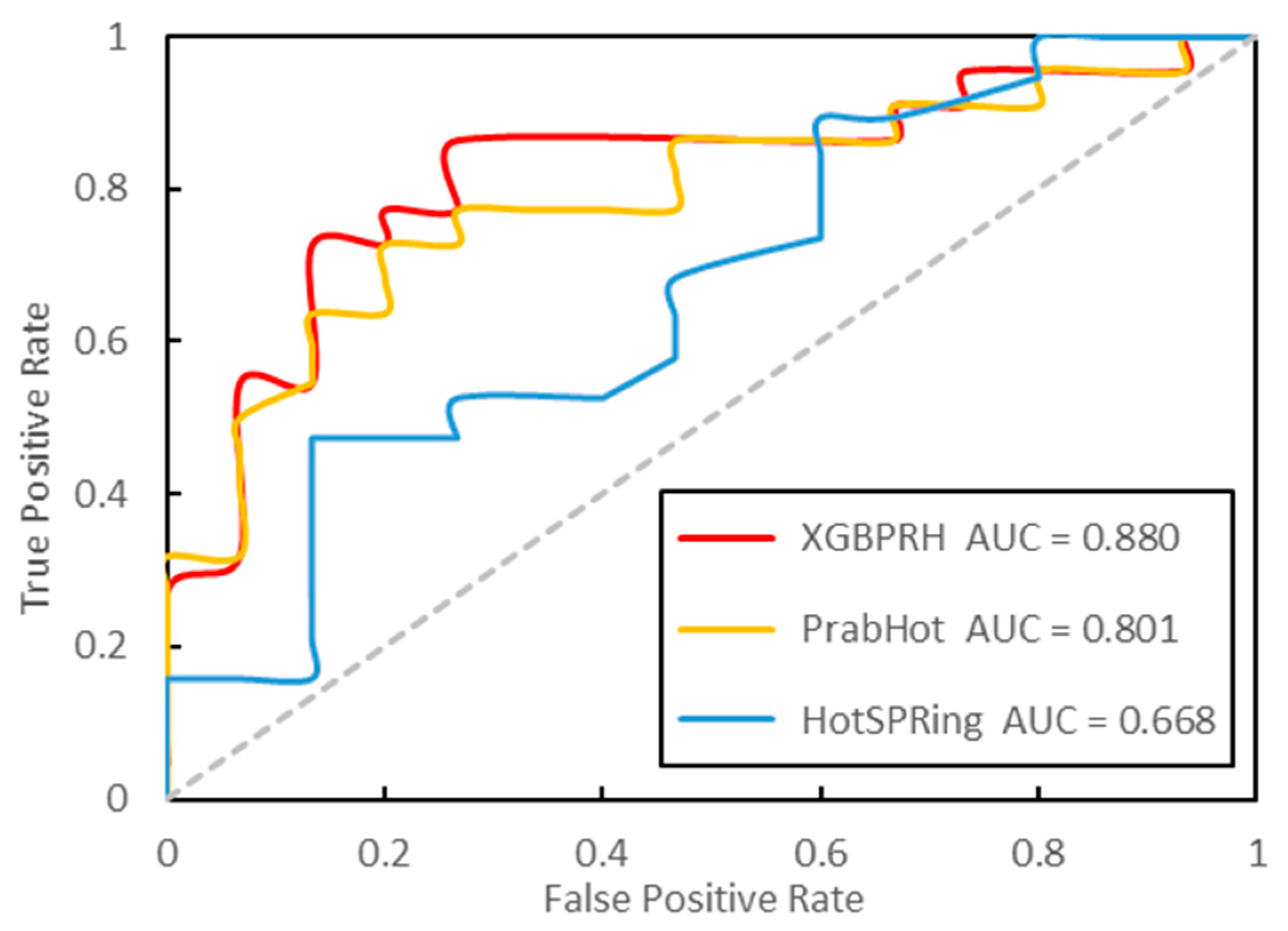
3.3. Performance Evaluation
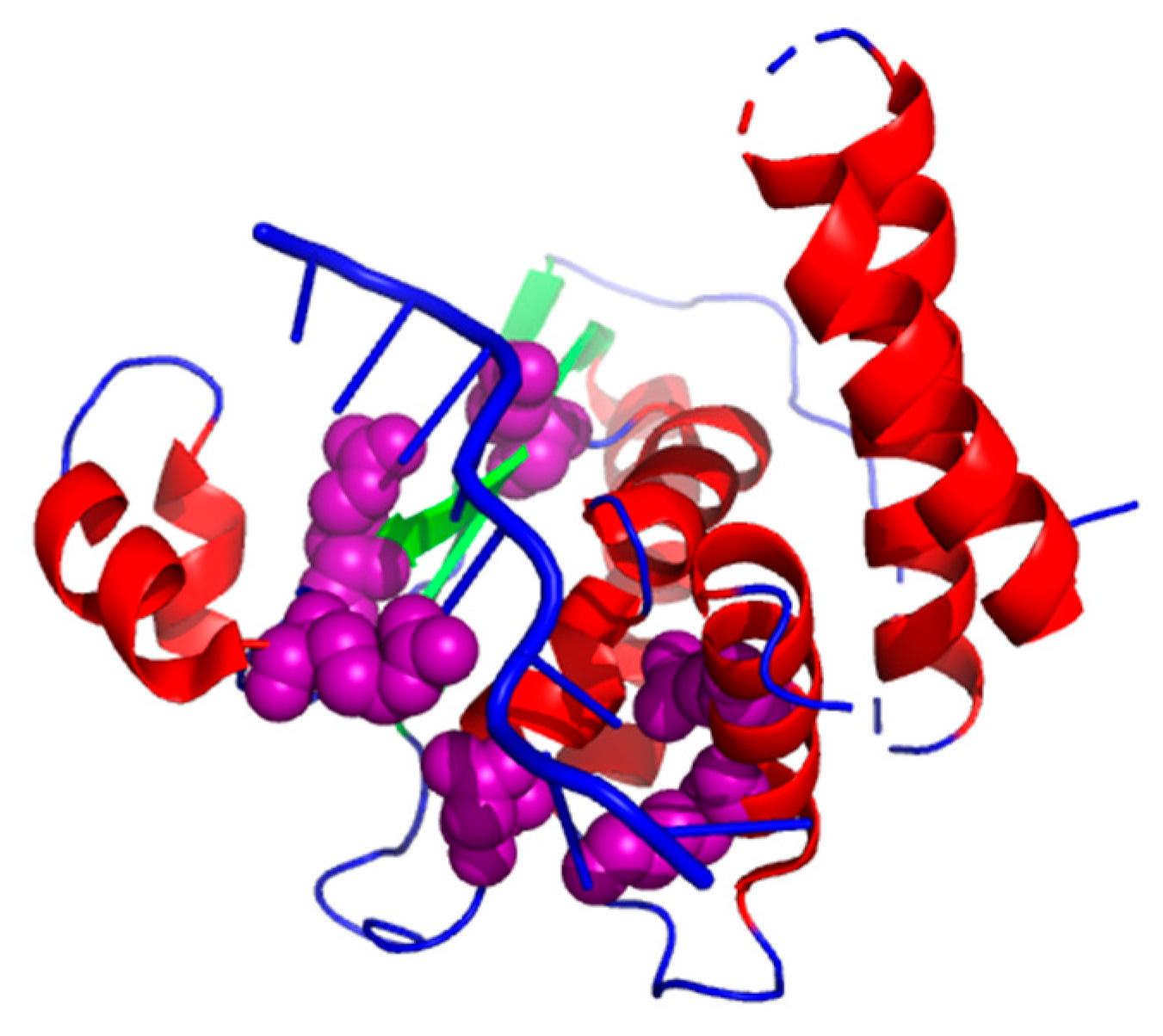
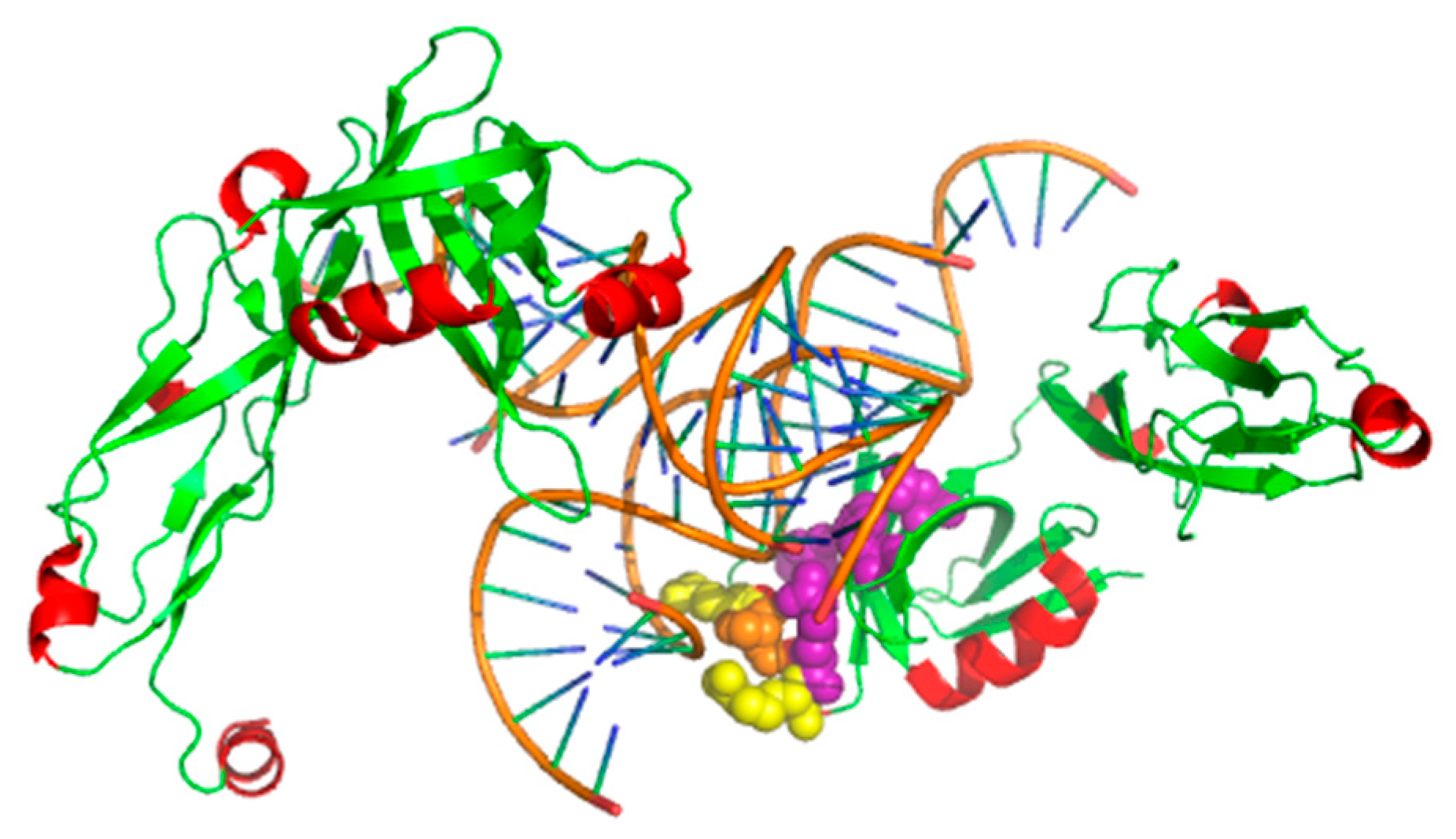
3.4. Case Study
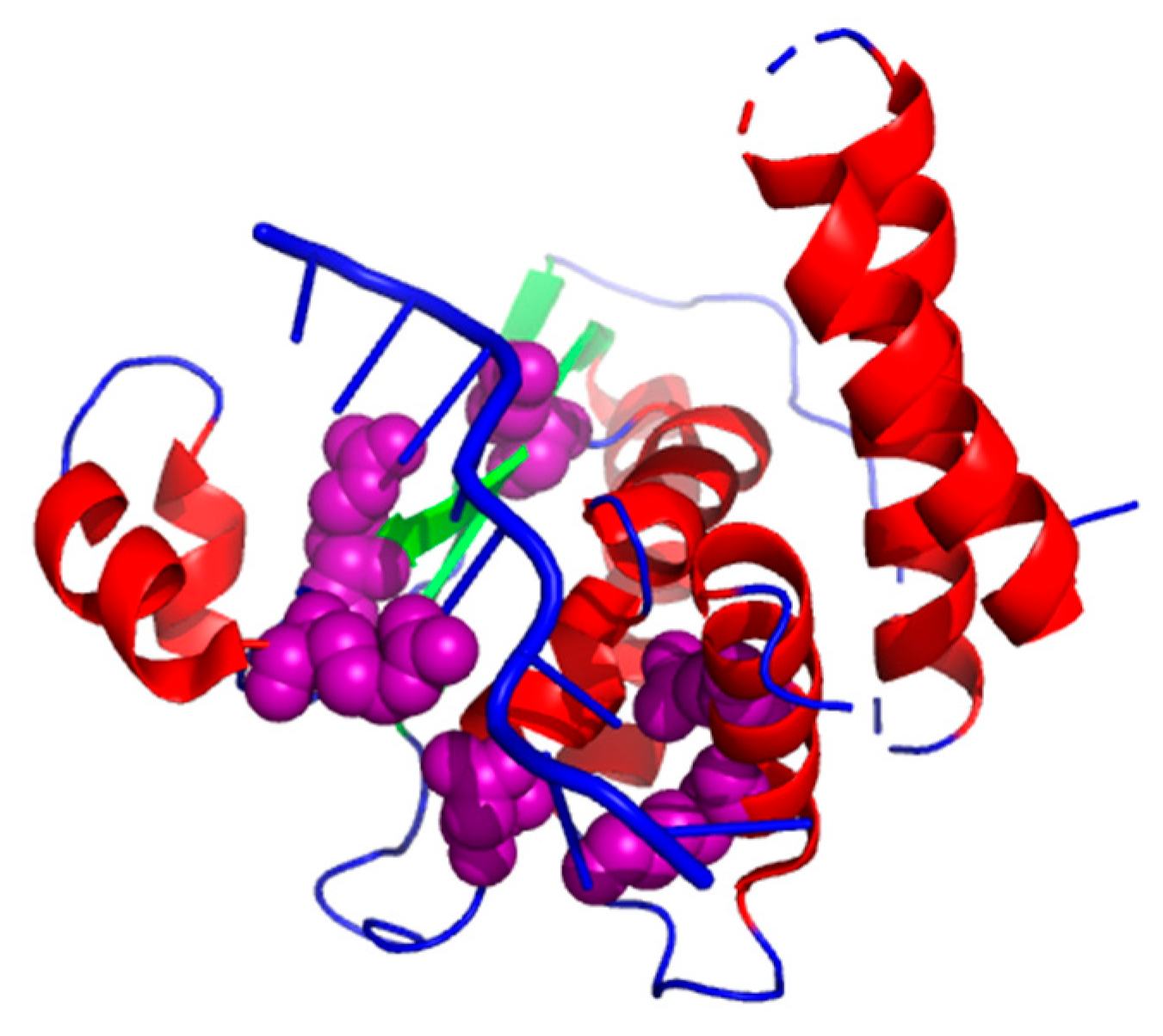
3.4.1. Structure of the Star Domain of Quaking Protein in Complex with RNA
3.4.2. The TL5 and Escherichia coli 5S RNA Complex
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, Z.; Zhao, X.; Chen, L. Identifying responsive functional modules from protein-protein interaction network. Mol. Cells 2009, 27, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Sun, B.; Tang, W.; Sun, P.; Ma, Z. Prediction of conformational B-cell epitope binding with individual antibodies using phage display peptides. Int. J. Clin. Exp. Med. 2016, 9, 2748–2757. [Google Scholar]
- Shen, C.; Ding, Y.; Tang, J.; Jiang, L.; Guo, F. LPI-KTASLP: Prediction of lncRNA-protein interaction by semi-supervised link learning with multivariate information. IEEE Access 2019, 7, 13486–13496. [Google Scholar] [CrossRef]
- Zou, Q.; Xing, P.; Wei, L.; Liu, B. Gene2vec: Gene subsequence embedding for prediction of mammalian N6-Methyladenosine sites from mRNA. RNA 2019, 25, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.; Wang, Z.; Liu, Y.; Deng, L. Ontological function annotation of long non-coding RNAs through hierarchical multi-label classification. Bioinformatics 2018, 34, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.I.; Kim, D.; Lee, D. A feature-based approach to modeling protein–protein interaction hot spots. Nucleic Acids Res. 2009, 37, 2672–2687. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chu, C.; Zhang, Y.H.; Zheng, M.Y.; Zhu, L.C.; Kong, X.Y.; Huang, T. Identification of drug-drug interactions using chemical interactions. Curr. Bioinform. 2017, 12, 526–534. [Google Scholar] [CrossRef]
- Deng, L.; Guan, J.; Dong, Q.; Zhou, S. Prediction of protein-protein interaction sites using an ensemble method. BMC Bioinform. 2009, 10, 426. [Google Scholar] [CrossRef]
- Wei, L.; Tang, J.; Zou, Q. Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information. Inf. Sci. 2017, 384, 135–144. [Google Scholar] [CrossRef]
- Xia, J.-F.; Zhao, X.-M.; Song, J.; Huang, D.-S. APIS: Accurate prediction of hot spots in protein interfaces by combining protrusion index with solvent accessibility. BMC Bioinform. 2010, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zhang, Q.C.; Chen, Z.; Meng, Y.; Guan, J.; Zhou, S. PredHS: A web server for predicting protein–protein interaction hot spots by using structural neighborhood properties. Nucleic Acids Res. 2014, 42, W290–W295. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Guan, J.-H.; Dong, Q.-W.; Zhou, S.-G. SemiHS: an iterative semi-supervised approach for predicting proteinprotein interaction hot spots. Protein Pept. Lett. 2011, 18, 896–905. [Google Scholar] [PubMed]
- Ozdemir, E.S.; Gursoy, A.; Keskin, O. Analysis of single amino acid variations in singlet hot spots of protein–protein interfaces. Bioinformatics 2018, 34, i795–i801. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, C.; Deng, L. Enhanced prediction of hot spots at protein-protein interfaces using extreme gradient boosting. Sci. Rep. 2018, 8, 14285. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Vangone, A.; Folkers, G.E.; Xue, L.C.; Bonvin, A.M. iSEE: Interface structure, evolution, and energy-based machine learning predictor of binding affinity changes upon mutations. Proteins Struct. Funct. Bioinform. 2019, 87, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Koukos, P.I.; Melo, R.; Almeida, J.G.; Preto, A.J.; Schaarschmidt, J.; Trellet, M.; Gümüş, Z.H.; Costa, J.; Bonvin, A.M. SpotOn: High accuracy identification of protein-protein interface hot-spots. Sci. Rep. 2017, 7, 8007. [Google Scholar] [CrossRef] [PubMed]
- Barik, A.; Nithin, C.; Karampudi, N.B.; Mukherjee, S.; Bahadur, R.P. Probing binding hot spots at protein-RNA recognition sites. Nucleic Acids Res. 2015, 44, e9. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, Z.; Zhan, W.; Deng, L. Computational identification of binding energy hot spots in protein-RNA complexes using an ensemble approach. Bioinformatics 2017, 34, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, J.; Guo, F. Identification of residue-residue contacts using a novel coevolution- based method. Curr. Proteom. 2016, 13, 122–129. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, D.; Wang, Z.; Wen, T.; Lei, D. A boosting approach for prediction of protein-RNA binding residues. BMC Bioinform. 2017, 18, 465. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, J.; Guo, F. Identification of protein–ligand binding sites by sequence information and ensemble classifier. J. Chem. Inf. Modeling 2017, 57, 3149–3161. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the Acm sigkdd International Conference on Knowledge Discovery & Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Zou, Q.; Lin, G.; Jiang, X.; Liu, X.; Zeng, X. Sequence clustering in bioinformatics: An empirical study. Brief. Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlic, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D.; et al. The RCSB Protein Data Bank: Redesigned web site and web services. Nucleic Acids Res. 2011, 39, D392–D401. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Raicar, G.; Tsunoda, T.; Patil, A.; Sharma, A. OPAL: Prediction of MoRF regions in intrinsically disordered protein sequences. Bioinformatics 2018, 34, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Sharma, A.; Patil, A.; Tsunoda, T. Discovering MoRFs by trisecting intrinsically disordered protein sequence into terminals and middle regions. BMC Bioinform. 2019, 19, 378. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Sharma, A.; Raicar, G.; Tsunoda, T.; Patil, A. OPAL+: Length-specific MoRF prediction in intrinsically disordered protein sequences. Proteomics 2018, e1800058. [Google Scholar] [CrossRef]
- Zheng, N.; Wang, K.; Zhan, W.; Deng, L. Targeting virus-host protein interactions: Feature extraction and machine learning approaches. Curr. Drug Metab. 2018. [Google Scholar] [CrossRef]
- Liu, S.; Liu, C.; Deng, L. Machine learning approaches for protein–protein interaction hot spot prediction: Progress and comparative assessment. Molecules 2018, 23, 2535. [Google Scholar] [CrossRef]
- Chakrabarty, B.; Parekh, N. NAPS: Network analysis of protein structures. Nucleic Acids Res 2016, 44, W375–W382. [Google Scholar] [CrossRef]
- Hamelryck, T. An amino acid has two sides: A new 2D measure provides a different view of solvent exposure. Proteins Struct. Funct. Bioinform. 2005, 59, 38–48. [Google Scholar] [CrossRef]
- Song, J.; Tan, H.; Takemoto, K.; Akutsu, T. HSEpred: Predict half-sphere exposure from protein sequences. Bioinformatics 2008, 24, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Šikić, M.; Tomić, S.; Vlahoviček, K. Prediction of protein–protein interaction sites in sequences and 3D structures by Random Forests. PLoS Comput. Biol. 2009, 5, e1000278. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Richards, F.M. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 1971, 55, 379–400. [Google Scholar] [CrossRef]
- Mihel, J.; Šikić, M.; Tomić, S.; Jeren, B.; Vlahoviček, K. PSAIA—Protein structure and interaction analyzer. BMC Struct. Biol. 2008, 8, 21. [Google Scholar] [CrossRef]
- Hubbard, S.J. NACCESS: Program for Calculating Accessibilities; Department of Biochemistry and Molecular Biology, University College of London: London, UK, 1992. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, R.; Paliwal, K.; Lyons, J.; Dehzangi, A.; Sharma, A.; Wang, J.; Sattar, A.; Yang, Y.; Zhou, Y. Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Sci. Rep. 2015, 5, 11476. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Grishin, N.V. Effective scoring function for protein sequence design. Proteins 2010, 54, 271–281. [Google Scholar] [CrossRef]
- Liang, S.; Meroueh, S.O.; Wang, G.; Qiu, C.; Zhou, Y. Consensus scoring for enriching near-native structures from protein-protein docking decoys. Proteins 2009, 75, 397–403. [Google Scholar] [CrossRef]
- Mcdonald, I.K.; Thornton, J.M.J. Satisfying hydrogen bonding potential in proteins. Mol. Biol. 1994, 238, 777–793. [Google Scholar] [CrossRef]
- Northey, T.; Barešic, A.; Martin, A.C. IntPred: A structure-based predictor of protein-protein interaction sites. Bioinformatics 2017, 34, 223–229. [Google Scholar] [CrossRef]
- Cilia, E.; Pancsa, R.; Tompa, P.; Lenaerts, T.; Vranken, W.F. From protein sequence to dynamics and disorder with DynaMine. Nat. Commun. 2013, 4, 2741. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.; Liang, H.K.; Hsiao, N.W.; Ko, M.T.; Lyu, P.C.; Hwang, J.K. Relationship between local structural entropy and protein thermostability. Proteins 2004, 57, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Capra, J.A.; Singh, M. Predicting Functionally Important residues from Sequence Conservation; Oxford University Press: Oxford, UK, 2007; pp. 1875–1882. [Google Scholar]
- Kawashima, S.; Pokarowski, P.; Pokarowska, M.; Kolinski, A.; Katayama, T.; Kanehisa, M. AAindex: Amino acid index database, progress report 2008. Nucleic Acids Res. 2012, 36, D202–D205. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef]
- Petersen, B.; Petersen, T.N.; Andersen, P.; Nielsen, M.; Lundegaard, C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 2009, 9, 51. [Google Scholar] [CrossRef]
- Yang, Y.; Heffernan, R.; Paliwal, K.; Lyons, J.; Dehzangi, A.; Sharma, A.; Wang, J.; Sattar, A.; Zhou, Y. SPIDER2: A Package to Predict Secondary Structure, Accessible Surface Area, and Main-Chain Torsional Angles by Deep Neural Networks; Springer: New York, NY, USA, 2017; p. 55. [Google Scholar]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, 72–76. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef]
- Yu, L.; Sun, X.; Tian, S.W.; Shi, X.Y.; Yan, Y.L. Drug and nondrug classification based on deep learning with various feature selection strategies. Curr. Bioinform. 2018, 13, 253–259. [Google Scholar] [CrossRef]
- Zou, Q.; Wan, S.; Ju, Y.; Tang, J.; Zeng, X. Pretata: Predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 2016, 10, 114. [Google Scholar] [CrossRef]
- Zou, Q.; Zeng, J.; Cao, L.; Ji, R. A novel features ranking metric with application to scalable visual and bioinformatics data classification. Neurocomputing 2016, 173, 346–354. [Google Scholar] [CrossRef]
- Ge, R.; Zhou, M.; Luo, Y.; Meng, Q.; Mai, G.; Ma, D.; Wang, G.; Zhou, F. McTwo: A two-step feature selection algorithm based on maximal information coefficient. BMC Bioinform. 2016, 17, 142. [Google Scholar] [CrossRef]
- Mundra, P.A.; Rajapakse, J.C. SVM-RFE with MRMR filter for gene selection. IEEE Trans. Nanobiosci. 2010, 9, 31–37. [Google Scholar] [CrossRef]
- Kursa, M.B.; Jankowski, A.; Rudnicki, W.R. Boruta—A System for Feature Selection. Fundam. Inform. 2010, 101, 271–285. [Google Scholar]
- Granitto, P.M.; Furlanello, C.; Biasioli, F.; Gasperi, F.J.C.; Systems, I.L. Recursive feature elimination with random forest for PTR-MS analysis of agroindustrial products. Chemom. Intell. Lab. Syst. 2006, 83, 83–90. [Google Scholar] [CrossRef]
- Yaqub, M.; Javaid, M.K.; Cooper, C.; Noble, J.A. Improving the Classification Accuracy of the Classic RF Method by Intelligent Feature Selection and Weighted Voting of Trees with Application to Medical Image Segmentation. In Proceedings of the International Conference on Machine Learning in Medical Imaging, Toronto, ON, Canada, 18 September 2011; pp. 184–192. [Google Scholar]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Babajide Mustapha, I.; Saeed, F. Bioactive molecule prediction using extreme gradient boosting. Molecules 2016, 21, 983. [Google Scholar] [CrossRef]
- Guo, H.; Liu, B.; Cai, D.; Lu, T. Predicting protein–protein interaction sites using modified support vector machine. Int. J. Mach. Learn. Cybern. 2018, 9, 393–398. [Google Scholar] [CrossRef]
- Teplova, M.; Hafner, M.; Teplov, D.; Essig, K.; Tuschl, T.; Patel, D.J. Structure-function studies of STAR family Quaking proteins bound to their in vivo RNA target sites. Genes Dev. 2013, 27, 928–940. [Google Scholar] [CrossRef]
- Fedorov, R.; Meshcheryakov, V.; Gongadze, G.; Fomenkova, N.; Nevskaya, N.; Selmer, M.; Laurberg, M.; Kristensen, O.; Al-Karadaghi, S.; Liljas, A.; et al. Structure of ribosomal protein TL5 complexed with RNA provides new insights into the CTC family of stress proteins. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001, 57, 968–976. [Google Scholar] [CrossRef]
- Gongadze, G.M.; Korepanov, A.P.; Stolboushkina, E.A.; Zelinskaya, N.V.; Korobeinikova, A.V.; Ruzanov, M.V.; Eliseev, B.D.; Nikonov, O.S.; Nikonov, S.V.; Garber, M.B. The crucial role of conserved intermolecular H-bonds inaccessible to the solvent in formation and stabilization of the TL5·5 SrRNA complex. J. Biol. Chem. 2005, 280, 16151–16156. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Training dataset | 1ASY | 1B23 | 1JBS | 1U0B | 1URN | 1YVP | 2BX2 | 2IX1 |
| 2M8D | 2PJP | 2Y8W | 2ZI0 | 2ZKO | 2ZZN | 3EQT | 3K5Q | |
| 3L25 | 3MOJ | 3OL6 | 3VYX | 4ERD | 4MDX | 4NGB | 4NKU | |
| 4OOG | 4PMW | 4QVC | 4YVI | 5AWH | 5DNO | 5IP2 | 5UDZ | |
| Independent testing dataset | 1FEU | 1WNE | 1ZDI | 2KXN | 2XB2 | 3AM1 | 3UZS | 3VYY |
| 4CIO | 4GOA | 4JVH | 4NL3 | 5EN1 | 5EV1 | 5HO4 |
| Method | ACC | SENS | SPEC | PRE | F1 | MCC | AUC |
|---|---|---|---|---|---|---|---|
| Boruta | 0.65 | 0.603 | 0.733 | 0.733 | 0.634 | 0.337 | 0.730 |
| mRMR | 0.667 | 0.661 | 0.663 | 0.726 | 0.662 | 0.347 | 0.760 |
| RFE | 0.692 | 0.671 | 0.702 | 0.725 | 0.678 | 0.366 | 0.768 |
| RF | 0.708 | 0.698 | 0.727 | 0.767 | 0.711 | 0.435 | 0.821 |
| Two-step | 0.733 | 0.732 | 0.770 | 0.797 | 0.743 | 0.505 | 0.889 |
| Rank | Feature Name | Symbol | F-Score |
|---|---|---|---|
| 1 | atom depth | RDa | 0.693 |
| 2 | Closeness | Closeness | 0.679 |
| 3 | Eccentricity | Eccentricity | 0.675 |
| 4 | Enrich conservation | Enrich_conserv | 0.634 |
| 5 | The number of atoms in the lower half sphere | HSEBD | 0.588 |
| 6 | ASA (relative total_side) | ASA_rts | 0.587 |
| Method | ACC | SENS | SPEC | PRE | F1 | MCC | AUC |
|---|---|---|---|---|---|---|---|
| RF | 0.710 | 0.650 | 0.781 | 0.779 | 0.690 | 0.430 | 0.783 |
| SVM | 0.741 | 0.738 | 0.741 | 0.775 | 0.741 | 0.480 | 0.802 |
| GTB | 0.740 | 0.728 | 0.755 | 0.784 | 0.739 | 0.481 | 0.810 |
| XGBoost | 0.744 | 0.740 | 0.755 | 0.785 | 0.744 | 0.494 | 0.822 |
| Method | SENS | SPEC | PRE | F1 | MCC | AUC |
|---|---|---|---|---|---|---|
| XGBPRH | 0.909 | 0.733 | 0.833 | 0.870 | 0.661 | 0.868 |
| XGBPRH-50 | 0.880 | 0.537 | 0.739 | 0.802 | 0.454 | 0.817 |
| PrabHot | 0.793 | 0.655 | 0.697 | 0.742 | 0.453 | 0.804 |
| PrabHot-50 | 0.695 | 0.690 | 0.703 | 0.733 | 0.389 | 0.771 |
| HotSPRing | 0.655 | 0.552 | 0.604 | 0.633 | 0.258 | 0.658 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, L.; Sui, Y.; Zhang, J. XGBPRH: Prediction of Binding Hot Spots at Protein–RNA Interfaces Utilizing Extreme Gradient Boosting. Genes 2019, 10, 242. https://doi.org/10.3390/genes10030242
Deng L, Sui Y, Zhang J. XGBPRH: Prediction of Binding Hot Spots at Protein–RNA Interfaces Utilizing Extreme Gradient Boosting. Genes. 2019; 10(3):242. https://doi.org/10.3390/genes10030242
Chicago/Turabian StyleDeng, Lei, Yuanchao Sui, and Jingpu Zhang. 2019. "XGBPRH: Prediction of Binding Hot Spots at Protein–RNA Interfaces Utilizing Extreme Gradient Boosting" Genes 10, no. 3: 242. https://doi.org/10.3390/genes10030242
APA StyleDeng, L., Sui, Y., & Zhang, J. (2019). XGBPRH: Prediction of Binding Hot Spots at Protein–RNA Interfaces Utilizing Extreme Gradient Boosting. Genes, 10(3), 242. https://doi.org/10.3390/genes10030242