Cell-Selective Regulation of CFTR Gene Expression: Relevance to Gene Editing Therapeutics


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Common Features of the CFTR Locus in All Cell Types
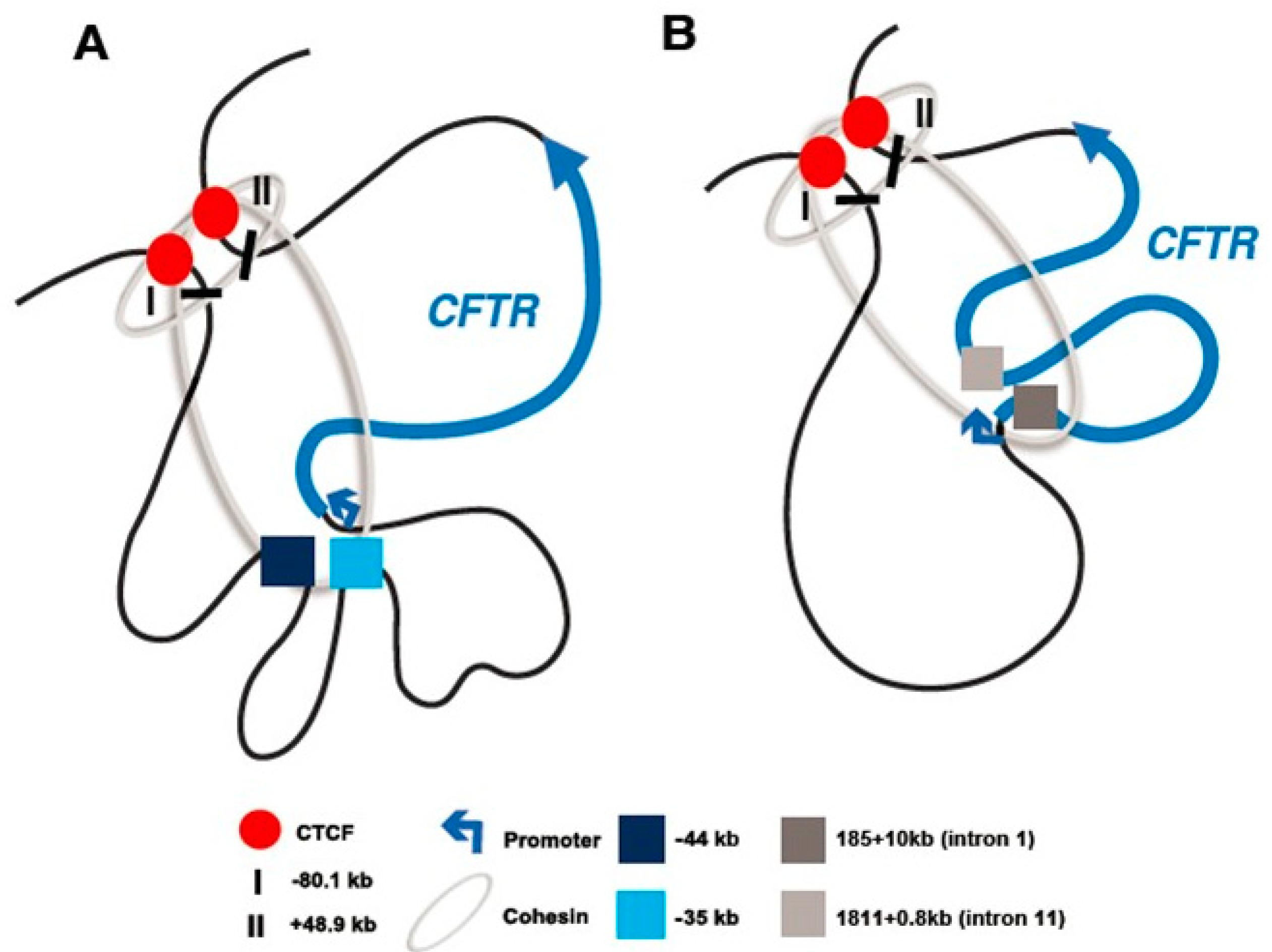
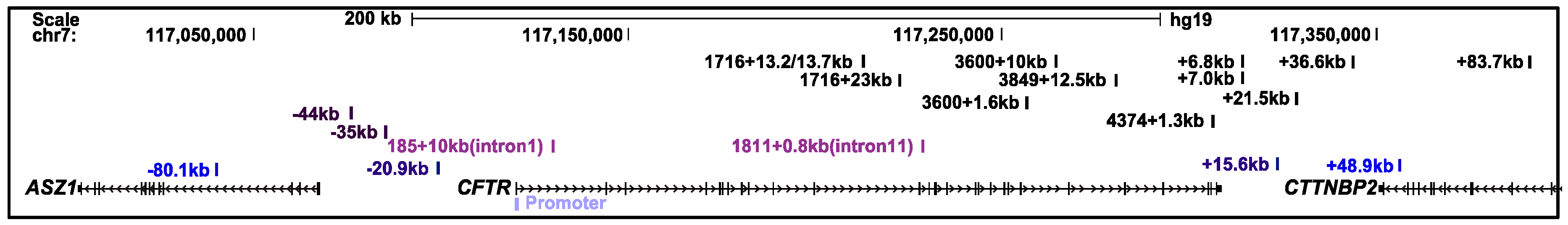
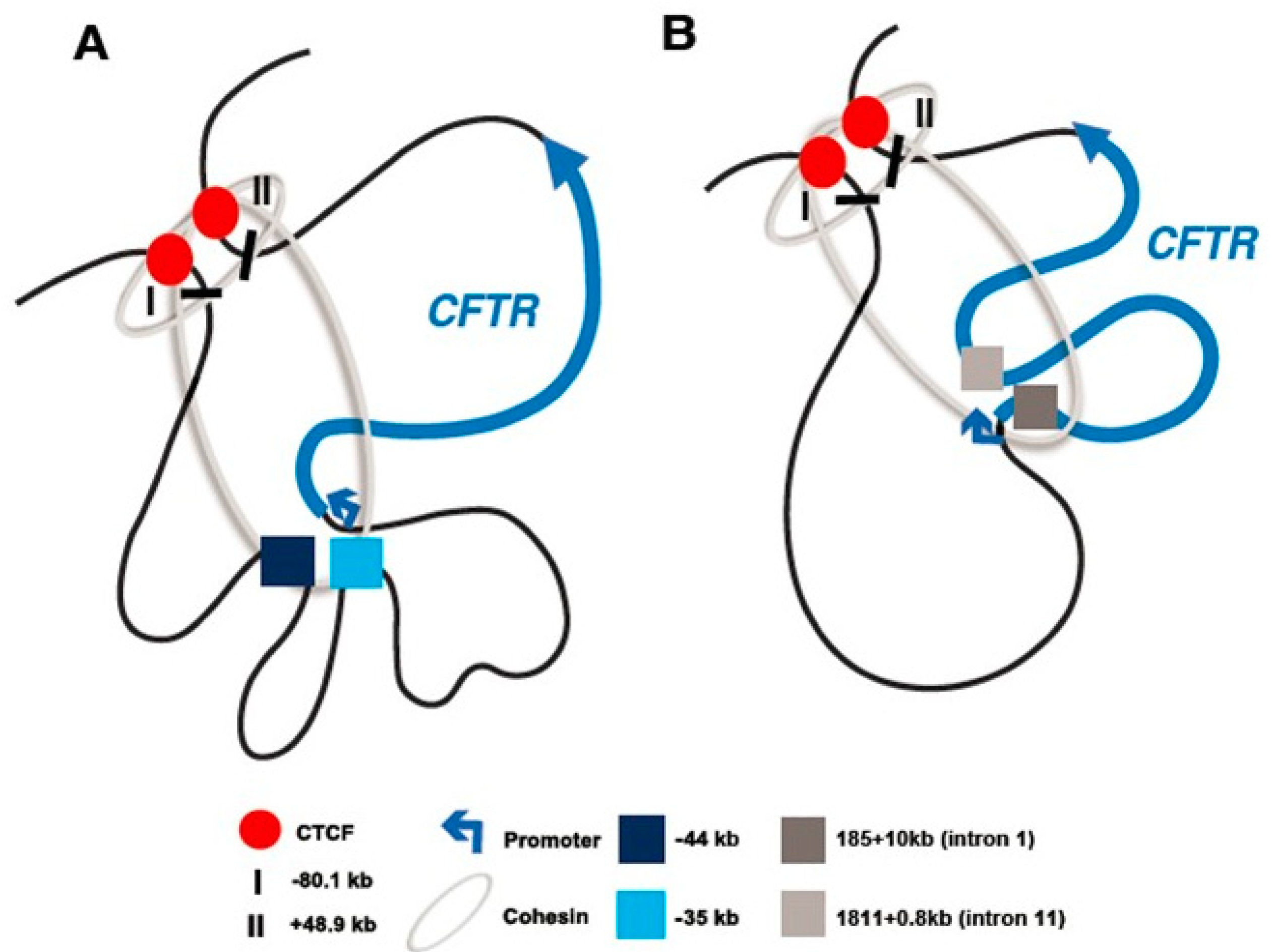
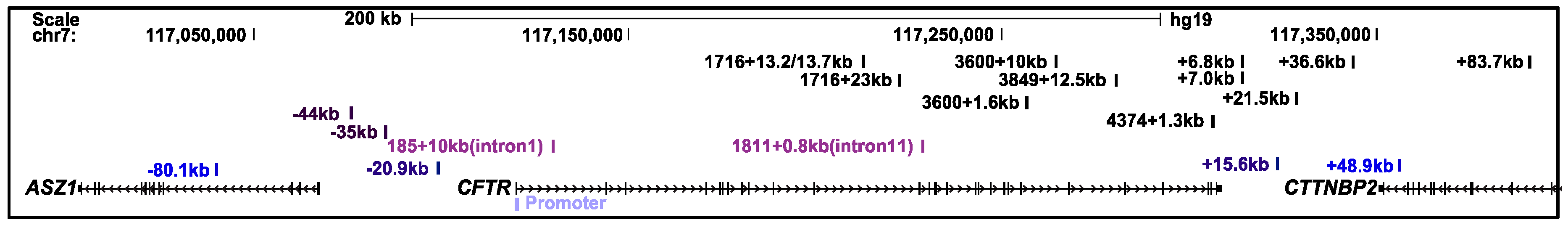
2.1. The CFTR Locus Is Organized Within a Topologically Associating Domain
2.2. The CFTR Locus Contains CTCF-Bound Insulator Elements
3. Cell-Type-Selective CFTR Regulatory Mechanisms
3.1. Airway
3.2. Intestine
3.3. Pancreas and Liver
3.4. Male Reproductive Tract
4. Regulation of CFTR Expression and its Impact on Gene Editing
Author Contributions
Funding
Conflicts of Interest
References
- Nuthall, H.N.; Moulin, D.S.; Huxley, C.; Harris, A. Analysis of DNase-I-hypersensitive sites at the 3′ end of the cystic fibrosis transmembrane conductance regulator gene (CFTR). Biochem. J. 1999, 341 Pt 3, 601–611. [Google Scholar] [CrossRef]
- Rowntree, R.K.; Vassaux, G.; McDowell, T.L.; Howe, S.; McGuigan, A.; Phylactides, M.; Huxley, C.; Harris, A. An element in intron 1 of the CFTR gene augments intestinal expression in vivo. Hum. Mol. Genet. 2001, 10, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.N.; Barth, M.L.; McDowell, T.L.; Moulin, D.S.; Nuthall, H.N.; Hollingsworth, M.A.; Harris, A. A regulatory element in intron 1 of the cystic fibrosis transmembrane conductance regulator gene. J. Biol. Chem. 1996, 271, 9947–9954. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Nuthall, H.N.; Majetti, M.E.; Harris, A. Multiple potential intragenic regulatory elements in the CFTR gene. Genomics 2000, 64, 90–96. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef] [PubMed]
- An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [CrossRef] [PubMed]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N.; et al. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef]
- Chou, J.L.; Rozmahel, R.; Tsui, L.C. Characterization of the promoter region of the cystic fibrosis transmembrane conductance regulator gene. J. Biol. Chem. 1991, 266, 24471–24476. [Google Scholar]
- Koh, J.; Sferra, T.J.; Collins, F.S. Characterization of the cystic fibrosis transmembrane conductance regulator promoter region. Chromatin context and tissue-specificity. J. Biol. Chem. 1993, 268, 15912–15921. [Google Scholar]
- Yoshimura, K.; Nakamura, H.; Trapnell, B.C.; Dalemans, W.; Pavirani, A.; Lecocq, J.P.; Crystal, R.G. The cystic fibrosis gene has a “housekeeping”-type promoter and is expressed at low levels in cells of epithelial origin. J. Biol. Chem. 1991, 266, 9140–9144. [Google Scholar] [PubMed]
- Broackes-Carter, F.C.; Mouchel, N.; Gill, D.; Hyde, S.; Bassett, J.; Harris, A. Temporal regulation of CFTR expression during ovine lung development: Implications for CF gene therapy. Hum. Mol. Genet. 2002, 11, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Crawford, I.; Maloney, P.C.; Zeitlin, P.L.; Guggino, W.B.; Hyde, S.C.; Turley, H.; Gatter, K.C.; Harris, A.; Higgins, C.F. Immunocytochemical localization of the cystic fibrosis gene product CFTR. Proc. Natl. Acad. Sci. USA 1991, 88, 9262–9266. [Google Scholar] [CrossRef] [PubMed]
- Gosalia, N.; Harris, A. Chromatin Dynamics in the Regulation of CFTR Expression. Genes 2015, 6, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Trezise, A.E.; Buchwald, M. In vivo cell-specific expression of the cystic fibrosis transmembrane conductance regulator. Nature 1991, 353, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Denning, G.M.; Ostedgaard, L.S.; Cheng, S.H.; Smith, A.E.; Welsh, M.J. Localization of cystic fibrosis transmembrane conductance regulator in chloride secretory epithelia. J. Clin. Investig. 1992, 89, 339–349. [Google Scholar] [CrossRef]
- Fernald, G.W.; Roberts, M.W.; Boat, T.F. Cystic fibrosis: A current review. Pediatr. Dent. 1990, 12, 72–78. [Google Scholar]
- Harris, A.; Chalkley, G.; Goodman, S.; Coleman, L. Expression of the cystic fibrosis gene in human development. Development 1991, 113, 305–310. [Google Scholar]
- Quinton, P.M. Physiological basis of cystic fibrosis: A historical perspective. Physiol. Rev. 1999, 79, S3–S22. [Google Scholar] [CrossRef]
- Kulka, M.; Gilchrist, M.; Duszyk, M.; Befus, A.D. Expression and functional characterization of CFTR in mast cells. J. Leukoc. Biol. 2002, 71, 54–64. [Google Scholar]
- Yoshimura, K.; Nakamura, H.; Trapnell, B.C.; Chu, C.S.; Dalemans, W.; Pavirani, A.; Lecocq, J.P.; Crystal, R.G. Expression of the cystic fibrosis transmembrane conductance regulator gene in cells of non-epithelial origin. Nucleic Acids Res. 1991, 19, 5417–5423. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Su, M.; McNutt, M.A.; Gu, J. Expression and distribution of cystic fibrosis transmembrane conductance regulator in neurons of the human brain. J. Histochem. Cytochem. 2009, 57, 1113–1120. [Google Scholar] [CrossRef]
- Guo, Y.; Su, M.; Su, M.; McNutt, M.A.; Gu, J. Expression and distribution of cystic fibrosis transmembrane conductance regulator in neurons of the spinal cord. J. Neurosci. Res. 2009, 87, 3611–3619. [Google Scholar] [CrossRef] [PubMed]
- Johannesson, M.; Bogdanovic, N.; Nordqvist, A.C.; Hjelte, L.; Schalling, M. Cystic fibrosis mRNA expression in rat brain: Cerebral cortex and medial preoptic area. Neuroreport 1997, 8, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Mulberg, A.E.; Resta, L.P.; Wiedner, E.B.; Altschuler, S.M.; Jefferson, D.M.; Broussard, D.L. Expression and localization of the cystic fibrosis transmembrane conductance regulator mRNA and its protein in rat brain. J. Clin. Investig. 1995, 96, 646–652. [Google Scholar] [CrossRef]
- Mulberg, A.E.; Weyler, R.T.; Altschuler, S.M.; Hyde, T.M. Cystic fibrosis transmembrane conductance regulator expression in human hypothalamus. Neuroreport 1998, 9, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Mulberg, A.E.; Wiedner, E.B.; Bao, X.; Marshall, J.; Jefferson, D.M.; Altschuler, S.M. Cystic fibrosis transmembrane conductance regulator protein expression in brain. Neuroreport 1994, 5, 1684–1688. [Google Scholar] [CrossRef]
- Niu, N.; Zhang, J.; Guo, Y.; Yang, C.; Gu, J. Cystic fibrosis transmembrane conductance regulator expression in human spinal and sympathetic ganglia. Lab. Investig. 2009, 89, 636–644. [Google Scholar] [CrossRef]
- Pan, J.; Bear, C.; Farragher, S.; Cutz, E.; Yeger, H. Cystic fibrosis transmembrane conductance regulator modulates neurosecretory function in pulmonary neuroendocrine cell-related tumor cell line models. Am. J. Respir. Cell Mol. Biol. 2002, 27, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Reznikov, L.R.; Dong, Q.; Chen, J.H.; Moninger, T.O.; Park, J.M.; Zhang, Y.; Du, J.; Hildebrand, M.S.; Smith, R.J.; Randak, C.O.; et al. CFTR-deficient pigs display peripheral nervous system defects at birth. Proc. Natl. Acad. Sci. USA 2013, 110, 3083–3088. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Guo, Y.; Zhao, Y.; Korteweg, C.; Gu, J. Expression of cystic fibrosis transmembrane conductance regulator in paracervical ganglia. Biochem. Cell Biol. 2010, 88, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Gu, H.; Qiu, Y.; Guo, Y.; Korteweg, C.; Huang, J.; Gu, J. Expression of Cystic Fibrosis Transmembrane Conductance Regulator in Ganglia of Human Gastrointestinal Tract. Sci. Rep. 2016, 6, 30926. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef]
- Splinter, E.; de Wit, E.; van de Werken, H.J.; Klous, P.; de Laat, W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: From fixation to computation. Methods 2012, 58, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Dostie, J.; Richmond, T.A.; Arnaout, R.A.; Selzer, R.R.; Lee, W.L.; Honan, T.A.; Rubio, E.D.; Krumm, A.; Lamb, J.; Nusbaum, C.; et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006, 16, 1299–1309. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Ott, C.J.; Gillen, A.E.; Harris, A. An insulator element 3′ to the CFTR gene binds CTCF and reveals an active chromatin hub in primary cells. Nucleic Acids Res. 2009, 37, 1086–1094. [Google Scholar] [CrossRef]
- Ott, C.J.; Suszko, M.; Blackledge, N.P.; Wright, J.E.; Crawford, G.E.; Harris, A. A complex intronic enhancer regulates expression of the CFTR gene by direct interaction with the promoter. J. Cell Mol. Med. 2009, 13, 680–692. [Google Scholar] [CrossRef]
- Ott, C.J.; Blackledge, N.P.; Kerschner, J.L.; Leir, S.H.; Crawford, G.E.; Cotton, C.U.; Harris, A. Intronic enhancers coordinate epithelial-specific looping of the active CFTR locus. Proc. Natl. Acad. Sci. USA 2009, 106, 19934–19939. [Google Scholar] [CrossRef] [PubMed]
- Gheldof, N.; Smith, E.M.; Tabuchi, T.M.; Koch, C.M.; Dunham, I.; Stamatoyannopoulos, J.A.; Dekker, J. Cell-type-specific long-range looping interactions identify distant regulatory elements of the CFTR gene. Nucleic Acids Res. 2010, 38, 4325–4336. [Google Scholar] [CrossRef]
- Yang, R.; Kerschner, J.L.; Gosalia, N.; Neems, D.; Gorsic, L.K.; Safi, A.; Crawford, G.E.; Kosak, S.T.; Leir, S.H.; Harris, A. Differential contribution of cis-regulatory elements to higher order chromatin structure and expression of the CFTR locus. Nucleic Acids Res. 2016, 44, 3082–3094. [Google Scholar] [CrossRef]
- Smith, E.M.; Lajoie, B.R.; Jain, G.; Dekker, J. Invariant TAD Boundaries Constrain Cell-Type-Specific Looping Interactions between Promoters and Distal Elements around the CFTR Locus. Am. J. Hum. Genet. 2016, 98, 185–201. [Google Scholar] [CrossRef]
- Moisan, S.; Berlivet, S.; Ka, C.; Le Gac, G.; Dostie, J.; Ferec, C. Analysis of long-range interactions in primary human cells identifies cooperative CFTR regulatory elements. Nucleic Acids Res. 2016, 44, 2564–2576. [Google Scholar] [CrossRef] [PubMed]
- Gosalia, N.; Neems, D.; Kerschner, J.L.; Kosak, S.T.; Harris, A. Architectural proteins CTCF and cohesin have distinct roles in modulating the higher order structure and expression of the CFTR locus. Nucleic Acids Res. 2014, 42, 9612–9622. [Google Scholar] [CrossRef]
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Ghirlando, R.; Felsenfeld, G. CTCF: Making the right connections. Genes Dev. 2016, 30, 881–891. [Google Scholar] [CrossRef]
- Ong, C.T.; Corces, V.G. CTCF: An architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014, 15, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Blackledge, N.P.; Carter, E.J.; Evans, J.R.; Lawson, V.; Rowntree, R.K.; Harris, A. CTCF mediates insulator function at the CFTR locus. Biochem. J. 2007, 408, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ott, C.J.; Lewandowska, M.A.; Leir, S.H.; Harris, A. Molecular mechanisms controlling CFTR gene expression in the airway. J. Cell Mol. Med. 2012, 16, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Parelho, V.; Hadjur, S.; Spivakov, M.; Leleu, M.; Sauer, S.; Gregson, H.C.; Jarmuz, A.; Canzonetta, C.; Webster, Z.; Nesterova, T.; et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 2008, 132, 422–433. [Google Scholar] [CrossRef]
- Rubio, E.D.; Reiss, D.J.; Welcsh, P.L.; Disteche, C.M.; Filippova, G.N.; Baliga, N.S.; Aebersold, R.; Ranish, J.A.; Krumm, A. CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. USA 2008, 105, 8309–8314. [Google Scholar] [CrossRef]
- Wendt, K.S.; Yoshida, K.; Itoh, T.; Bando, M.; Koch, B.; Schirghuber, E.; Tsutsumi, S.; Nagae, G.; Ishihara, K.; Mishiro, T.; et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 2008, 451, 796–801. [Google Scholar] [CrossRef]
- Zhang, Z.; Leir, S.H.; Harris, A. Immune mediators regulate CFTR expression through a bifunctional airway-selective enhancer. Mol. Cell Biol. 2013, 33, 2843–2853. [Google Scholar] [CrossRef]
- Zhang, Z.; Leir, S.H.; Harris, A. Oxidative stress regulates CFTR gene expression in human airway epithelial cells through a distal antioxidant response element. Am. J. Respir. Cell Mol. Biol. 2015, 52, 387–396. [Google Scholar] [CrossRef]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar] [CrossRef]
- Hector, A.; Griese, M.; Hartl, D. Oxidative stress in cystic fibrosis lung disease: An early event, but worth targeting? Eur. Respir. J. 2014, 44, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Mutolo, M.J.; Leir, S.H.; Fossum, S.L.; Browne, J.A.; Harris, A. A transcription factor network represses CFTR gene expression in airway epithelial cells. Biochem. J. 2018, 475, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.F.; Yankaskas, J.R.; Ernst, S.A.; Yang, Y.; Marino, C.R.; Boucher, R.C.; Cohn, J.A.; Wilson, J.M. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat. Genet. 1992, 2, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Ameen, N.A.; Ardito, T.; Kashgarian, M.; Marino, C.R. A unique subset of rat and human intestinal villus cells express the cystic fibrosis transmembrane conductance regulator. Gastroenterology 1995, 108, 1016–1023. [Google Scholar] [CrossRef]
- Trezise, A.E.; Chambers, J.A.; Wardle, C.J.; Gould, S.; Harris, A. Expression of the cystic fibrosis gene in human foetal tissues. Hum. Mol. Genet. 1993, 2, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef]
- Plasschaert, L.W.; Zilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Mouchel, N.; Henstra, S.A.; McCarthy, V.A.; Williams, S.H.; Phylactides, M.; Harris, A. HNF1alpha is involved in tissue-specific regulation of CFTR gene expression. Biochem. J. 2004, 378, 909–918. [Google Scholar] [CrossRef]
- Kerschner, J.L.; Harris, A. Transcriptional networks driving enhancer function in the CFTR gene. Biochem. J. 2012, 446, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Kerschner, J.L.; Gosalia, N.; Leir, S.H.; Harris, A. Chromatin remodeling mediated by the FOXA1/A2 transcription factors activates CFTR expression in intestinal epithelial cells. Epigenetics 2014, 9, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, V.A.; Ott, C.J.; Phylactides, M.; Harris, A. Interaction of intestinal and pancreatic transcription factors in the regulation of CFTR gene expression. Biochim. Biophys. Acta 2009, 1789, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.A.; Strong, T.V.; Picciotto, M.R.; Nairn, A.C.; Collins, F.S.; Fitz, J.G. Localization of the cystic fibrosis transmembrane conductance regulator in human bile duct epithelial cells. Gastroenterology 1993, 105, 1857–1864. [Google Scholar] [CrossRef]
- Colombo, C.; Battezzati, P.M.; Strazzabosco, M.; Podda, M. Liver and biliary problems in cystic fibrosis. Semin. Liver Dis. 1998, 18, 227–235. [Google Scholar] [CrossRef]
- Cornwall, G.A. New insights into epididymal biology and function. Hum. Reprod. Updat. 2009, 15, 213–227. [Google Scholar] [CrossRef]
- Bischof, J.M.; Gillen, A.E.; Song, L.; Gosalia, N.; London, D.; Furey, T.S.; Crawford, G.E.; Harris, A. A genome-wide analysis of open chromatin in human epididymis epithelial cells reveals candidate regulatory elements for genes coordinating epididymal function. Biol. Reprod. 2013, 89, 104. [Google Scholar] [CrossRef]
- Leir, S.H.; Browne, J.A.; Eggener, S.E.; Harris, A. Characterization of primary cultures of adult human epididymis epithelial cells. Fertil. Steril. 2015, 103, 647–654.e641. [Google Scholar] [CrossRef] [PubMed]
- Browne, J.A.; Yang, R.; Leir, S.H.; Eggener, S.E.; Harris, A. Expression profiles of human epididymis epithelial cells reveal the functional diversity of caput, corpus and cauda regions. Mol. Hum. Reprod. 2016, 22, 69–82. [Google Scholar] [CrossRef]
- Browne, J.A.; Yang, R.; Eggener, S.E.; Leir, S.H.; Harris, A. HNF1 regulates critical processes in the human epididymis epithelium. Mol. Cell. Endocrinol. 2016, 425, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Browne, J.A.; Eggener, S.E.; Leir, S.H.; Harris, A. A novel transcriptional network for the androgen receptor in human epididymis epithelial cells. Mol. Hum. Reprod. 2018, 24, 433–443. [Google Scholar] [CrossRef]
- Rochwerger, L.; Buchwald, M. Stimulation of the cystic fibrosis transmembrane regulator expression by estrogen in vivo. Endocrinology 1993, 133, 921–930. [Google Scholar] [CrossRef]
- Rochwerger, L.; Dho, S.; Parker, L.; Foskett, J.K.; Buchwald, M. Estrogen-dependent expression of the cystic fibrosis transmembrane regulator gene in a novel uterine epithelial cell line. J. Cell Sci. 1994, 107 Pt 9, 2439–2448. [Google Scholar]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Hess, G.T.; Tycko, J.; Yao, D.; Bassik, M.C. Methods and Applications of CRISPR-Mediated Base Editing in Eukaryotic Genomes. Mol. Cell 2017, 68, 26–43. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Komor, A.C.; Yeh, W.H.; Caetano-Lopes, J.; Warman, M.; Edge, A.S.B.; Liu, D.R. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun. 2017, 8, 15790. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.; Alshareef, S.; Mahfouz, M.M. CRISPR base editors: Genome editing without double-stranded breaks. Biochem. J. 2018, 475, 1955–1964. [Google Scholar] [CrossRef]
- Marx, V. Base editing a CRISPR way. Nat. Methods 2018, 15, 767–770. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.A.; Strug, L.J.; Doshi, V.K.; Commander, C.W.; Blackman, S.M.; Sun, L.; Berthiaume, Y.; Cutler, D.; Cojocaru, A.; Collaco, J.M.; et al. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat. Genet. 2011, 43, 539–546. [Google Scholar] [CrossRef]
- Corvol, H.; Blackman, S.M.; Boelle, P.Y.; Gallins, P.J.; Pace, R.G.; Stonebraker, J.R.; Accurso, F.J.; Clement, A.; Collaco, J.M.; Dang, H.; et al. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat. Commun. 2015, 6, 8382. [Google Scholar] [CrossRef]
- Fossum, S.L.; Mutolo, M.J.; Tugores, A.; Ghosh, S.; Randell, S.H.; Jones, L.C.; Leir, S.H.; Harris, A. Ets homologous factor (EHF) has critical roles in epithelial dysfunction in airway disease. J. Biol. Chem. 2017, 292, 10938–10949. [Google Scholar] [CrossRef]
- Fossum, S.L.; Mutolo, M.J.; Yang, R.; Dang, H.; O’Neal, W.K.; Knowles, M.R.; Leir, S.H.; Harris, A. Ets homologous factor regulates pathways controlling response to injury in airway epithelial cells. Nucleic Acids Res. 2014, 42, 13588–13598. [Google Scholar] [CrossRef]
- Stolzenburg, L.R.; Yang, R.; Kerschner, J.L.; Fossum, S.; Xu, M.; Hoffmann, A.; Lamar, K.M.; Ghosh, S.; Wachtel, S.; Leir, S.H.; et al. Regulatory dynamics of 11p13 suggest a role for EHF in modifying CF lung disease severity. Nucleic Acids Res. 2017, 45, 8773–8784. [Google Scholar] [CrossRef] [PubMed]
- Strug, L.J.; Stephenson, A.L.; Panjwani, N.; Harris, A. Recent advances in developing therapeutics for cystic fibrosis. Hum. Mol. Genet. 2018, 27, R173–R186. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swahn, H.; Harris, A. Cell-Selective Regulation of CFTR Gene Expression: Relevance to Gene Editing Therapeutics. Genes 2019, 10, 235. https://doi.org/10.3390/genes10030235
Swahn H, Harris A. Cell-Selective Regulation of CFTR Gene Expression: Relevance to Gene Editing Therapeutics. Genes. 2019; 10(3):235. https://doi.org/10.3390/genes10030235
Chicago/Turabian StyleSwahn, Hannah, and Ann Harris. 2019. "Cell-Selective Regulation of CFTR Gene Expression: Relevance to Gene Editing Therapeutics" Genes 10, no. 3: 235. https://doi.org/10.3390/genes10030235
APA StyleSwahn, H., & Harris, A. (2019). Cell-Selective Regulation of CFTR Gene Expression: Relevance to Gene Editing Therapeutics. Genes, 10(3), 235. https://doi.org/10.3390/genes10030235