Novel Insights into Plant Genome Evolution and Adaptation as Revealed through Transposable Elements and Non-Coding RNAs in Conifers


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Transposable Elements: A Source of Genetic Innovation
3. Transposable Elements–Epigenetics Components Interplay: An Evolutionary Force in Adaptation
4. Genome-Purging Mechanism: Another Evolutionary Force to Counterbalance Transposable Element Increase
5. Transposable Elements in Conifer Genome Architecture
6. Non-Coding RNA Features in Conifers
7. Outstanding Questions
- To what extent do TEs contribute to conifer genetic variation and population divergence? Based on TE polymorphisms, what are TE dynamics in natural populations and how are these loci under selection?
- ⇨
- Given the availability of reference genome and TE consensus sequences, computational approaches (e.g., [154]) provide a possibility of addressing this question.
- How does the landscape of ncRNAs and DNA methylation alter over time (e.g., reproduction vs. vegetative growth) in the region of TEs (or different types of TEs), genes, and introns of focus conifers?
- ⇨
- We could additionally focus on polymorphic TE loci identified in the previous question and genes involved in known pathways to explore how epigenetic components work in synergy in those “hotspots”.
- What are the features of TEs–epigenetics components (ncRNA and DNA methylation) interactions when individuals are exposed to stressful vs. benign environmental conditions? How are these features associated with local adaptation?
- ⇨
- By identifying polymorphic TEs among populations (e.g., [155]), we are able to infer whether different types of TEs have undergone differential expansion or contraction, hence playing a role in adaptive evolution. Then, contrasting these results with their DNA methylation landscape permits testing whether and how the two mechanisms jointly contribute to adaptation.
- Is there evidence supporting genome-purging mechanisms in conifers?
8. Closing Remarks
9. Terminology
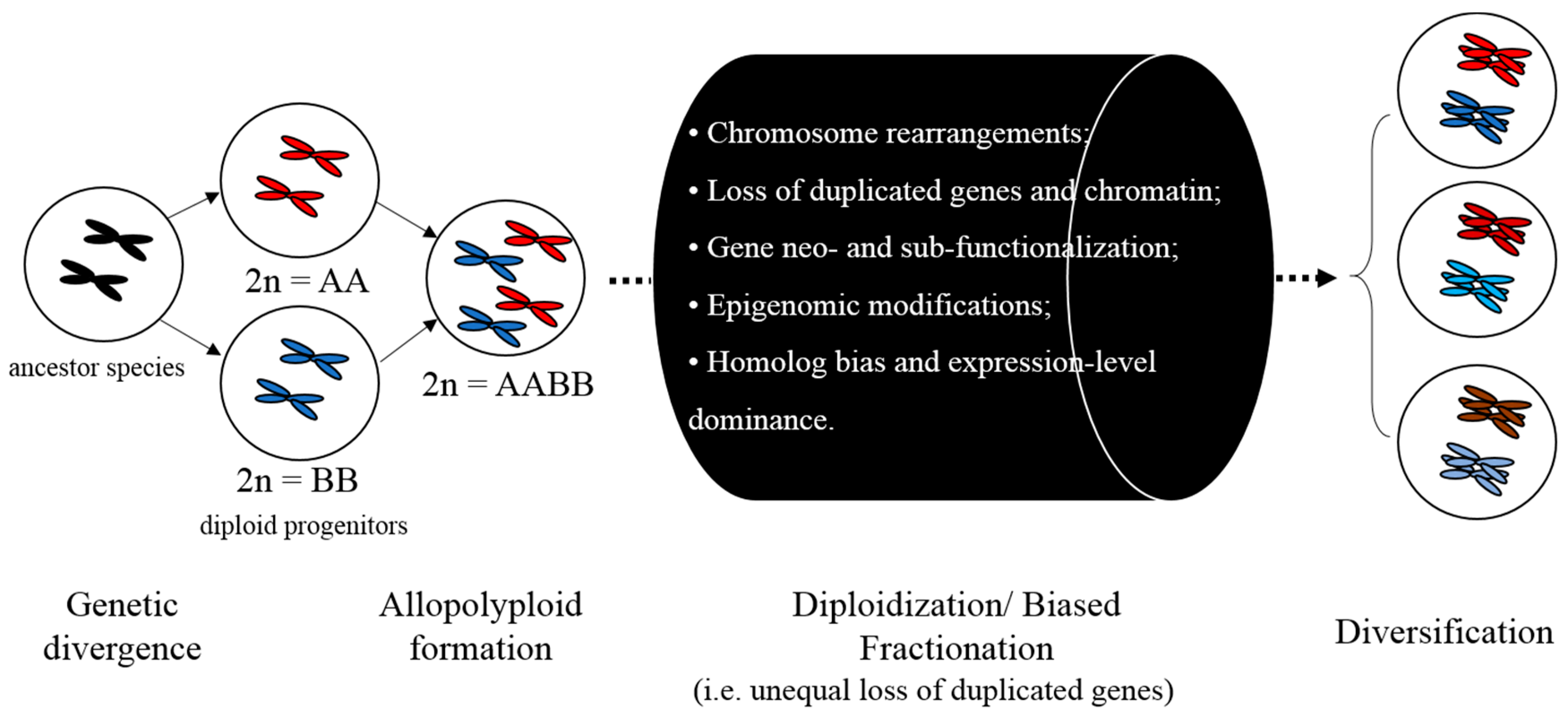
- Biased fractionation: the unequal loss of genes from ancestral progenitor genomes, which is a frequent event after polyploidy in many lineages.
- Class II elements of TEs: DNA transposons; the DNA genome of the element itself serves as the template for transposition either by a “cut and paste” mechanism or using a rolling circle process.
- Long terminal repeats (LTRs): identical DNA sequence that can be repeated at the ends of retrotransposons.
- Non-coding RNAs (ncRNAs): randomly grouped into short (<200 nt) and long (>200 nt) types [160]. Members of short ncRNAs involved in plant transcriptional (indirect and low) and post-transcriptional (major) regulations have been well documented (e.g., review in [52]). These short ncRNAs chiefly consist of microRNAs (prevalence of 21 or 22 nt long in suppressing target mRNAs), heterochromatic small interfering RNAs (hc-siRNAs; 24 nt mediators in silencing DNA methylation and histone modifications), and trans-acting siRNAs (tasiRNAs or phasiRNAs; 22 (or 21) nt with a phased configuration, playing similar roles as microRNAs or other uncharacterized functions).
- Whole-genome duplication (WGD) or polyploidization: an event in which the entire genome of an organism is copied once or multiple times. A widely accepted hypothesis for WGD events is based on a hexaploidization of all eudicots (ancestral γ events), first put forth by Jaillon et al. [161].
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paton, A.J.; Brummitt, N.; Govaerts, R.; Harman, K.; Hinchcliffe, S.; Allkin, B.; Lughadha, E.N. Towards target 1 of the global strategy for plant conservation: A working list of all known plant species — progress and prospects. Taxon 2008, 57, 602–611. [Google Scholar] [CrossRef]
- Willis, K.J.; McElwain, J.C. The Evolution of Plants; Oxford University Press: New York, NY, USA, 2002. [Google Scholar]
- Hilton, J.; Bateman, R.M. Pteridosperms are the backbone of seed-plant phylogeny. J. Torrey Bot. Soc. 2006, 133, 119–168. [Google Scholar] [CrossRef]
- Bateman, R.M.; Hilton, J.; Rudall, P.J. Morphological and molecular phylogenetic context of the angiosperms: Contrasting the ’top-down’ and ’bottom-up’ approaches used to infer the likely characteristics of the first flowers. J. Exp. Bot. 2006, 57, 3471–3503. [Google Scholar] [CrossRef]
- Friis, E.M.; Pedersen, K.R.; Crane, P.R. Diversity in obscurity: Fossil flowers and the early history of angiosperms. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 369–382. [Google Scholar] [CrossRef]
- Friedman, W.E. The meaning of Darwin’s “abominable mystery”. Am. J. Bot. 2009, 96, 5–21. [Google Scholar] [CrossRef]
- Farjón, A. A Handbook of the World Conifers; Brill Academic Publishers: Leiden, The Netherlands, 2010. [Google Scholar]
- Eckenwalder, J.E. Conifers of the World: The Complete Reference; Timber Press: London, UK, 2009. [Google Scholar]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef]
- Birol, I.; Raymond, A.; Jackman, S.D.; Pleasance, S.; Coope, R.; Taylor, G.A.; Yuen, M.M.; Keeling, C.I.; Brand, D.; Vandervalk, B.P.; et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics 2013, 29, 1492–1497. [Google Scholar] [CrossRef]
- Warren, R.L.; Keeling, C.I.; Yuen, M.M.; Raymond, A.; Taylor, G.A.; Vandervalk, B.P.; Mohamadi, H.; Paulino, D.; Chiu, R.; Jackman, S.D.; et al. Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. 2015, 83, 189–212. [Google Scholar] [CrossRef]
- Neale, D.B.; Wegrzyn, J.L.; Stevens, K.A.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; Holtz-Morris, A.E.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15, R59. [Google Scholar] [CrossRef]
- Wegrzyn, J.L.; Liechty, J.D.; Stevens, K.A.; Wu, L.S.; Loopstra, C.A.; Vasquez-Gross, H.A.; Dougherty, W.M.; Lin, B.Y.; Zieve, J.J.; Martinez-Garcia, P.J.; et al. Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics 2014, 196, 891–909. [Google Scholar] [CrossRef]
- Zimin, A.; Stevens, K.A.; Crepeau, M.W.; Holtz-Morris, A.; Koriabine, M.; Marcais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.L.; de Jong, P.J.; et al. Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef]
- Stevens, K.A.; Wegrzyn, J.L.; Zimin, A.; Puiu, D.; Crepeau, M.; Cardeno, C.; Paul, R.; Gonzalez-Ibeas, D.; Koriabine, M.; Holtz-Morris, A.E.; et al. Sequence of the sugar pine megagenome. Genetics 2016, 204, 1613–1626. [Google Scholar] [CrossRef]
- Liu, Y.; El-Kassaby, Y.A. Landscape of fluid sets of hairpin-derived 21-/24-nt-long small RNAs at seed set uncovers special epigenetic features in Picea glauca. Genome Biol. Evol. 2017, 9, 82–92. [Google Scholar] [CrossRef]
- Xia, R.; Xu, J.; Arikit, S.; Meyers, B.C. Extensive families of miRNAs and PHAS loci in Norway spruce demonstrate the origins of complex phasiRNA networks in seed plants. Mol. Biol. Evol. 2015, 32, 2905–2918. [Google Scholar] [CrossRef]
- Casacuberta, E.; González, J. The impact of transposable elements in environmental adaptation. Mol. Ecol. 2013, 22, 1503–1517. [Google Scholar] [CrossRef]
- Bukhari, A.I.; Shapiro, J.A.; Adhya, S.L.; Cold Spring Harbor Laboratory. DNA Insertion Elements, Plasmids, and Episomes; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1977; p. 782. [Google Scholar]
- Lynch, M. The Origins of Genome Architecture; Sinauer Associates: Sunderland, MA, USA, 2007. [Google Scholar]
- Vicient, C.M.; Casacuberta, J.M. Impact of transposable elements on polyploid plant genomes. Ann. Bot. 2017, 120, 195–207. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef]
- Zhao, D.Y.; Ferguson, A.A.; Jiang, N. What makes up plant genomes: The vanishing line between transposable elements and genes. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2016, 1859, 366–380. [Google Scholar] [CrossRef]
- Chénais, B.; Caruso, A.; Hiard, S.; Casse, N. The impact of transposable elements on eukaryotic genomes: From genome size increase to genetic adaptation to stressful environments. Gene 2012, 509, 7–15. [Google Scholar] [CrossRef]
- Bhattacharyya, M.K.; Smith, A.M.; Ellis, T.H.N.; Hedley, C.; Martin, C. The wrinkled-seed character of pea described by Mendel is caused by a transposon-like insertion in a gene encoding starch-branching enzyme. Cell 1990, 60, 115–122. [Google Scholar] [CrossRef]
- Liu, B.; Kanazawa, A.; Matsumura, H.; Takahashi, R.; Harada, K.; Abe, J. Genetic redundancy in soybean photoresponses associated with duplication of the phytochrome A gene. Genetics 2008, 180, 995–1007. [Google Scholar] [CrossRef]
- Kanazawa, A.; Liu, B.H.; Kong, F.J.; Arase, S.; Abe, J. Adaptive evolution involving gene duplication and insertion of a novel Ty1/copia-like retrotransposon in soybean. J. Mol. Evol. 2009, 69, 164–175. [Google Scholar] [CrossRef]
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Doebley, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 2011, 43, 1160–1163. [Google Scholar] [CrossRef]
- Lisch, D.R. Transposons in plant gene regulation. In Plant Transposons and Genome Dynamics in Evolution; Fedoroff, N.V., Ed.; Willey-Blackwell: Ames, IA, USA, 2013; pp. 93–116. [Google Scholar]
- Springer, N.M.; Lisch, D.; Li, Q. Creating order from chaos: Epigenome dynamics in plants with complex genomes. Plant Cell 2016, 28, 314–325. [Google Scholar] [CrossRef]
- Diez, C.M.; Roessler, K.; Gaut, B.S. Epigenetics and plant genome evolution. Curr. Opin. Plant Biol. 2014, 18, 1–8. [Google Scholar] [CrossRef]
- Mita, P.; Boeke, J.D. How retrotransposons shape genome regulation. Curr. Opin. Genet. Dev. 2016, 37, 90–100. [Google Scholar] [CrossRef]
- McClintock, B. The Discovery and Characterization of Transposable Elements: The Collected Papers of Barbara Mcclintock; Garland: New York, NY, USA, 1987. [Google Scholar]
- Bennetzen, J.L. Transposable elements, gene creation and genome rearrangement in flowering plants. Curr. Opin. Genet. Dev. 2005, 15, 621–627. [Google Scholar] [CrossRef]
- Parisod, C.; Alix, K.; Just, J.; Petit, M.; Sarilar, V.; Mhiri, C.; Ainouche, M.; Chalhoub, B.; Grandbastien, M.A. Impact of transposable elements on the organization and function of allopolyploid genomes. New Phytol. 2010, 186, 37–45. [Google Scholar] [CrossRef]
- Houle, D.; Nuzhdin, S.V. Mutation accumulation and the effect of copia insertions in Drosophila melanogaster. Genet. Res. 2004, 83, 7–18. [Google Scholar] [CrossRef]
- Papaceit, M.; Avila, V.; Aguadé, M.; García-Dorado, A. The dynamics of the roo transposable element in mutation-accumulation lines and segregating populations of Drosophila melanogaster. Genetics 2007, 177, 511–522. [Google Scholar] [CrossRef]
- Vinogradov, A.E. Genome size and extinction risk in vertebrates. Proc. R. Soc. Lond. Ser. B Biol. Sci. 2004, 271, 1701–1705. [Google Scholar] [CrossRef]
- Vinogradov, A.E. Evolution of genome size: Multilevel selection, mutation bias or dynamical chaos? Curr. Opin. Genet. Dev. 2004, 14, 620–626. [Google Scholar] [CrossRef]
- Vinogradov, A.E. Selfish DNA is maladaptive: Evidence from the plant Red List. Trends Genet. 2003, 19, 609–614. [Google Scholar] [CrossRef]
- Brookfield, J.F. The ecology of the genome - Mobile DNA elements and their hosts. Nat. Rev. Genet. 2005, 6, 128–136. [Google Scholar] [CrossRef]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.S. The genomes and transposable elements in plants: Are they friends or foes? Genes Genom. 2017, 39, 359–370. [Google Scholar] [CrossRef]
- Schnable, J.C.; Springer, N.M.; Freeling, M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. USA 2011, 108, 4069–4074. [Google Scholar] [CrossRef]
- Steige, K.A.; Slotte, T. Genomic legacies of the progenitors and the evolutionary consequences of allopolyploidy. Curr. Opin. Plant Biol. 2016, 30, 88–93. [Google Scholar] [CrossRef]
- Woodhouse, M.R.; Cheng, F.; Pires, J.C.; Lisch, D.; Freeling, M.; Wang, X.W. Origin, inheritance, and gene regulatory consequences of genome dominance in polyploids. Proc. Natl. Acad. Sci. USA 2014, 111, 5283–5288. [Google Scholar] [CrossRef]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef]
- Hollister, J.D.; Smith, L.M.; Guo, Y.L.; Ott, F.; Weigel, D.; Gaut, B.S. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2011, 108, 2322–2327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Meyers, B.C.; Cai, C.; Xu, W.; Ma, J. Evolutionary patterns and coevolutionary consequences of MIRNA genes and microRNA targets triggered by multiple mechanisms of genomic duplications in soybean. Plant Cell 2015, 27, 546–562. [Google Scholar] [CrossRef] [PubMed]
- Piriyapongsa, J.; Jordan, I.K. Dual coding of siRNAs and miRNAs by plant transposable elements. RNA 2008, 14, 814–821. [Google Scholar] [CrossRef]
- Sun, J.; Zhou, M.; Mao, Z.T.; Li, C.X. Characterization and evolution of microRNA genes derived from repetitive elements and duplication events in plants. PLoS ONE 2012, 7, e34092. [Google Scholar] [CrossRef]
- Wendel, J.F.; Jackson, S.A.; Meyers, B.C.; Wing, R.A. Evolution of plant genome architecture. Genome Biol. 2016, 17, 37. [Google Scholar] [CrossRef]
- Galhardo, R.S.; Hastings, P.J.; Rosenberg, S.M. Mutation as a stress response and the regulation of evolvability. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 399–435. [Google Scholar] [CrossRef]
- Zeh, D.W.; Zeh, J.A.; Ishida, Y. Transposable elements and an epigenetic basis for punctuated equilibria. Bioessays 2009, 31, 715–726. [Google Scholar] [CrossRef]
- Rey, O.; Danchin, E.; Mirouze, M.; Loot, C.; Blanchet, S. Adaptation to global change: A transposable element–epigenetics perspective. Trends Ecol. Evol. 2016, 31, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Schrader, L.; Kim, J.W.; Ence, D.; Zimin, A.; Klein, A.; Wyschetzki, K.; Weichselgartner, T.; Kemena, C.; Stokl, J.; Schultner, E.; et al. Transposable element islands facilitate adaptation to novel environments in an invasive species. Nature Commun. 2014, 5, 5495. [Google Scholar] [CrossRef]
- Stapley, J.; Santure, A.W.; Dennis, S.R. Transposable elements as agents of rapid adaptation may explain the genetic paradox of invasive species. Mol. Ecol. 2015, 24, 2241–2252. [Google Scholar] [CrossRef] [PubMed]
- Schrader, L.; Schmitz, J. The impact of transposable elements in adaptive evolution. Mol. Ecol. 2018, 0, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Lisch, D. Epigenetic regulation of transposable elements in plants. Annu. Rev. Plant Biol. 2009, 60. [Google Scholar] [CrossRef] [PubMed]
- Cubas, P.; Vincent, C.; Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 1999, 401, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P.; et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009, 5, e1000530. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.L.; Schrey, A.W.; Pigliucci, M. Invasion of diverse habitats by few Japanese knotweed genotypes is correlated with epigenetic differentiation. Ecol. Lett. 2012, 15, 1016–1025. [Google Scholar] [CrossRef]
- Reinders, J.; Wulff, B.B.H.; Mirouze, M.; Marí-Ordóñez, A.; Dapp, M.; Rozhon, W.; Bucher, E.; Theiler, G.; Paszkowski, J. Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev. 2009, 23, 939–950. [Google Scholar] [CrossRef]
- Cortijo, S.; Wardenaar, R.; Colome-Tatché, M.; Gilly, A.; Etcheverry, M.; Labadie, K.; Caillieux, E.; Hospital, F.; Aury, J.M.; Wincker, P.; et al. Mapping the epigenetic basis of complex traits. Science 2014, 343, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Fedoroff, N.V. Transposable elements, epigenetics, and genome evolution. Science 2012, 338, 758–767. [Google Scholar] [CrossRef]
- Fablet, M.; Vieira, C. Evolvability, epigenetics and transposable elements. Biomol. Concepts 2011, 2, 333–341. [Google Scholar] [CrossRef]
- Oliver, K.R.; Greene, W.K. Mobile DNA and the TE-Thrust hypothesis: Supporting evidence from the primates. Mobil. DNA 2011, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Mirouze, M.; Paszkowski, J. Epigenetic contribution to stress adaptation in plants. Curr. Opin. Plant Biol. 2011, 14, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Oliver, K.R.; Greene, W.K. Transposable elements: Powerful facilitators of evolution. Bioessays 2009, 31, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The significance of responses of the genome to challenge. Science 1984, 226, 792–801. [Google Scholar] [CrossRef]
- Ito, H.; Kim, J.M.; Matsunaga, W.; Saze, H.; Matsui, A.; Endo, T.A.; Harukawa, Y.; Takagi, H.; Yaegashi, H.; Masuta, Y.; et al. A stress-activated transposon in Arabidopsis induces transgenerational abscisic acid insensitivity. Sci. Rep. 2016, 6, srep23181. [Google Scholar] [CrossRef]
- Grandbastien, M.A. LTR retrotransposons, handy hitchhikers of plant regulation and stress response. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2015, 1849, 403–416. [Google Scholar] [CrossRef]
- Kawakami, T.; Strakosh, S.C.; Zhen, Y.; Ungerer, M.C. Different scales of Ty1/copia-like retrotransposon proliferation in the genomes of three diploid hybrid sunflower species. Heredity 2010, 104, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Senerchia, N.; Felber, F.; Parisod, C. Genome reorganization in F1 hybrids uncovers the role of retrotransposons in reproductive isolation. P. Roy. Soc. B Biol. Sci. 2015, 282, 20142874. [Google Scholar] [CrossRef]
- Miousse, I.R.; Chalbot, M.C.G.; Lumen, A.; Ferguson, A.; Kavouras, I.G.; Koturbash, I. Response of transposable elements to environmental stressors. Mutat. Res. Rev. Mut. Res. 2015, 765, 19–39. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.J.; Yang, X.J.; Wang, H.Y.; Shi, F.X.; Liu, Y.; Liu, J.S.; Li, L.F.; Wang, D.L.; Liu, B. Cytosine methylation alteration in natural populations of Leymus chinensis induced by multiple abiotic stresses. PLoS ONE 2013, 8, e55772. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Zhang, Z.; Wu, H.; Huang, C.; Shuai, P.; Ye, C.Y.; Tang, S.; Wang, Y.; Yang, L.; Wang, J.; et al. Single-base-resolution methylomes of Populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 2014, 15 Suppl 1, S9. [Google Scholar] [CrossRef]
- Wang, D.; Qu, Z.; Yang, L.; Zhang, Q.; Liu, Z.H.; Do, T.; Adelson, D.L.; Wang, Z.Y.; Searle, I.; Zhu, J.K. Transposable elements (TEs) contribute to stress-related long intergenic noncoding RNAs in plants. Plant J. 2017, 90, 133–146. [Google Scholar] [CrossRef]
- Naito, K.; Zhang, F.; Tsukiyama, T.; Saito, H.; Hancock, C.N.; Richardson, A.O.; Okumoto, Y.; Tanisaka, T.; Wessler, S.R. Unexpected consequences of a sudden and massive transposon amplification on rice gene expression. Nature 2009, 461, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Makarevitch, I.; Waters, A.J.; West, P.T.; Stitzer, M.; Hirsch, C.N.; Ross-Ibarra, J.; Springer, N.M. Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genet. 2015, 11. [Google Scholar] [CrossRef]
- Woodrow, P.; Pontecorvo, G.; Ciarmiello, L.F.; Fuggi, A.; Carillo, P. Ttd1a promoter is involved in DNA-protein binding by salt and light stresses. Mol. Biol. Rep. 2011, 38, 3787–3794. [Google Scholar] [CrossRef] [PubMed]
- Suoniemi, A.; Anamthawat-Jónsson, K.; Arna, T.; Schulman, A.H. Retrotransposon BARE-1 is a major, dispersed component of the barley (Hordeum vulgare L) genome. Plant Mol. Biol. 1996, 30, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Beguiristain, T.; Grandbastien, M.A.; Puigdomènech, P.; Casacuberta, J.M. Three Tnt1 subfamilies show different stress-associated patterns of expression in tobacco. Consequences for retrotransposon control and evolution in plants. Plant Physiol. 2001, 127, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Sugimoto, K.; Otsuki, H.; Hirochika, H. A 13-bp cis-regulatory element in the LTR promoter of the tobacco retrotransposon Tto1 is involved in responsiveness to tissue culture, wounding, methyl jasmonate and fungal elicitors. Plant J. 1999, 18, 383–393. [Google Scholar] [CrossRef]
- Egue, F.; Chénais, B.; Tastard, E.; Marchand, J.; Hiard, S.; Gateau, H.; Hermann, D.; Morant-Manceau, A.; Casse, N.; Caruso, A. Expression of the retrotransposons Surcouf and Blackbeard in the marine diatom Phaeodactylum tricornutum under thermal stress. Phycologia 2015, 54, 617–627. [Google Scholar] [CrossRef]
- Horváth, V.; Merenciano, M.; González, J. Revisiting the relationship between transposable elements and the eukaryotic stress response. Trends Genet. 2017, 33, 832–841. [Google Scholar] [CrossRef]
- Mao, H.D.; Wang, H.W.; Liu, S.X.; Li, Z.; Yang, X.H.; Yan, J.B.; Li, J.S.; Tran, L.S.P.; Qin, F. A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nature Commun. 2015, 6, 8326. [Google Scholar] [CrossRef]
- Hunter, R.G.; Murakami, G.; Dewell, S.; Seligsohn, M.a.; Baker, M.E.R.; Datson, N.A.; McEwen, B.S.; Pfaff, D.W. Acute stress and hippocampal histone H3 lysine 9 trimethylation, a retrotransposon silencing response. Proc. Natl. Acad. Sci. USA 2012, 109, 17657–17662. [Google Scholar] [CrossRef]
- Secco, D.; Wang, C.; Shou, H.X.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.R.; Whelan, J.; Lister, R. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. eLife 2015, 4, e09343. [Google Scholar] [CrossRef]
- Rutherford, S.L.; Henikoff, S. Quantitative epigenetics. Nat. Genet. 2003, 33, 6–8. [Google Scholar] [CrossRef]
- True, H.L.; Berlin, I.; Lindquist, S.L. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature 2004, 431, 184–187. [Google Scholar] [CrossRef]
- Folse, H.J.; Roughgarden, J. Direct benefits of genetic mosaicism and intraorganismal selection: Modeling coevolution between a long-lived tree and a short-lived herbivore. Evolution 2012, 66, 1091–1113. [Google Scholar] [CrossRef]
- Baulcombe, D.C.; Dean, C. Epigenetic regulation in plant responses to the environment. Cold Spring Harb. Perspect. Biol. 2014, 6, a019471. [Google Scholar] [CrossRef]
- Fultz, D.; Choudury, S.G.; Slotkin, R.K. Silencing of active transposable elements in plants. Curr. Opin. Plant Biol. 2015, 27, 67–76. [Google Scholar] [CrossRef]
- Friedli, M.; Trono, D. The developmental control of transposable elements and the evolution of higher species. Annu. Rev. Cell. Dev. Biol. 2015, 31, 429–451. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- McCue, A.D.; Nuthikattu, S.; Reeder, S.H.; Slotkin, R.K. Gene expression and stress response mediated by the epigenetic regulation of a transposable element small RNA. PLoS Genet. 2012, 8, e1002474. [Google Scholar] [CrossRef] [PubMed]
- Buchon, N.; Vaury, C. RNAi: A defensive RNA-silencing against viruses and transposable elements. Heredity 2006, 96, 195–202. [Google Scholar] [CrossRef]
- Hu, T.T.; Pattyn, P.; Bakker, E.G.; Cao, J.; Cheng, J.F.; Clark, R.M.; Fahlgren, N.; Fawcett, J.A.; Grimwood, J.; Gundlach, H.; et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 2011, 43, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Laclette, E.; Lyons, E.; Hernández-Guzmán, G.; Pérez-Torres, C.A.; Carretero-Paulet, L.; Chang, T.-H.; Lan, T.; Welch, A.J.; Juárez, M.J.A.; Simpson, J.; et al. Architecture and evolution of a minute plant genome. Nature 2013, 498, 94. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, P.; Goedecke, W.; Obe, G. Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis 2000, 15, 289–302. [Google Scholar] [CrossRef]
- Schuermann, D.; Molinier, J.; Fritsch, O.; Hohn, B. The dual nature of homologous recombination in plants. Trends Genet. 2005, 21, 172–181. [Google Scholar] [CrossRef]
- Argueso, J.L.; Westmoreland, J.; Mieczkowski, P.A.; Gawel, M.; Petes, T.D.; Resnick, M.A. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc. Natl. Acad. Sci. USA 2008, 105, 11845–11850. [Google Scholar] [CrossRef]
- Bilinski, P.; Albert, P.S.; Berg, J.J.; Birchler, J.A.; Grote, M.N.; Lorant, A.; Quezada, J.; Swarts, K.; Yang, J.; Ross-Ibarra, J. Parallel altitudinal clines reveal trends in adaptive evolution of genome size in Zea mays. PLoS Genet. 2018, 14, e1007162. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Díez, C.M.; Gaut, B.S.; Meca, E.; Scheinvar, E.; Montes-Hernandez, S.; Eguiarte, L.E.; Tenaillon, M.I. Genome size variation in wild and cultivated maize along altitudinal gradients. New Phytol. 2013, 199, 264–276. [Google Scholar] [CrossRef]
- Rayburn, A.L.; Dudley, J.W.; Biradar, D.P. Selection for early flowering results in simultaneous selection for reduced nuclear DNA content in maize. Plant Breeding 1994, 112, 318–322. [Google Scholar] [CrossRef]
- Higgins, D.M.; Lowry, E.G.; Kanizay, L.B.; Becraft, P.W.; Hall, D.W.; Dawe, R.K. Fitness costs and variation in transmission distortion associated with the abnormal chromosome 10 meiotic drive system in maize. Genetics 2018, 208, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Kupiec, M.; Petes, T.D. Allelic and ectopic recombination between Ty elements in yeast. Genetics 1988, 119, 549–559. [Google Scholar] [PubMed]
- Han, K.; Lee, J.; Meyer, T.J.; Remedios, P.; Goodwin, L.; Batzer, M.A. L1 recombination-associated deletions generate human genomic variation. Proc. Natl. Acad. Sci. USA 2008, 105, 19366–19371. [Google Scholar] [CrossRef]
- Michael, T.P. Plant genome size variation: Bloating and purging DNA. Brief. Funct. Genom. 2014, 13, 308–317. [Google Scholar] [CrossRef]
- Grover, C.E.; Wendel, J.F. Recent insights into mechanisms of genome size change in plants. J. Bot. 2010, 2010, 382732. [Google Scholar] [CrossRef]
- Hawkins, J.S.; Proulx, S.R.; Rapp, R.A.; Wendel, J.F. Rapid DNA loss as a counterbalance to genome expansion through retrotransposon proliferation in plants. Proc. Natl. Acad. Sci. USA 2009, 106, 17811–17816. [Google Scholar] [CrossRef]
- Devos, K.M.; Brown, J.K.; Bennetzen, J.L. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 2002, 12, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Devos, K.M.; Bennetzen, J.L. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004, 14, 860–869. [Google Scholar] [CrossRef]
- Vitte, C.; Panaud, O.; Quesneville, H. LTR retrotransposons in rice (Oryza sativa L.): Recent burst amplifications followed by rapid DNA loss. BMC Genom. 2007, 8, 218. [Google Scholar] [CrossRef] [PubMed]
- Vitte, C.; Panaud, O. Formation of solo-LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice Oryza sativa L. Mol. Biol. Evol. 2003, 20, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; McCarthy, E.M.; Ganko, E.W.; McDonald, J.F. Evolutionary history of Oryza sativa LTR retrotransposons: A preliminary survey of the rice genome sequences. BMC Genom. 2004, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.S.; Kim, H.; Nason, J.D.; Wing, R.A.; Wendel, J.F. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006, 16, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Q.; Gao, D.; Wang, J.; Lang, Y.; Liu, T.; Li, B.; Bai, Z.; Luis Goicoechea, J.; Liang, C.; et al. Whole-genome sequencing of Oryza brachyantha reveals mechanisms underlying Oryza genome evolution. Nature Commun. 2013, 4, 1595. [Google Scholar] [CrossRef]
- Schubert, I.; Vu, G.T.H. Genome stability and evolution: Attempting a holistic view. Trends Plant Sci. 2016, 21, 749–757. [Google Scholar] [CrossRef]
- Fawcett, J.A.; Rouzé, P.; Van de Peer, Y. Higher intron loss rate in Arabidopsis thaliana than A. lyrata is consistent with stronger selection for a smaller genome. Mol. Biol. Evol. 2012, 29, 849–859. [Google Scholar] [CrossRef]
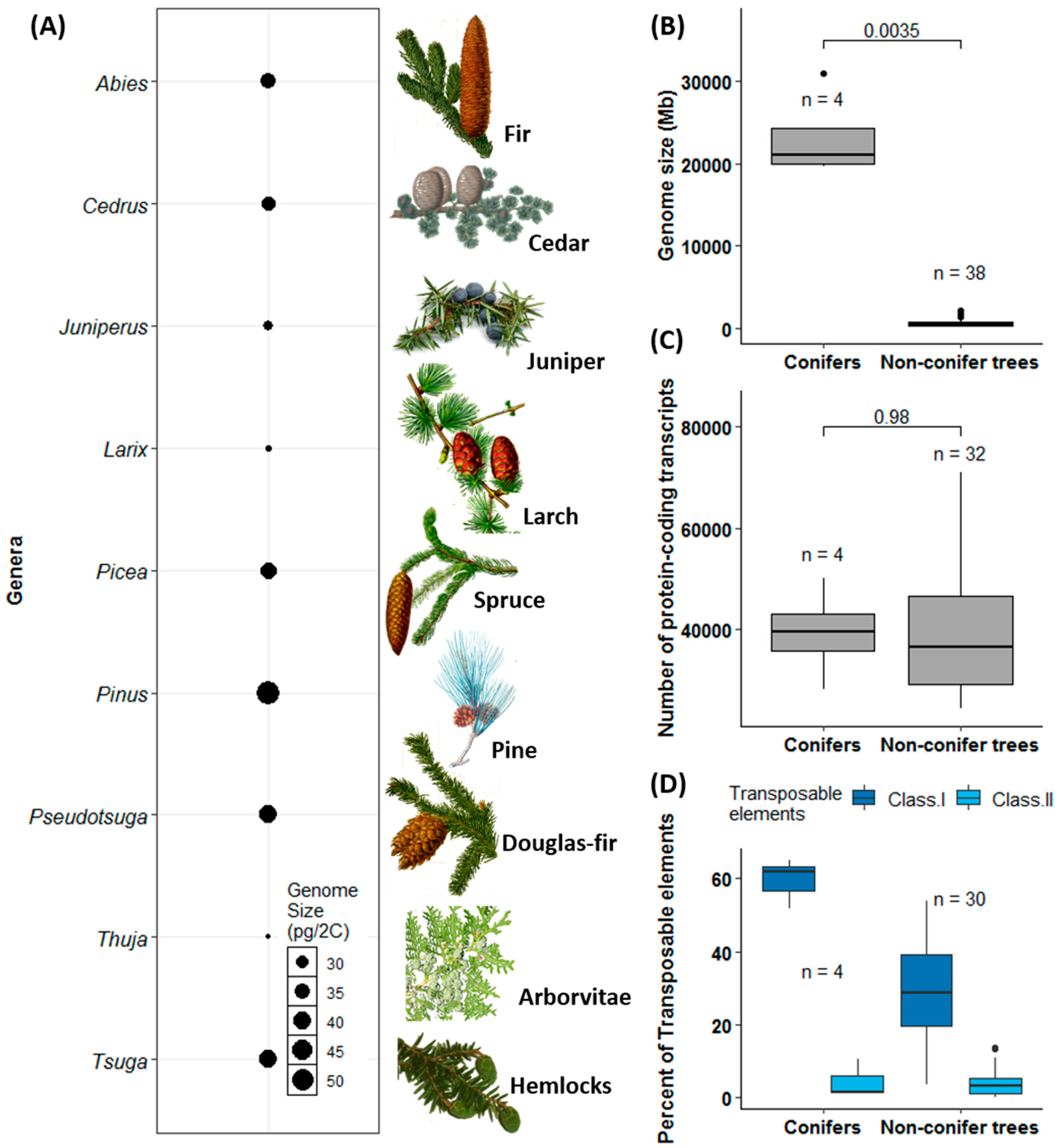
- De La Torre, A.R.; Birol, I.; Bousquet, J.; Ingvarsson, P.K.; Jansson, S.; Jones, S.J.M.; Keeling, C.I.; MacKay, J.; Nilsson, O.; Ritland, K.; et al. Insights into conifer giga-genomes. Plant Physiol. 2014, 166, 1724–1732. [Google Scholar] [CrossRef]
- Kovach, A.; Wegrzyn, J.L.; Parra, G.; Holt, C.; Bruening, G.E.; Loopstra, C.A.; Hartigan, J.; Yandell, M.; Langley, C.H.; Korf, I.; et al. The Pinus taeda genome is characterized by diverse and highly diverged repetitive sequences. BMC Genom. 2010, 11, 420. [Google Scholar] [CrossRef]
- Hamberger, B.; Hall, D.; Yuen, M.; Oddy, C.; Keeling, C.I.; Ritland, C.; Ritland, K.; Bohlmann, J. Targeted isolation, sequence assembly and characterization of two white spruce (Picea glauca) BAC clones for terpenoid synthase and cytochrome P450 genes involved in conifer defence reveal insights into a conifer genome. BMC Plant Biol. 2009, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Ling, J.J.; Xiao, Y.; Zhang, H.G.; Ouyang, F.Q.; Wang, J.H. ConTEdb: A comprehensive database of transposable elements in conifers. Database J. Biol. Databases Curation 2018. [Google Scholar] [CrossRef]
- Morse, A.M.; Peterson, D.G.; Islam-Faridi, M.N.; Smith, K.E.; Magbanua, Z.; Garcia, S.A.; Kubisiak, T.L.; Amerson, H.V.; Carlson, J.E.; Nelson, C.D.; et al. Evolution of genome size and complexity in Pinus. PLoS ONE 2009, 4, 1–11. [Google Scholar] [CrossRef]
- Wegrzyn, J.L.; Lin, B.Y.; Zieve, J.J.; Dougherty, W.M.; Martínez-García, P.J.; Koriabine, M.; Holtz-Morris, A.; de Jong, P.; Crepeau, M.; Langley, C.H.; et al. Insights into the loblolly pine genome: Characterization of BAC and fosmid sequences. PLoS ONE 2013, 8, e72439. [Google Scholar] [CrossRef] [PubMed]
- Cossu, R.M.; Casola, C.; Giacomello, S.; Vidalis, A.; Scofield, D.G.; Zuccolo, A. LTR retrotransposons show low levels of unequal recombination and high rates of intraelement gene conversion in large plant genomes. Genome Biol. Evol. 2017, 9, 3449–3462. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo-Correa, J.P.; Verdú, M.; González-Martínez, S.C. The contribution of recombination to heterozygosity differs among plant evolutionary lineages and life-forms. BMC Evol. Biol. 2010, 10, 22. [Google Scholar] [CrossRef]
- Stapley, J.; Feulner, P.G.D.; Johnston, S.E.; Santure, A.W.; Smadja, C.M. Recombination: The good, the bad and the variable. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20170279. [Google Scholar] [CrossRef]
- Leitch, I.J.; Leitch, A.R. Genome size diversity and evolution in land plants. In Diversity of Plant Genomes Volume 2: Physical Structure, Behaviour and Evolution of Plant Genomes; Greilhuber, J., Dolezel, J., Wendel, J.F., Eds.; Springer-Verlag: New York, NY, USA, 2012. [Google Scholar]
- Fawcett, J.A.; Van de Peer, Y.; Maere, S. Significance and Biological Consequences of Polyploidization in Land Plants; Springer-Verlag: Wien, Austria, 2013. [Google Scholar]
- Williams, C.G. Conifer Reproductive Biology; Springer: Dordrecht, The Netherlands, 2009. [Google Scholar]
- Brown, G.R.; Gill, G.P.; Kuntz, R.J.; Langley, C.H.; Neale, D.B. Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc. Natl. Acad. Sci. USA 2004, 101, 15255–15260. [Google Scholar] [CrossRef] [PubMed]
- Pavy, N.; Namroud, M.C.; Gagnon, F.; Isabel, N.; Bousquet, J. The heterogeneous levels of linkage disequilibrium in white spruce genes and comparative analysis with other conifers. Heredity 2012, 108, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Namroud, M.C.; Guillet-Claude, C.; Mackay, J.; Isabel, N.; Bousquet, J. Molecular evolution of regulatory genes in spruces from different species and continents: Heterogeneous patterns of linkage disequilibrium and selection but correlated recent demographic changes. J. Mol. Evol. 2010, 70, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, O.; Pyhäjärvi, T.; Knürr, T. Gene flow and local adaptation in trees. Annu. Rev. Ecol. Evol. Syst. 2007, 38, 595–619. [Google Scholar] [CrossRef]
- Prunier, J.; Verta, J.P.; MacKay, J.J. Conifer genomics and adaptation: At the crossroads of genetic diversity and genome function. New Phytol. 2016, 209, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Sena, J.S.; Giguère, I.; Boyle, B.; Rigault, P.; Birol, I.; Zuccolo, A.; Ritland, K.; Ritland, C.; Bohlmann, J.; Jones, S.; et al. Evolution of gene structure in the conifer Picea glauca: A comparative analysis of the impact of intron size. BMC Plant Biol. 2014, 14, 95. [Google Scholar] [CrossRef]
- Castillo-Davis, C.I.; Mekhedov, S.L.; Hartl, D.L.; Koonin, E.V.; Kondrashov, F.A. Selection for short introns in highly expressed genes. Nat. Genet. 2002, 31, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Gregory, T.R. The case for junk DNA. PLoS Genet. 2014, 10, e1004351. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Appels, R.; Eversole, K.; Feuillet, C.; Keller, B.; Rogers, J.; Stein, N.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef] [PubMed]
- Soltis, D.E.; Soltis, P.S.; Bennett, M.D.; Leitch, I.J. Evolution of genome size in the angiosperms. Am. J. Bot. 2003, 90, 1596–1603. [Google Scholar] [CrossRef]
- Nosaka, M.; Itoh, J.I.; Nagato, Y.; Ono, A.; Ishiwata, A.; Sato, Y. Role of transposon-derived small RNAs in the interplay between genomes and parasitic DNA in rice. PLoS Genet. 2012, 8, e1002953. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.; Allshire, R.C. RNA silencing and genome regulation. Trends Cell Biol. 2005, 15, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Kasschau, K.D.; Fahlgren, N.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Carrington, J.C. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007, 5, e57. [Google Scholar] [CrossRef]
- Zhang, X.; Henderson, I.R.; Lu, C.; Green, P.J.; Jacobsen, S.E. Role of RNA polymerase IV in plant small RNA metabolism. Proc. Natl. Acad. Sci. USA 2007, 104, 4536–4541. [Google Scholar] [CrossRef]
- Takuno, S.; Ran, J.H.; Gaut, B.S. Evolutionary patterns of genic DNA methylation vary across land plants. Nature Plants 2016, 2, 15222. [Google Scholar] [CrossRef]
- Zhuang, J.L.; Wang, J.; Theurkauf, W.; Weng, Z.P. TEMP: A computational method for analyzing transposable element polymorphism in populations. Nucleic Acids Res. 2014, 42, 6826–6838. [Google Scholar] [CrossRef]
- Platzer, A.; Nizhynska, V.; Long, Q. TE-Locate: A tool to locate and group transposable element occurrences using paired-end next-generation sequencing data. Biology (Basel) 2012, 1, 395–410. [Google Scholar] [CrossRef]
- Steinbiss, S.; Willhoeft, U.; Gremme, G.; Kurtz, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 2009, 37, 7002–7013. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat. Rev. Genet. 2008, 9, 411–412. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; El-Kassaby, Y.A. Novel Insights into Plant Genome Evolution and Adaptation as Revealed through Transposable Elements and Non-Coding RNAs in Conifers. Genes 2019, 10, 228. https://doi.org/10.3390/genes10030228
Liu Y, El-Kassaby YA. Novel Insights into Plant Genome Evolution and Adaptation as Revealed through Transposable Elements and Non-Coding RNAs in Conifers. Genes. 2019; 10(3):228. https://doi.org/10.3390/genes10030228
Chicago/Turabian StyleLiu, Yang, and Yousry A. El-Kassaby. 2019. "Novel Insights into Plant Genome Evolution and Adaptation as Revealed through Transposable Elements and Non-Coding RNAs in Conifers" Genes 10, no. 3: 228. https://doi.org/10.3390/genes10030228
APA StyleLiu, Y., & El-Kassaby, Y. A. (2019). Novel Insights into Plant Genome Evolution and Adaptation as Revealed through Transposable Elements and Non-Coding RNAs in Conifers. Genes, 10(3), 228. https://doi.org/10.3390/genes10030228