Population Genomics and Structure of the Critically Endangered Mariana Crow (Corvus kubaryi)

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Crow Samples
2.2. Genome Sequencing and Assembly
2.3. Hybridization Capture Assay Design
2.4. DNA Extraction, Library Preparation, Capture and Next Generation Sequencing
2.5. Sequence Alignment and Genotyping
2.6. Population Structure
2.7. Kinship and Inbreeding Analysis
3. Results
3.1. Genome
3.2. Single Nucleotide Variants Data
3.3. Principal Component Analysis
3.4. Population Structure
3.5. Kinship and Inbreeding Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Steadman, D.W. Extinction and Biogeography of Tropical Pacific Birds; University of Chicago Press: Chicago, IL, USA, 2006; ISBN 0-226-77142-3. [Google Scholar]
- Kueffer, C.; Drake, D.R.; Fernandez-Palacios, J.M. Island biology: Looking towards the future. Biol. Lett. 2014, 10, 20140719. [Google Scholar] [CrossRef] [PubMed]
- Zarones, L.; Sussman, A.; Morton, J.M.; Plentovich, S.; Faegre, S.; Aguon, C.; Amar, A.; Ha, R.R. Population status and nest success of the Critically Endangered Mariana Crow (Corvus kubaryi) on Rota, Northern Mariana Islands. Bird Conserv. Int. 2015, 25, 220–233. [Google Scholar] [CrossRef]
- Newton, I. Population Limitation in Birds; Academic Press: Cambridge, CA, USA, 1998; ISBN 0-08-087923-3. [Google Scholar]
- Wiles, G.J.; Lemke, T.O.; Payne, N.H. Population Estimates of Fruit Bats (Pteropus mariannus) in the Mariana Islands. Conserv. Biol. 1989, 3, 66–76. [Google Scholar] [CrossRef]
- Steadman, D.W. Extinct and extirpated birds from Rota, Mariana Islands. Micronesica 1992, 25, 71–84. [Google Scholar]
- Steadman, D.W. Prehistoric Extinctions of Pacific Island Birds: Biodiversity Meets Zooarchaeology. Science 1995, 267, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Steadman, D.W. The prehistory of vertebrates, especially birds, on Tinian, Aguiguan, and Rota, Northern Mariana Islands. Micronesica 1999, 31, 319–345. [Google Scholar]
- Savidge, J.A. Extinction of an Island Forest Avifauna by an Introduced Snake. Ecology 1987, 68, 660–668. [Google Scholar] [CrossRef]
- Fritts, T.H.; Rodda, G.H. The role of introduced species in the degradation of island ecosystems: A case history of Guam. Annu. Rev. Ecol. Syst. 1998, 29, 113–140. [Google Scholar] [CrossRef]
- Wiles, G.J.; Bart, J.; Beck, R.E.; Aguon, C.F. Impacts of the Brown Tree Snake: Patterns of Decline and Species Persistence in Guam’s Avifauna. Conserv. Biol. 2003, 17, 1350–1360. [Google Scholar] [CrossRef]
- NRC. The Scientific Bases for the Preservation of the Mariana Crow; National Academies Press: Washington, DC, USA, 1997. [Google Scholar]
- Haig, S.M.; Ballou, J.D.; Derrickson, S.R. Management Options for Preserving Genetic Diversity: Reintroduction of Guam Rails to the Wild. Conserv. Biol. 1990, 4, 290–300. [Google Scholar] [CrossRef]
- Kesler, D.C.; Haig, S.M. Multiscale Habitat Use and Selection in Cooperatively Breeding Micronesian Kingfishers. J. Wildl. Manag. 2007, 71, 765–772. [Google Scholar] [CrossRef]
- Cruz, J.B.; Kremer, S.R.; Martin, G.; Williams, L.L.; Camacho, V.A. Relative abundance and distribution of Mariana swiftlets (Aves: Apodidae) in the Northern Mariana Islands. Pac. Sci. 2008, 62, 233–246. [Google Scholar] [CrossRef]
- U.S. Fish and Wildlife Service. Draft Revised Recovery Plan for the Aga or Mariana Crow, Corvus Kubaryi; U.S. Fish and Wildlife Service: Portland, OR, USA, 2005.
- MAC Working Group. Marianas Avifauna Conservation (MAC) Plan: Long-Term Conservation Plan for the Native Forest Birds of the Northern Marian Islands; CNMI Division of Fish and Wildlife: Tanapag, Saipan; U.S. Fish and Wildlife Service: Honolulu, HI, USA, 2013.
- Kroner, A.; Ha, R.R. An update of the breeding population status of the critically endangered Mariana Crow (Corvus kubaryi) on Rota, Northern Mariana Islands 2013–2014. Bird Conserv. Int. 2018, 28, 416–422. [Google Scholar] [CrossRef]
- Plentovich, S.; Morton, J.M.; Bart, J.; Camp, R.J.; Lusk, M.; Johnson, N.; Vanderwerf, E. Population trends of Mariana Crow (Corvus kubaryi) on Rota, Commonwealth of the Northern Mariana Islands. Bird Conserv. Int. 2005, 15. [Google Scholar] [CrossRef]
- Engbring, J.; Ramsey, F.L.; Wildman, V.J. Micronesian forest bird survey, 1982: Saipan, Tinian, Agiguan, and Rota; Fish and Wildlife Service, US Department of the Interior: Washington, DC, USA, 1986.
- Tarr, C.L.; Fleischer, R.C. Primers for polymorphic GT microsatellites isolated from the Mariana Crow, (Corvus kubaryi). Mol. Ecol. 1998, 7, 253–255. [Google Scholar] [PubMed]
- Beck, R.E.; Department of Aquatic and Wildlife Resources (DAWR), Guam; Pimm, S. (Univ. of Tennessee, Knoxville, TN, USA). Personal communication. 1998.
- Lusk, M. (Draft memo on analysis of Rota crow population survey from M. Lusk to K. Rosa, Chief Recovery Branch, US FWS Pacific Region). Personal communication. 2005.
- Fancy, S.G.; Lusk, M.R.; Grout, D. Status of the Mariana crow population on Rota, Mariana Islands. Micronesica 1999, 32, 3–10. [Google Scholar]
- Camp., R.J.; United States Geological Survey (USGS). Personal Communication. 2001.
- Amar, A.; Amidon, F.; Arroyo, B.; Esselstyn, J.A.; Marshall, A.P. Population trends of the forest bird community on the Pacific island of Rota, Mariana Islands. Condor 2008, 110, 421–427. [Google Scholar] [CrossRef]
- Camp, R.J.; Brinck, K.W.; Gorresen, P.M.; Amidon, F.A.; Radley, P.M.; Berkowitz, S.P.; Banko, P.C. Current land bird distribution and trends in population abundance between 1982 and 2012 on Rota, Mariana Islands. J. Fish Wildlife Manag. 2015, 2, 511–540. [Google Scholar] [CrossRef]
- Hoeck, P.E.A.; Wolak, M.E.; Switzer, R.A.; Kuehler, C.M.; Lieberman, A.A. Effects of inbreeding and parental incubation on captive breeding success in Hawaiian crows. Biol. Conserv. 2015, 184, 357–364. [Google Scholar] [CrossRef]
- Hedrick, P.W.; Hoeck, P.E.; Fleischer, R.C.; Farabaugh, S.; Masuda, B.M. The influence of captive breeding management on founder representation and inbreeding in the Alalā, the Hawaiian crow. Conserv. Genet. 2016, 17, 369–378. [Google Scholar] [CrossRef]
- Sutton, J.; Helmkampf, M.; Steiner, C.; Bellinger, M.R.; Korlach, J.; Hall, R.; Baybayan, P.; Muehling, J.; Gu, J.; Kingan, S.; et al. A high-quality, long-read de novo genome assembly to aid conservation of Hawaii’s last remaining crow species. Genes 2018, 9, 393. [Google Scholar] [CrossRef] [PubMed]
- Tarr, C.L.; Fleischer, R. Population boundaries and genetic diversity in the endangered Mariana crow (Corvus kubaryi). Mol. Ecol. 1999, 8, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data, version 0.11; Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 December 2018).
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2015. Available online: http://www.repeatmasker.org (accessed on 10 December 2018).
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, 2013; arXiv:13033997. [Google Scholar]
- Broad Institute Picard Tools. Broad Institute, GitHub repository 2018. Available online: https://broadinstitute.github.io/picard/ (accessed on 10 December 2018).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Campana, M.G. BaitsTools: Software for hybridization capture bait design. Mol. Ecol. Resour. 2018, 18, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Renaud, G.; Stenzel, U.; Maricic, T.; Wiebe, V.; Kelso, J. deML: Robust demultiplexing of Illumina sequences using a likelihood-based approach. Bioinformatics 2015, 31, 770–772. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next- generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Lischer, H.E.L.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2012, 28, 298–299. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015; ISBN 3-900051-07-0. [Google Scholar]
- Dray, S.; Dufour, A.-B. The ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat. Softw. 2007, 22. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Campana, M.G.; Hunt, H.V.; Jones, H.; White, J. CorrSieve: Software for summarizing and evaluating Structure output: Computer Program Note. Mol. Ecol. Resour. 2011, 11, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.K.; McKelvey, K.S. Why sampling scheme matters: The effect of sampling scheme on landscape genetic results. Conserv. Genet. 2009, 10, 441–452. [Google Scholar] [CrossRef]
- Rosenberg, N.A. Distruct: A program for the graphical display of population structure: Program note. Mol. Ecol. Notes 2003, 4, 137–138. [Google Scholar] [CrossRef]
- Gattepaille, L.M.; Jakobsson, M.; Blum, M.G. Inferring population size changes with sequence and SNP data: Lessons from human bottlenecks. Heredity 2013, 110, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.M.; Plentovich, S.; Sharp, T. Reproduction and Juvenile Dispersal of Mariana Crows (Corvus kubayi) on Rota, 1996–1999; U.S. Fish and Wildlife Service: Honolulu, HI, USA, 1999.
- U.S. Fish and Wildlife Service. Structured Decision Making for the Mariana Crow; U.S. Fish and Wildlife Service: Honolulu, HI, USA, 2014.
- Faegre, S.; Nietmann, L.; Hubl, D.; Ha, J.C.; Ha, R.R. Spatial ecology of the Mariana Crow (Corvus kubaryi): Implications for management strategies. Bird Conserv. Int. 2018. [Google Scholar] [CrossRef]
- Yaremych, S.A.; Novak, R.J.; Raim, A.J.; Mankin, P.C.; Warner, R.E. Home range and habitat use by American Crows in relation to transmission of West Nile virus. Wilson Bull. 2004, 116, 232–239. [Google Scholar] [CrossRef]
- Webb, W.C.; Boarman, W.I.; Rotenberry, J.T. Movements of Juvenile Common Ravens in an Arid Landscape. J. Wildl. Manag. 2009, 73, 72–81. [Google Scholar] [CrossRef]
- Holzhaider, J.C.; Sibley, M.D.; Taylor, A.H.; Singh, P.J.; Gray, R.D.; Hunt, G.R. The social structure of New Caledonian crows. Anim. Behav. 2011, 81, 83–92. [Google Scholar] [CrossRef]
- Schoener, T.W. Sizes of feeding territories among birds. Ecology 1968, 49, 123–141. [Google Scholar] [CrossRef]
- Ha, R.R.; Faegre, S.; Ha, J.C. Rules of Dispersal in the Endangered Mariana Crow (Corvus kubaryi). In Proceedings of the Animal Behavior Conference, Williamsburg, VA, USA, 2010. [Google Scholar]
- Omland, K.E.; Baker, J.M.; Peters, J.L. Genetic signatures of intermediate divergence: Population history of Old and New World Holarctic ravens (Corvus corax): Genetic signatures of intermediate divergence. Mol. Ecol. 2006, 15, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, R.C.; Boarman, W.I.; Gonzalez, E.G.; Godinez, A.; Omland, K.E.; Young, S.; Helgen, L.; Syed, G.; Mcintosh, C.E. As the raven flies: Using genetic data to infer the history of invasive common raven (Corvus corax) populations in the Mojave Desert. Mol. Ecol. 2008, 17, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Rutz, C.; Ryder, T.B.; Fleischer, R.C. Restricted gene flow and fine-scale population structuring in tool using New Caledonian crows. Naturwissenschaften 2012, 99, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Morinha, F.; Dávila, J.A.; Bastos, E.; Cabral, J.A.; Frías, Ó.; González, J.L.; Travassos, P.; Carvalho, D.; Milá, B.; Blanco, G. Extreme genetic structure in a social bird species despite high dispersal capacity. Mol. Ecol. 2017, 26, 2812–2825. [Google Scholar] [CrossRef] [PubMed]
- St Clair, J.J.H.; Burns, Z.T.; Bettaney, E.M.; Morrissey, M.B.; Otis, B.; Ryder, T.B.; Fleischer, R.C.; James, R.; Rutz, C. Experimental resource pulses influence social-network dynamics and the potential for information flow in tool-using crows. Nat. Commun. 2015, 6, 7197. [Google Scholar] [CrossRef] [PubMed]
- St Clair, J.J.H.; Klump, B.C.; van der Wal, J.E.M.; Sugasawa, S.; Rutz, C. Strong between-site variation in New Caledonian crows’ use of hook-tool-making materials. Biol. J. Linn. Soc. 2016, 118, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.C.; Butler, A.; Robinette Ha, R. Reduction of first-year survival threatens the viability of the Mariana Crow (Corvus kubaryi) population on Rota, CNMI. Bird Conserv. Int. 2010, 20, 335–342. [Google Scholar] [CrossRef]
- Pratt, T.K.; Atkinson, C.T.; Banko, P.C. Conservation Biology of Hawaiian Forest Birds: Implications for Island Avifauna; Yale University Press: New Haven, CT, USA, 2009; ISBN 0-300-14108-4. [Google Scholar]






{kind=link}
{kind=link}
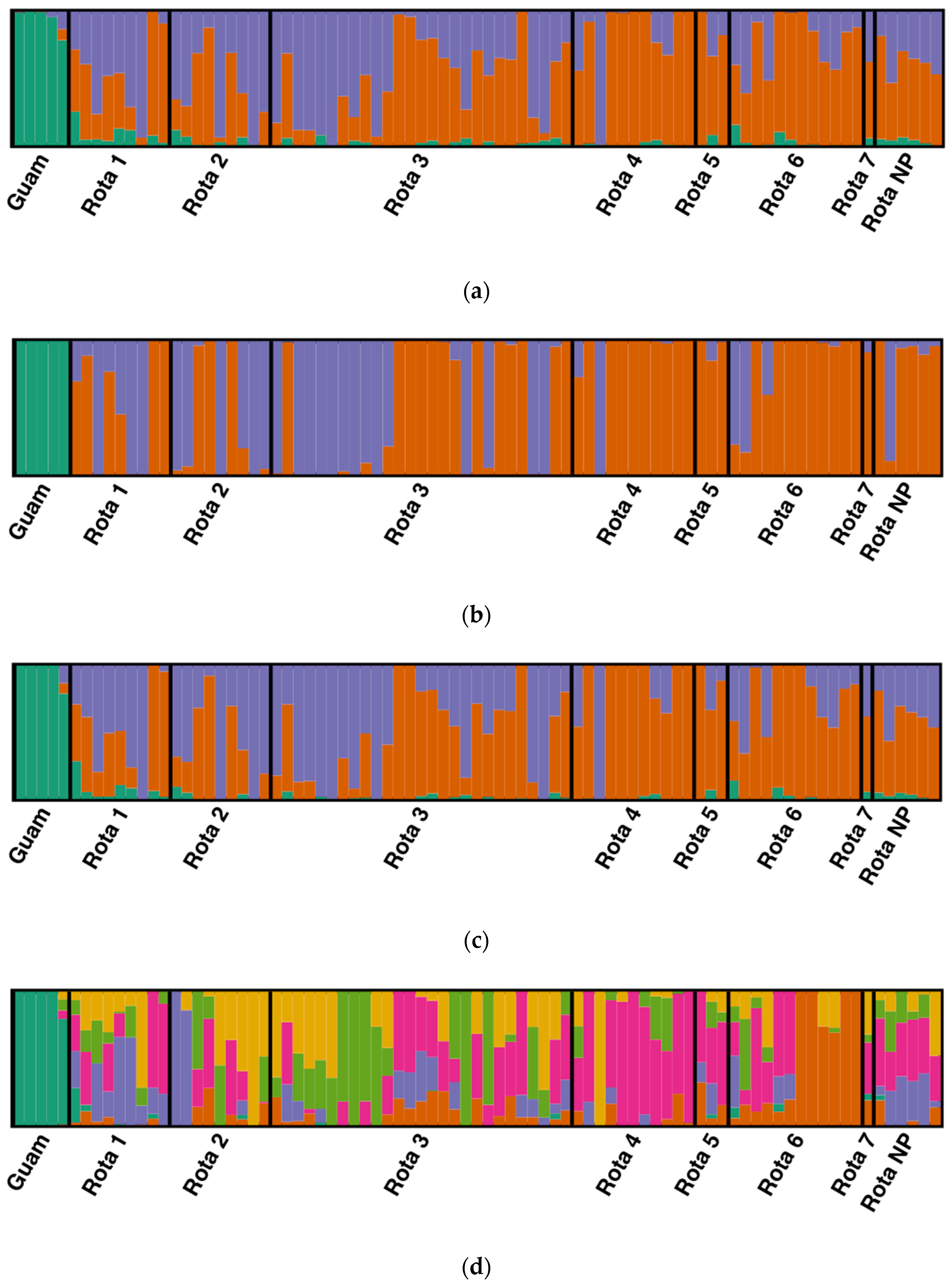{kind=link}
{kind=link}
{kind=link}
| Year | Est. Number (and/or Range of Individuals | Survey Method | Source |
|---|---|---|---|
| 1982 | 1318 (1136–1564) | Off-road VCP | [20] |
| 1988 | (600–1000) | Informal Estimate | [22] |
| 1992 | (447–931) | Roadside VCP | [23] |
| 1993 | (336–454) | Roadside VCP | [23] |
| 1995 | 592 (474–729) | Off-road VCP | [24] |
| 1995 | (365–607) | Off-road VCP | [25] |
| 1998 | (138–504) | Off-road VCP | [25] |
| 1999 | 234 breeding adults | Extrapolated from known pairs and density estimates | [19] |
| 2004 | 170 breeding adults | Extrapolated from known pairs and density estimates | [26] |
| 2007 | 120 breeding adults | Direct count of breeding pairs | [3] |
| 2012 | (30–202) | Off-road VCP | [27] |
| 2014 | 108 breeding adults; 178 | Direct Count of pairs; Chapman estimate of total population | [18] |
| Statistic | Statistic | Statistic |
|---|---|---|
| Total base pairs: 1,073,754,616 | N10: 229,841 | L10: 363 |
| Total number contigs: 54,052 | N20: 168,264 | L20: 915 |
| Mean contig length: 19,865 | N30: 128,954 | L30: 1651 |
| Median contig length: 2383 | N40: 100,208 | L40: 2597 |
| Longest contig: 677,288 | N50: 79,541 | L50: 3800 |
| Shortest contig: 182 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortes-Rodriguez, N.; Campana, M.G.; Berry, L.; Faegre, S.; Derrickson, S.R.; Ha, R.R.; Dikow, R.B.; Rutz, C.; Fleischer, R.C. Population Genomics and Structure of the Critically Endangered Mariana Crow (Corvus kubaryi). Genes 2019, 10, 187. https://doi.org/10.3390/genes10030187
Cortes-Rodriguez N, Campana MG, Berry L, Faegre S, Derrickson SR, Ha RR, Dikow RB, Rutz C, Fleischer RC. Population Genomics and Structure of the Critically Endangered Mariana Crow (Corvus kubaryi). Genes. 2019; 10(3):187. https://doi.org/10.3390/genes10030187
Chicago/Turabian StyleCortes-Rodriguez, Nandadevi, Michael G. Campana, Lainie Berry, Sarah Faegre, Scott R. Derrickson, Renee Robinette Ha, Rebecca B. Dikow, Christian Rutz, and Robert C. Fleischer. 2019. "Population Genomics and Structure of the Critically Endangered Mariana Crow (Corvus kubaryi)" Genes 10, no. 3: 187. https://doi.org/10.3390/genes10030187
APA StyleCortes-Rodriguez, N., Campana, M. G., Berry, L., Faegre, S., Derrickson, S. R., Ha, R. R., Dikow, R. B., Rutz, C., & Fleischer, R. C. (2019). Population Genomics and Structure of the Critically Endangered Mariana Crow (Corvus kubaryi). Genes, 10(3), 187. https://doi.org/10.3390/genes10030187