Progression of Cystic Fibrosis Lung Disease from Childhood to Adulthood: Neutrophils, Neutrophil Extracellular Trap (NET) Formation, and NET Degradation


Abstract
:1. Introduction
2. Cystic Fibrosis
2.1. Cystic Fibrosis Transmembrane Conductance Regulator
2.2. Classes of Cystic Fibrosis Transmembrane Conductance Regulator Mutations
2.3. Cystic Fibrosis Disease Pathophysiology: Mucus Dehydration and Declining Lung Function
2.4. Infection and Inflammation in Cystic Fibrosis
3. Neutrophils
3.1. Neutrophil Development and Maturation
3.2. Cystic Fibrosis Transmembrane Conductance Regulator in Neutrophils
3.3. Cystic Fibrosis Neutrophil Dysfunction
4. Neutrophil Anti-Microbial Properties
4.1. Neutrophil Extracellular Trap Formation (NETosis)
4.1.1. NOX-Dependent NETosis
4.1.2. NOX-Independent NETosis
4.1.3. NETs and the Vicious Cycle of Cystic Fibrosis lung Disease
5. Clearing Neutrophil Extracellular Traps (NETs)—DNases and Macrophages
6. Suppressing NETs: Future Directions and Therapeutic Potential
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Briganti, D.F.; D’Ovidio, F. Long-term management of patients with end-stage lung diseases. Best Pr. Res. Clin. Anaesthesiol. 2017, 31, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Rout-Pitt, N.; Farrow, N.; Parsons, D.; Donnelley, M. Epithelial mesenchymal transition (EMT): A universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir. Res. 2018, 19, 136. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Assael, B.M. Cystic fibrosis: A clinical view. Cell. Mol. Life Sci. 2017, 74, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.B.; Long, R.F.; Kissner, W.J.; Atieh, E.; Garbarine, I.C.; Markovetz, M.R.; Fontana, N.C.; Christy, M.; Habibpour, M.; Tarran, R.; et al. Pathological mucus and impaired mucus clearance in cystic fibrosis patients result from increased concentration, not altered pH. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef] [PubMed]
- Csanady, L.; Vergani, P.; Gadsby, D.C. Structure, Gating, and Regulation of the Cftr Anion Channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Salomon, J.J.; Sheppard, D.N.; Mall, M.A.; Galietta, L.J. Bypassing CFTR dysfunction in cystic fibrosis with alternative pathways for anion transport. Curr. Opin. Pharm. 2017, 34, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Wine, J.J.; Hansson, G.C.; Konig, P.; Joo, N.S.; Ermund, A.; Pieper, M. Progress in understanding mucus abnormalities in cystic fibrosis airways. J. Cyst. Fibros. 2018, 17, S35–S39. [Google Scholar] [CrossRef] [PubMed]
- Haq, I.J.; Gray, M.A.; Garnett, J.P.; Ward, C.; Brodlie, M. Airway surface liquid homeostasis in cystic fibrosis: Pathophysiology and therapeutic targets. Thorax 2016, 71, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Bezzerri, V.; Piacenza, F.; Caporelli, N.; Malavolta, M.; Provinciali, M.; Cipolli, M. Is cellular senescence involved in cystic fibrosis? Respir Res. 2019, 20, 32. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Yoo, D.G.; Kahlenberg, J.M.; Bridges, S.L., Jr.; Oni, O.; Huang, H.; Stecenko, A.; Rada, B. Systemic levels of anti-PAD4 autoantibodies correlate with airway obstruction in cystic fibrosis. J. Cyst. Fibros. 2019. [Google Scholar] [CrossRef] [PubMed]
- Margaroli, C.; Garratt, L.W.; Horati, H.; Dittrich, A.S.; Rosenow, T.; Montgomery, S.T.; Frey, D.L.; Brown, M.R.; Schultz, C.; Guglani, L.; et al. Elastase Exocytosis by Airway Neutrophils Associates with Early Lung Damage in Cystic Fibrosis Children. Am. J. Respir. Crit. Care Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53, S30–S50. [Google Scholar] [CrossRef] [PubMed]
- Forrest, O.A.; Ingersoll, S.A.; Preininger, M.K.; Laval, J.; Limoli, D.H.; Brown, M.R.; Lee, F.E.; Bedi, B.; Sadikot, R.T.; Goldberg, J.B.; et al. Frontline Science: Pathological conditioning of human neutrophils recruited to the airway milieu in cystic fibrosis. J. Leukoc. Biol. 2018, 104, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Laval, J.; Ralhan, A.; Hartl, D. Neutrophils in cystic fibrosis. Biol. Chem. 2016, 397, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Agraz-Cibrian, J.M.; Giraldo, D.M.; Mary, F.M.; Urcuqui-Inchima, S. Understanding the molecular mechanisms of NETs and their role in antiviral innate immunity. Virus Res. 2017, 228, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Law, S.M.; Gray, R.D. Neutrophil extracellular traps and the dysfunctional innate immune response of cystic fibrosis lung disease: A review. J. Inflamm 2017, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Rada, B. Neutrophil extracellular trap release driven by bacterial motility: Relevance to cystic fibrosis lung disease. Commun. Integr. Biol. 2017, 10, e1296610. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhao, G.; Song, N.; Li, P.; Chen, Y.; Guo, Y.; Li, J.; Du, L.; Jiang, S.; Guo, R.; et al. Blockade of the C5a-C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg. Microb. Infect. 2018, 7, 77. [Google Scholar] [CrossRef] [PubMed]
- Douda, D.N.; Grasemann, H.; Pace-Asciak, C.; Palaniyar, N. A lipid mediator hepoxilin A3 is a natural inducer of neutrophil extracellular traps in human neutrophils. Mediat. Inflamm. 2015, 2015, 520871. [Google Scholar] [CrossRef] [PubMed]
- Jennings, S.; Ng, H.P.; Wang, G. Establishment of a DeltaF508-CF promyelocytic cell line for cystic fibrosis research and drug screening. J. Cyst. Fibros. 2019, 18, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.T.; Dittrich, A.S.; Garratt, L.W.; Turkovic, L.; Frey, D.L.; Stick, S.M.; Mall, M.A.; Kicic, A.; Arest, C.F. Interleukin-1 is associated with inflammation and structural lung disease in young children with cystic fibrosis. J. Cyst. Fibros. 2018, 17, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Verhaeghe, C.; Delbecque, K.; de Leval, L.; Oury, C.; Bours, V. Early inflammation in the airways of a cystic fibrosis foetus. J. Cyst. Fibros. 2007, 6, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Eckrich, J.; Zissler, U.M.; Serve, F.; Leutz, P.; Smaczny, C.; Schmitt-Grohe, S.; Fussbroich, D.; Schubert, R.; Zielen, S.; Eickmeier, O. Airway inflammation in mild cystic fibrosis. J. Cyst. Fibros. 2017, 16, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Bonfield, T.L.; Chmiel, J.F.; Pearlman, E. Neutrophils from F508del cystic fibrosis patients produce IL-17A and express IL-23—Dependent IL-17RC. Clin. Immunol. 2016, 170, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Ermert, D.; Zychlinsky, A.; Urban, C. Fungal and bacterial killing by neutrophils. Methods Mol. Biol. 2009, 470, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rizo, V.; Martinez-Guzman, M.A.; Iniguez-Gutierrez, L.; Garcia-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Albrett, A.M.; Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J. Leukoc Biol 2012, 91, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, A.S.; Kuhbandner, I.; Gehrig, S.; Rickert-Zacharias, V.; Twigg, M.; Wege, S.; Taggart, C.C.; Herth, F.; Schultz, C.; Mall, M.A. Elastase activity on sputum neutrophils correlates with severity of lung disease in cystic fibrosis. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.D.; Margaroli, C.; Horati, H.; Kilgore, M.B.; Veltman, M.; Liu, H.K.; Taurone, A.J.; Peng, L.; Guglani, L.; Uppal, K.; et al. Myeloperoxidase oxidation of methionine associates with early cystic fibrosis lung disease. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef] [PubMed]
- Marcos, V.; Zhou-Suckow, Z.; Onder Yildirim, A.; Bohla, A.; Hector, A.; Vitkov, L.; Krautgartner, W.D.; Stoiber, W.; Griese, M.; Eickelberg, O.; et al. Free DNA in cystic fibrosis airway fluids correlates with airflow obstruction. Mediat. Inflamm. 2015, 2015, 408935. [Google Scholar] [CrossRef] [PubMed]
- Skopelja, S.; Hamilton, B.J.; Jones, J.D.; Yang, M.L.; Mamula, M.; Ashare, A.; Gifford, A.H.; Rigby, W.F. The role for neutrophil extracellular traps in cystic fibrosis autoimmunity. JCI Insight 2016, 1, e88912. [Google Scholar] [CrossRef] [PubMed]
- Dickerhof, N.; Pearson, J.F.; Hoskin, T.S.; Berry, L.J.; Turner, R.; Sly, P.D.; Kettle, A.J.; Arest, C.F. Oxidative stress in early cystic fibrosis lung disease is exacerbated by airway glutathione deficiency. Free Radic. Biol. Med. 2017, 113, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Cheng, O.Z.; Palaniyar, N. NET balancing: A problem in inflammatory lung diseases. Front. Immunol. 2013, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Nadesalingam, A.; Chen, J.H.K.; Farahvash, A.; Khan, M.A. Hypertonic Saline Suppresses NADPH Oxidase-Dependent Neutrophil Extracellular Trap Formation and Promotes Apoptosis. Front. Immunol. 2018, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Pillarisetti, N.; Williamson, E.; Linnane, B.; Skoric, B.; Robertson, C.F.; Robinson, P.; Massie, J.; Hall, G.L.; Sly, P.; Stick, S.; et al. Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Mott, L.S.; Park, J.; Murray, C.P.; Gangell, C.L.; de Klerk, N.H.; Robinson, P.J.; Robertson, C.F.; Ranganathan, S.C.; Sly, P.D.; Stick, S.M.; et al. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax 2012, 67, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.A.; Stahl, M.; Graeber, S.Y.; Sommerburg, O.; Kauczor, H.U.; Wielputz, M.O. Early detection and sensitive monitoring of CF lung disease: Prospects of improved and safer imaging. Pediatr. Pulmonol. 2016, 51, S49–S60. [Google Scholar] [CrossRef] [PubMed]
- VanDevanter, D.R.; Kahle, J.S.; O’Sullivan, A.K.; Sikirica, S.; Hodgkins, P.S. Cystic fibrosis in young children: A review of disease manifestation, progression, and response to early treatment. J. Cyst. Fibros. 2016, 15, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Marini, T.; Hobbs, S.K.; Chaturvedi, A.; Kaproth-Joslin, K. Beyond bronchitis: A review of the congenital and acquired abnormalities of the bronchus. Insights Imaging 2017, 8, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Kube, D.; Sontich, U.; Fletcher, D.; Davis, P.B. Proinflammatory cytokine responses to P. aeruginosa infection in human airway epithelial cell lines. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L493–L502. [Google Scholar] [CrossRef] [PubMed]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.S.; Prince, A. Cystic fibrosis: A mucosal immunodeficiency syndrome. Nat. Med. 2012, 18, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Garth, J.; Barnes, J.W.; Krick, S. Targeting Cytokines as Evolving Treatment Strategies in Chronic Inflammatory Airway Diseases. Int J. Mol. Sci 2018, 19, 3402. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Leach, S.T.; Katz, T.; Day, A.S.; Jaffe, A.; Ooi, C.Y. Update of faecal markers of inflammation in children with cystic fibrosis. Mediat. Inflamm. 2012, 2012, 948367. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.S.; Hook, S.M.; Jamsen, K.M.; Nixon, G.M.; Carzino, R.; Carlin, J.B.; Robertson, C.F.; Grimwood, K. Lower airway inflammation in infants with cystic fibrosis detected by newborn screening. Pediatr. Pulmonol. 2005, 40, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Saiman, L. Microbiology of early CF lung disease. Paediatr. Respir. Rev. 2004, 5 (Suppl. A), S367–S369. [Google Scholar] [CrossRef]
- Tridello, G.; Castellani, C.; Meneghelli, I.; Tamanini, A.; Assael, B.M. Early diagnosis from newborn screening maximises survival in severe cystic fibrosis. Erj Open Res. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Laguna, T.A.; Williams, C.B.; Nunez, M.G.; Welchlin-Bradford, C.; Moen, C.E.; Reilly, C.S.; Wendt, C.H. Biomarkers of inflammation in infants with cystic fibrosis. Respir Res. 2018, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.K.; Kazmierczak, B.I. Inflammation: A Double-Edged Sword in the Response to Pseudomonas aeruginosa Infection. J. Innate Immun. 2017, 9, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Bhagirath, A.Y.; Li, Y.; Somayajula, D.; Dadashi, M.; Badr, S.; Duan, K. Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. Bmc Pulm Med. 2016, 16, 174. [Google Scholar] [CrossRef] [PubMed]
- Jochmann, A.; Artusio, L.; Robson, K.; Nagakumar, P.; Collins, N.; Fleming, L.; Bush, A.; Saglani, S. Infection and inflammation in induced sputum from preschool children with chronic airways diseases. Pediatr. Pulmonol. 2016, 51, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Adler, F.R.; Aurora, P.; Barker, D.H.; Barr, M.L.; Blackwell, L.S.; Bosma, O.H.; Brown, S.; Cox, D.R.; Jensen, J.L.; Kurland, G.; et al. Lung transplantation for cystic fibrosis. Proc. Am. Thorac. Soc. 2009, 6, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Fitton, I.; Revel, M.P.; Burgel, P.R.; Hernigou, A.; Boussaud, V.; Guillemain, R.; Le Pimpec-Barthes, F.; Bennani, S.; Freche, G.; Frija, G.; et al. Cumulative radiation dose after lung transplantation in patients with cystic fibrosis. Diagn. Interv. Imaging 2019. [Google Scholar] [CrossRef] [PubMed]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Moraes, T.J.; Plumb, J.; Martin, R.; Vachon, E.; Cherepanov, V.; Koh, A.; Loeve, C.; Jongstra-Bilen, J.; Zurawska, J.H.; Kus, J.V.; et al. Abnormalities in the pulmonary innate immune system in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2006, 34, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Philip, L.M.; Cheung, G.; Vadakepeedika, S.; Grasemann, H.; Sweezey, N.; Palaniyar, N. Regulating NETosis: Increasing pH Promotes NADPH Oxidase-Dependent NETosis. Front. Med. (Lausanne) 2018, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.S.; Meyerholz, D.K.; Tang, X.X.; Reznikov, L.; Abou Alaiwa, M.; Ernst, S.E.; Karp, P.H.; Wohlford-Lenane, C.L.; Heilmann, K.P.; Leidinger, M.R.; et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science 2016, 351, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.; Lubamba, B.A. Role of IRE1alpha/XBP-1 in Cystic Fibrosis Airway Inflammation. Int. J. Mol. Sci. 2017, 18, 118. [Google Scholar] [CrossRef]
- Cant, N.; Pollock, N.; Ford, R.C. CFTR structure and cystic fibrosis. Int. J. Biochem. Cell Biol. 2014, 52, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, P.; Wilschanski, M.; Muallem, S.; Lukacs, G.L.; Sahin-Toth, M.; Uc, A.; Gray, M.A.; Rakonczay, Z., Jr.; Maleth, J. CFTR: A New Horizon in the Pathomechanism and Treatment of Pancreatitis. Rev. Physiol. Biochem. Pharm. 2016, 170, 37–66. [Google Scholar] [CrossRef]
- Fanen, P.; Wohlhuter-Haddad, A.; Hinzpeter, A. Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. Int. J. Biochem. Cell Biol. 2014, 52, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Soe, K.; Gregoire-Bottex, M.M. A rare CFTR mutation associated with severe disease progression in a 10-year-old Hispanic patient. Clin. Case Rep. 2017, 5, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Sosnay, P.R.; Siklosi, K.R.; Van Goor, F.; Kaniecki, K.; Yu, H.; Sharma, N.; Ramalho, A.S.; Amaral, M.D.; Dorfman, R.; Zielenski, J.; et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 2013, 45, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, L.W.; Zilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Z.; Csanady, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95e88. [Google Scholar] [CrossRef] [PubMed]
- Protasevich, I.; Yang, Z.; Wang, C.; Atwell, S.; Zhao, X.; Emtage, S.; Wetmore, D.; Hunt, J.F.; Brouillette, C.G. Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein Sci. 2010, 19, 1917–1931. [Google Scholar] [CrossRef] [PubMed]
- Chappe, V.; Irvine, T.; Liao, J.; Evagelidis, A.; Hanrahan, J.W. Phosphorylation of CFTR by PKA promotes binding of the regulatory domain. Embo J. 2005, 24, 2730–2740. [Google Scholar] [CrossRef] [PubMed]
- Bonadia, L.C.; de Lima Marson, F.A.; Ribeiro, J.D.; Paschoal, I.A.; Pereira, M.C.; Ribeiro, A.F.; Bertuzzo, C.S. CFTR genotype and clinical outcomes of adult patients carried as cystic fibrosis disease. Gene 2014, 540, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Registry—2017, T.C.C.F. Annual Data Report. 2017. Available online: https://www.cftr2.org/.
- Proesmans, M.; Vermeulen, F.; De Boeck, K. What’s new in cystic fibrosis? From treating symptoms to correction of the basic defect. Eur. J. Pediatr. 2008, 167, 839–849. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Bratcher, P.E.; Rowe, S.M.; Reeves, G.; Roberts, T.; Szul, T.; Harris, W.T.; Tirouvanziam, R.; Gaggar, A. Alterations in blood leukocytes of G551D-bearing cystic fibrosis patients undergoing treatment with ivacaftor. J. Cyst. Fibros. 2016, 15, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.P.; Valentine, V.G.; Wang, G. CFTR targeting during activation of human neutrophils. J. Leukoc. Biol. 2016, 100, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, H. CFTR Modulator Therapy for Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Klingel, M.; Black, P.; Powers, M.R.; Grasemann, H.; Solomon, M.; Sagel, S.D.; Donaldson, S.H.; Rowe, S.M.; Rosenfeld, M. Changes in Lung Clearance Index in Preschool-aged Patients with Cystic Fibrosis Treated with Ivacaftor (GOAL): A Clinical Trial. Am. J. Respir. Crit. Care Med. 2018, 198, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Skilton, M.; Krishan, A.; Patel, S.; Sinha, I.P.; Southern, K.W. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database Syst. Rev. 2019, 1, CD009841. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Sinha, I.P.; Dwan, K.; Echevarria, C.; Schechter, M.; Southern, K.W. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database Syst. Rev. 2015, CD009841. [Google Scholar] [CrossRef] [PubMed]
- Cebotaru, L.; Rapino, D.; Cebotaru, V.; Guggino, W.B. Correcting the cystic fibrosis disease mutant, A455E CFTR. PLoS ONE 2014, 9, e85183. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.T.; Yu, Y.C.; Hwang, T.C. Structural mechanisms for defective CFTR gating caused by the Q1412X mutation, a severe Class VI pathogenic mutation in cystic fibrosis. J. Physiol. 2019, 597, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Zhou-Suckow, Z.; Duerr, J.; Hagner, M.; Agrawal, R.; Mall, M.A. Airway mucus, inflammation and remodeling: Emerging links in the pathogenesis of chronic lung diseases. Cell Tissue Res. 2017, 367, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Araujo, F.G.; Novaes, F.C.; Santos, N.P.; Martins, V.C.; Souza, S.M.; Santos, S.E.; Ribeiro-dos-Santos, A.K. Prevalence of deltaF508, G551D, G542X, and R553X mutations among cystic fibrosis patients in the North of Brazil. Braz. J. Med. Biol. Res. 2005, 38, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.Z.; Haaf, J.B.; Leal, T.; Noel, S. Cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis: Current perspectives. Clin. Pharm. 2016, 8, 127–140. [Google Scholar] [CrossRef]
- Wu, H.X.; Zhu, M.; Xiong, X.F.; Wei, J.; Zhuo, K.Q.; Cheng, D.Y. Efficacy and Safety of CFTR Corrector and Potentiator Combination Therapy in Patients with Cystic Fibrosis for the F508del-CFTR Homozygous Mutation: A Systematic Review and Meta-analysis. Adv. Ther. 2019, 36, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Silvis, M.R.; Picciano, J.A.; Bertrand, C.; Weixel, K.; Bridges, R.J.; Bradbury, N.A. A mutation in the cystic fibrosis transmembrane conductance regulator generates a novel internalization sequence and enhances endocytic rates. J. Biol. Chem. 2003, 278, 11554–11560. [Google Scholar] [CrossRef] [PubMed]
- Boucher, R.C. Cystic fibrosis: A disease of vulnerability to airway surface dehydration. Trends Mol. Med. 2007, 13, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Collawn, J.F.; Lazrak, A.; Bebok, Z.; Matalon, S. The CFTR and ENaC debate: How important is ENaC in CF lung disease? Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L1141–L1146. [Google Scholar] [CrossRef] [PubMed]
- Althaus, M. ENaC inhibitors and airway re-hydration in cystic fibrosis: State of the art. Curr. Mol. Pharm. 2013, 6, 3–12. [Google Scholar] [CrossRef]
- Hobbs, C.A.; Da Tan, C.; Tarran, R. Does epithelial sodium channel hyperactivity contribute to cystic fibrosis lung disease? J. Physiol. 2013, 591, 4377–4387. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.J.; Tarran, R. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis lung disease. Expert Opin. Targets 2018, 22, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Duerr, J.; Johannesson, B.; Schubert, S.C.; Treis, D.; Harm, M.; Graeber, S.Y.; Dalpke, A.; Schultz, C.; Mall, M.A. The ENaC-overexpressing mouse as a model of cystic fibrosis lung disease. J. Cyst. Fibros. 2011, 10 (Suppl. 2), S172–S182. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Galietta, L. New pulmonary therapies directed at targets other than CFTR. Cold Spring Harb. Perspect. Med. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Boucher, R.C. Evidence for airway surface dehydration as the initiating event in CF airway disease. J. Intern. Med. 2007, 261, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, A.W. New horizons in the treatment of cystic fibrosis. Br. J. Pharm. 2011, 163, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Salomon, J.J.; Leitz, D.; Feigenbutz, D.; Korsch, L.; Lisewski, I.; Schrimpf, K.; Millar-Buchner, P.; Mall, M.A.; Frings, S.; et al. Expression and function of Anoctamin 1/TMEM16A calcium-activated chloride channels in airways of in vivo mouse models for cystic fibrosis research. Pflug. Arch. 2018, 470, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Wang, Y.C.; Chen, C.S.; Chin, W.C. Functionalized positive nanoparticles reduce mucin swelling and dispersion. PLoS ONE 2010, 5, e15434. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Hollinger, M.; Lachowicz-Scroggins, M.E.; Kerr, S.C.; Dunican, E.M.; Daniel, B.M.; Ghosh, S.; Erzurum, S.C.; Willard, B.; Hazen, S.L.; et al. Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci. Transl. Med. 2015, 7, 276ra227. [Google Scholar] [CrossRef] [PubMed]
- Ostedgaard, L.S.; Moninger, T.O.; McMenimen, J.D.; Sawin, N.M.; Parker, C.P.; Thornell, I.M.; Powers, L.S.; Gansemer, N.D.; Bouzek, D.C.; Cook, D.P.; et al. Gel-forming mucins form distinct morphologic structures in airways. Proc. Natl. Acad. Sci. USA 2017, 114, 6842–6847. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Paul, K.; van Koningsbruggen, S.; Breitenstein, S.; Rietschel, E.; Nikolaizik, W. DNA concentrations in BAL fluid of cystic fibrosis patients with early lung disease: Influence of treatment with dornase alpha. Pediatr. Pulmonol. 2005, 39, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Bayes, H.K.; Bicknell, S.; MacGregor, G.; Evans, T.J. T helper cell subsets specific for Pseudomonas aeruginosa in healthy individuals and patients with cystic fibrosis. PLoS ONE 2014, 9, e90263. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Bossard, F.; Goepp, J.; Verkman, A.S.; Galietta, L.J.; Hanrahan, J.W.; Lukacs, G.L. Proinflammatory cytokine secretion is suppressed by TMEM16A or CFTR channel activity in human cystic fibrosis bronchial epithelia. Mol. Biol. Cell 2012, 23, 4188–4202. [Google Scholar] [CrossRef] [PubMed]
- Paats, M.S.; Bergen, I.M.; Bakker, M.; Hoek, R.A.; Nietzman-Lammering, K.J.; Hoogsteden, H.C.; Hendriks, R.W.; van der Eerden, M.M. Cytokines in nasal lavages and plasma and their correlation with clinical parameters in cystic fibrosis. J. Cyst. Fibros. 2013, 12, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Regamey, N.; Brown, S.; Bush, A.; Lloyd, C.M.; Davies, J.C. The Th17 pathway in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2011, 184, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Oz, H.H.; Zhou, B.; Voss, P.; Carevic, M.; Schroth, C.; Frey, N.; Rieber, N.; Hector, A.; Hartl, D. Pseudomonas aeruginosa Airway Infection Recruits and Modulates Neutrophilic Myeloid-Derived Suppressor Cells. Front. Cell Infect. Microbiol. 2016, 6, 167. [Google Scholar] [CrossRef] [PubMed]
- Damlund, D.S.; Christophersen, L.; Jensen, P.O.; Alhede, M.; Hoiby, N.; Moser, C. Activation of pulmonary and lymph node dendritic cells during chronic Pseudomonas aeruginosa lung infection in mice. APMIS 2016, 124, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, R.; Gagnon, S.; Sweezey, N.B. Intrinsic predisposition of naive cystic fibrosis T cells to differentiate towards a Th17 phenotype. Respir. Res. 2013, 14, 138. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, R.; Gagnon, S.; Sweezey, N.B. T cell unresponsiveness in a pediatric cystic fibrosis patient: A case report. Allergy Asthma Clin. Immunol. 2014, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Ingersoll, S.A.; Laval, J.; Forrest, O.A.; Preininger, M.; Brown, M.R.; Arafat, D.; Gibson, G.; Tangpricha, V.; Tirouvanziam, R. Mature cystic fibrosis airway neutrophils suppress T cell function: Evidence for a role of arginase 1 but not programmed death-ligand 1. J. Immunol. 2015, 194, 5520–5528. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, L.; Schelstraete, P.; Van Simaey, L.; Van Braeckel, E.; Willekens, J.; Van Daele, S.; De Baets, F.; Vaneechoutte, M. Establishing the diagnosis of chronic colonization with Pseudomonas aeruginosa of cystic fibrosis patients: Comparison of the European consensus criteria with genotyping of P. aeruginosa isolates. J. Cyst. Fibros. 2018, 17, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, A.M. Chronic Pseudomonas aeruginosa airway colonization in cystic fibrosis patients: Prevention concepts. Internist 2017, 58, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Murray, T.S.; Egan, M.; Kazmierczak, B.I. Pseudomonas aeruginosa chronic colonization in cystic fibrosis patients. Curr. Opin. Pediatr. 2007, 19, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.; Gibson, R.L.; McNamara, S.; Emerson, J.; Burns, J.L.; Castile, R.; Hiatt, P.; McCoy, K.; Wilson, C.B.; Inglis, A.; et al. Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr. Pulmonol. 2001, 32, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Regamey, N.; Jeffery, P.K.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling and its relationship to inflammation in cystic fibrosis. Thorax 2011, 66, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Lammertyn, E.J.; Vandermeulen, E.; Bellon, H.; Everaerts, S.; Verleden, S.E.; Van Den Eynde, K.; Bracke, K.R.; Brusselle, G.G.; Goeminne, P.C.; Verbeken, E.K.; et al. End-stage cystic fibrosis lung disease is characterised by a diverse inflammatory pattern: An immunohistochemical analysis. Respir. Res. 2017, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H. Infection and inflammation in cystic fibrosis: A short review. J. Cyst. Fibros. 2005, 4 (Suppl. 2), 3–5. [Google Scholar] [CrossRef]
- Cohen-Cymberknoh, M.; Kerem, E.; Ferkol, T.; Elizur, A. Airway inflammation in cystic fibrosis: Molecular mechanisms and clinical implications. Thorax 2013, 68, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Sly, P.D.; Brennan, S.; Gangell, C.; de Klerk, N.; Murray, C.; Mott, L.; Stick, S.M.; Robinson, P.J.; Robertson, C.F.; Ranganathan, S.C.; et al. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am. J. Respir Crit Care Med. 2009, 180, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.L.; Logie, K.M.; Parsons, F.; Schulzke, S.M.; Nolan, G.; Murray, C.; Ranganathan, S.; Robinson, P.; Sly, P.D.; Stick, S.M.; et al. Air trapping on chest CT is associated with worse ventilation distribution in infants with cystic fibrosis diagnosed following newborn screening. PLoS ONE 2011, 6, e23932. [Google Scholar] [CrossRef] [PubMed]
- Elizur, A.; Cannon, C.L.; Ferkol, T.W. Airway inflammation in cystic fibrosis. Chest 2008, 133, 489–495. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, K.; Bradley, J.M.; Reid, A.; Downey, D.G.; Rendall, J.; McCaughan, J.; Moore, J.E.; Tunney, M.M.; Elborn, J.S. Airway infection, systemic inflammation and lung clearance index in children and adults with cystic fibrosis. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef] [PubMed]
- Adam, D.; Roux-Delrieu, J.; Luczka, E.; Bonnomet, A.; Lesage, J.; Merol, J.C.; Polette, M.; Abely, M.; Coraux, C. Cystic fibrosis airway epithelium remodelling: Involvement of inflammation. J. Pathol. 2015, 235, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Cowland, J.B.; Borregaard, N. Granulopoiesis and granules of human neutrophils. Immunol Rev. 2016, 273, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Hoffman, H.M.; Kubes, P.; Cassatella, M.A.; Zychlinsky, A.; Hedrick, C.C.; Catz, S.D. Neutrophils: New insights and open questions. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol 2010, 31, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Farahvash, A.; Douda, D.N.; Licht, J.C.; Grasemann, H.; Sweezey, N.; Palaniyar, N. JNK Activation Turns on LPS- and Gram-Negative Bacteria-Induced NADPH Oxidase-Dependent Suicidal NETosis. Sci. Rep. 2017, 7, 3409. [Google Scholar] [CrossRef] [PubMed]
- Carestia, A.; Kaufman, T.; Rivadeneyra, L.; Landoni, V.I.; Pozner, R.G.; Negrotto, S.; D’Atri, L.P.; Gomez, R.M.; Schattner, M. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J. Leukoc. Biol. 2016, 99, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Keshari, R.S.; Jyoti, A.; Dubey, M.; Kothari, N.; Kohli, M.; Bogra, J.; Barthwal, M.K.; Dikshit, M. Cytokines induced neutrophil extracellular traps formation: Implication for the inflammatory disease condition. PLoS ONE 2012, 7, e48111. [Google Scholar] [CrossRef] [PubMed]
- Sabbione, F.; Keitelman, I.A.; Iula, L.; Ferrero, M.; Giordano, M.N.; Baldi, P.; Rumbo, M.; Jancic, C.; Trevani, A.S. Neutrophil Extracellular Traps Stimulate Proinflammatory Responses in Human Airway Epithelial Cells. J. Innate Immun. 2017, 9, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Kasama, T.; Miwa, Y.; Isozaki, T.; Odai, T.; Adachi, M.; Kunkel, S.L. Neutrophil-derived cytokines: Potential therapeutic targets in inflammation. Curr Drug Targets Inflamm. Allergy 2005, 4, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Feng, C.; Zhang, X.; Lu, J.; Zhao, Y. The Diverse Biological Functions of Neutrophils, Beyond the Defense Against Infections. Inflammation 2017, 40, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Zychlinsky, A. Beneficial suicide: Why neutrophils die to make NETs. Nat. Rev. Microbiol. 2007, 5, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Suratt, B.T.; Petty, J.M.; Young, S.K.; Malcolm, K.C.; Lieber, J.G.; Nick, J.A.; Gonzalo, J.A.; Henson, P.M.; Worthen, G.S. Role of the CXCR4/SDF-1 chemokine axis in circulating neutrophil homeostasis. Blood 2004, 104, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Lakschevitz, F.S.; Hassanpour, S.; Rubin, A.; Fine, N.; Sun, C.; Glogauer, M. Identification of neutrophil surface marker changes in health and inflammation using high-throughput screening flow cytometry. Exp. Cell Res. 2016, 342, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Invest. 2010, 120, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.R.; Heath, P.R.; Catto, J.W.; Whyte, M.K.; Milo, M.; Renshaw, S.A. Regulation of neutrophil senescence by microRNAs. PLoS ONE 2011, 6, e15810. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.L.; Soehnlein, O.; Viola, J.R. Organ-Specific Mechanisms of Transendothelial Neutrophil Migration in the Lung, Liver, Kidney, and Aorta. Front. Immunol. 2018, 9, 2739. [Google Scholar] [CrossRef] [PubMed]
- Prame Kumar, K.; Nicholls, A.J.; Wong, C.H.Y. Partners in crime: Neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Makam, M.; Diaz, D.; Laval, J.; Gernez, Y.; Conrad, C.K.; Dunn, C.E.; Davies, Z.A.; Moss, R.B.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Activation of critical, host-induced, metabolic and stress pathways marks neutrophil entry into cystic fibrosis lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 5779–5783. [Google Scholar] [CrossRef] [PubMed]
- Carevic, M.; Singh, A.; Rieber, N.; Eickmeier, O.; Griese, M.; Hector, A.; Hartl, D. CXCR4+ granulocytes reflect fungal cystic fibrosis lung disease. Eur. Respir. J. 2015, 46, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, K.; Painter, R.G.; Aiken, M.; Reiser, J.; Stanton, B.A.; Nauseef, W.M.; Wang, G. Cystic fibrosis transmembrane conductance regulator recruitment to phagosomes in neutrophils. J. Innate Immun. 2013, 5, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.G.; Valentine, V.G.; Lanson, N.A., Jr.; Leidal, K.; Zhang, Q.; Lombard, G.; Thompson, C.; Viswanathan, A.; Nauseef, W.M.; Wang, G.; et al. CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry 2006, 45, 10260–10269. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.L.; Hampton, M.B.; Senthilmohan, R.; Winterbourn, C.C.; Kettle, A.J. Chlorination of bacterial and neutrophil proteins during phagocytosis and killing of Staphylococcus aureus. J. Biol. Chem. 2002, 277, 9757–9762. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.D.; Hardisty, G.; Regan, K.H.; Smith, M.; Robb, C.T.; Duffin, R.; Mackellar, A.; Felton, J.M.; Paemka, L.; McCullagh, B.N.; et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax 2018, 73, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Hayes, E.; Pohl, K.; McElvaney, N.G.; Reeves, E.P. The cystic fibrosis neutrophil: A specialized yet potentially defective cell. Arch. Immunol. Exp. 2011, 59, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Houston, N.; Stewart, N.; Smith, D.S.; Bell, S.C.; Champion, A.C.; Reid, D.W. Sputum neutrophils in cystic fibrosis patients display a reduced respiratory burst. J. Cyst. Fibros. 2013, 12, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Cifani, N.; Pompili, B.; Anile, M.; Patella, M.; Diso, D.; Venuta, F.; Cimino, G.; Quattrucci, S.; Di Domenico, E.G.; Ascenzioni, F.; et al. Reactive-oxygen-species-mediated P. aeruginosa killing is functional in human cystic fibrosis macrophages. PLoS ONE 2013, 8, e71717. [Google Scholar] [CrossRef] [PubMed]
- Yonker, L.M.; Cigana, C.; Hurley, B.P.; Bragonzi, A. Host-pathogen interplay in the respiratory environment of cystic fibrosis. J. Cyst. Fibros. 2015, 14, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Bedi, P.; Davidson, D.J.; McHugh, B.J.; Rossi, A.G.; Hill, A.T. Blood Neutrophils Are Reprogrammed in Bronchiectasis. Am. J. Respir. Crit. Care Med. 2018, 198, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Novakowski, K.E.; Loukov, D.; Chawla, V.; Bowdish, D.M. Bacterial Binding, Phagocytosis, and Killing: Measurements Using Colony Forming Units. Methods Mol. Biol. 2017, 1519, 297–309. [Google Scholar] [CrossRef]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A. Priming of the neutrophil NADPH oxidase activation: Role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin. Immunopathol. 2008, 30, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Ralhan, A.; Laval, J.; Lelis, F.; Ballbach, M.; Grund, C.; Hector, A.; Hartl, D. Current Concepts and Controversies in Innate Immunity of Cystic Fibrosis Lung Disease. J. Innate Immun. 2016, 8, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; Gaggar, A.; Bruscia, E.; Hector, A.; Marcos, V.; Jung, A.; Greene, C.; McElvaney, G.; Mall, M.; Doring, G. Innate immunity in cystic fibrosis lung disease. J. Cyst Fibros. 2012, 11, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Marin-Esteban, V.; Turbica, I.; Dufour, G.; Semiramoth, N.; Gleizes, A.; Gorges, R.; Beau, I.; Servin, A.L.; Lievin-Le Moal, V.; Sandre, C.; et al. Afa/Dr diffusely adhering Escherichia coli strain C1845 induces neutrophil extracellular traps that kill bacteria and damage human enterocyte-like cells. Infect. Immun. 2012, 80, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Kuijpers, T.W.; Wirawan, E.; Lippens, S.; Vandenabeele, P.; Vanden Berghe, T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011, 18, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N.; Sorensen, O.E.; Theilgaard-Monch, K. Neutrophil granules: A library of innate immunity proteins. Trends Immunol. 2007, 28, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Branzk, N.; Papayannopoulos, V. Molecular mechanisms regulating NETosis in infection and disease. Semin. Immunopathol. 2013, 35, 513–530. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Palaniyar, N. Transcriptional firing helps to drive NETosis. Sci. Rep. 2017, 7, 41749. [Google Scholar] [CrossRef] [PubMed]
- Douda, D.N.; Yip, L.; Khan, M.A.; Grasemann, H.; Palaniyar, N. Akt is essential to induce NADPH-dependent NETosis and to switch the neutrophil death to apoptosis. Blood 2014, 123, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Chicca, I.J.; Milward, M.R.; Chapple, I.L.C.; Griffiths, G.; Benson, R.; Dietrich, T.; Cooper, P.R. Development and Application of High-Content Biological Screening for Modulators of NET Production. Front. Immunol. 2018, 9, 337. [Google Scholar] [CrossRef] [PubMed]
- Hamam, H.J.; Khan, M.A.; Palaniyar, N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules 2019, 9, 32. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Wong, C.H.; Zemp, F.J.; McDonald, B.; Rahman, M.M.; Forsyth, P.A.; McFadden, G.; Kubes, P. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 2013, 13, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F. Recent advances in cystic fibrosis. Paediatr. Respir. Rev. 2008, 9, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Dubois, A.V.; Gauthier, A.; Brea, D.; Varaigne, F.; Diot, P.; Gauthier, F.; Attucci, S. Influence of DNA on the activities and inhibition of neutrophil serine proteases in cystic fibrosis sputum. Am. J. Respir. Cell Mol. Biol. 2012, 47, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.G.; Floyd, M.; Winn, M.; Moskowitz, S.M.; Rada, B. NET formation induced by Pseudomonas aeruginosa cystic fibrosis isolates measured as release of myeloperoxidase-DNA and neutrophil elastase-DNA complexes. Immunol. Lett. 2014, 160, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Aleman, S.R.; Campos-Garcia, L.; Palma-Nicolas, J.P.; Hernandez-Bello, R.; Gonzalez, G.M.; Sanchez-Gonzalez, A. Understanding the Entanglement: Neutrophil Extracellular Traps (NETs) in Cystic Fibrosis. Front. Cell Infect. Microbiol. 2017, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Rohm, M.; Grimm, M.J.; D’Auria, A.C.; Almyroudis, N.G.; Segal, B.H.; Urban, C.F. NADPH oxidase promotes neutrophil extracellular trap formation in pulmonary aspergillosis. Infect. Immun 2014, 82, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes-Costa, A.B.; Nascimento, M.T.; Froment, G.S.; Soares, R.P.; Morgado, F.N.; Conceicao-Silva, F.; Saraiva, E.M. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. USA 2009, 106, 6748–6753. [Google Scholar] [CrossRef] [PubMed]
- Drescher, B.; Bai, F. Neutrophil in viral infections, friend or foe? Virus Res. 2013, 171, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Neeli, I.; Radic, M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front. Immunol. 2013, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.; Dupuis, A.; Aaron, S.D.; Ratjen, F. The effect of chronic infection with Aspergillus fumigatus on lung function and hospitalization in patients with cystic fibrosis. Chest 2010, 137, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Rochael, N.C.; Guimaraes-Costa, A.B.; Nascimento, M.T.; DeSouza-Vieira, T.S.; Oliveira, M.P.; Garcia e Souza, L.F.; Oliveira, M.F.; Saraiva, E.M. Classical ROS-dependent and early/rapid ROS-independent release of Neutrophil Extracellular Traps triggered by Leishmania parasites. Sci. Rep. 2015, 5, 18302. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Vanden Berghe, T.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Azzouz, D.; Khan, M.A.; Sweezey, N.; Palaniyar, N. Two-in-one: UV radiation simultaneously induces apoptosis and NETosis. Cell Death Discov. 2018, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Pace-Asciak, C.; Al-Hassan, J.M.; Afzal, M.; Liu, Y.F.; Oommen, S.; Paul, B.M.; Nair, D.; Palaniyar, N. Furanoid F-Acid F6 Uniquely Induces NETosis Compared to C16 and C18 Fatty Acids in Human Neutrophils. Biomolecules 2018, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- Naffah de Souza, C.; Breda, L.C.D.; Khan, M.A.; de Almeida, S.R.; Camara, N.O.S.; Sweezey, N.; Palaniyar, N. Alkaline pH Promotes NADPH Oxidase-Independent Neutrophil Extracellular Trap Formation: A Matter of Mitochondrial Reactive Oxygen Species Generation and Citrullination and Cleavage of Histone. Front. Immunol. 2017, 8, 1849. [Google Scholar] [CrossRef] [PubMed]
- Chatfield, S.M.; Grebe, K.; Whitehead, L.W.; Rogers, K.L.; Nebl, T.; Murphy, J.M.; Wicks, I.P. Monosodium Urate Crystals Generate Nuclease-Resistant Neutrophil Extracellular Traps via a Distinct Molecular Pathway. J. Immunol. 2018, 200, 1802–1816. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Witsch, T.; Farley, K.; Gallant, M.; Remold-O’Donnell, E.; Wagner, D.D. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J. Thromb. Haemost. 2016, 14, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, C.; Palaniyar, N.; Otulakowski, G.; Khan, M.A.; Post, M.; Kuebler, W.M.; Tanswell, K.; Belcastro, R.; Masood, A.; Engelberts, D.; et al. Mechanical ventilation induces neutrophil extracellular trap formation. Anesthesiology 2015, 122, 864–875. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Favia, M.; Bobba, A.; Guerra, L.; Casavola, V.; Reshkin, S.J. Characterization of mitochondrial function in cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. J. Bioenerg. Biomembr. 2016, 48, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Robledo-Avila, F.H.; Ruiz-Rosado, J.D.; Brockman, K.L.; Kopp, B.T.; Amer, A.O.; McCoy, K.; Bakaletz, L.O.; Partida-Sanchez, S. Dysregulated Calcium Homeostasis in Cystic Fibrosis Neutrophils Leads to Deficient Antimicrobial Responses. J. Immunol. 2018, 201, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kinter, M.; Shank, S.; Cotton, C.; Kelley, T.J.; Ziady, A.G. Dysfunction of Nrf-2 in CF epithelia leads to excess intracellular H2O2 and inflammatory cytokine production. PLoS ONE 2008, 3, e3367. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.; Shan, Q.; D’Ortona, S.; Maurer, R.; Mitchell, R.; Olesen, H.; Thiel, S.; Huebner, J.; Gadjeva, M. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J. Innate Immun. 2014, 6, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Rada, B. Interactions between Neutrophils and Pseudomonas aeruginosa in Cystic Fibrosis. Pathogens 2017, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Coburn, B.; Wang, P.W.; Diaz Caballero, J.; Clark, S.T.; Brahma, V.; Donaldson, S.; Zhang, Y.; Surendra, A.; Gong, Y.; Elizabeth Tullis, D.; et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci. Rep. 2015, 5, 10241. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Gadjeva, M. Does NETosis Contribute to the Bacterial Pathoadaptation in Cystic Fibrosis? Front. Immunol. 2014, 5, 378. [Google Scholar] [CrossRef] [PubMed]
- Young, R.L.; Malcolm, K.C.; Kret, J.E.; Caceres, S.M.; Poch, K.R.; Nichols, D.P.; Taylor-Cousar, J.L.; Saavedra, M.T.; Randell, S.H.; Vasil, M.L.; et al. Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: Evidence of acquired resistance within the CF airway, independent of CFTR. PLoS ONE 2011, 6, e23637. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kitagawa, S. Regulation of neutrophil functions by proinflammatory cytokines. Int J. Hematol. 2006, 84, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Claudius, C.; Perner, A.; Moller, M.H. Nebulised dornase alfa versus placebo or hypertonic saline in adult critically ill patients: A systematic review of randomised clinical trials with meta-analysis and trial sequential analysis. Syst Rev. 2015, 4, 153. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Eckhart, L.; Mildner, M.; Jaeger, K.; Buchberger, M.; Ghannadan, M.; Tschachler, E. DNase1L2 degrades nuclear DNA during corneocyte formation. J. Invest. Derm. 2007, 127, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Alcazar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renne, C.; Renne, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.B.H.; Shteinberg, M.; Efrati, O.; Alon, S.; Dekel, E.; Amit, B.C.; Raul, C.; Brill-Almon, E.; Fux, L.; et al. WS01. 2 Phase II clinical trial results of alidornase alfa for the treatment of cystic fibrosis. J. Cyst. Fibros. 2017, 16 (Suppl. 1), S1. [Google Scholar] [CrossRef]
- Konstan, M.W.; Ratjen, F. Effect of dornase alfa on inflammation and lung function: Potential role in the early treatment of cystic fibrosis. J. Cyst Fibros. 2012, 11, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, D.; Shida, H.; Kusunoki, Y.; Miyoshi, A.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. The responses of macrophages in interaction with neutrophils that undergo NETosis. J. Autoimmun 2016, 67, 19–28. [Google Scholar] [CrossRef] [PubMed]
- McQuattie-Pimentel, A.C.; Budinger, G.R.S.; Ballinger, M.N. Monocyte-derived Alveolar Macrophages: The Dark Side of Lung Repair? Am. J. Respir Cell Mol. Biol 2018, 58, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Bruscia, E.M.; Bonfield, T.L. Cystic Fibrosis Lung Immunity: The Role of the Macrophage. J. Innate Immun 2016, 8, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Sorio, C.; Montresor, A.; Bolomini-Vittori, M.; Caldrer, S.; Rossi, B.; Dusi, S.; Angiari, S.; Johansson, J.E.; Vezzalini, M.; Leal, T.; et al. Mutations of Cystic Fibrosis Transmembrane Conductance Regulator Gene Cause a Monocyte-Selective Adhesion Deficiency. Am. J. Respir Crit Care Med. 2016, 193, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.M.; McElvaney, N.G. Proteases and antiproteases in chronic neutrophilic lung disease—Relevance to drug discovery. Br. J. Pharm. 2009, 158, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Invest. 2012, 122, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Tangsombatvisit, S.; Rosenberg, J.M.; Mandelbaum, G.; Gillespie, E.C.; Gozani, O.P.; Alizadeh, A.A.; Utz, P.J. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res. 2012, 14, R25. [Google Scholar] [CrossRef] [PubMed]
- Kovach, K.; Davis-Fields, M.; Irie, Y.; Jain, K.; Doorwar, S.; Vuong, K.; Dhamani, N.; Mohanty, K.; Touhami, A.; Gordon, V.D. Evolutionary adaptations of biofilms infecting cystic fibrosis lungs promote mechanical toughness by adjusting polysaccharide production. Npj Biofilms Microbiomes 2017, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.; Rumbaugh, K.P. Approaches to Dispersing Medical Biofilms. Microorganisms 2017, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, H.; Ratjen, F. Nitric oxide and L-arginine deficiency in cystic fibrosis. Curr. Pharm. Des. 2012, 18, 726–736. [Google Scholar] [CrossRef] [PubMed]
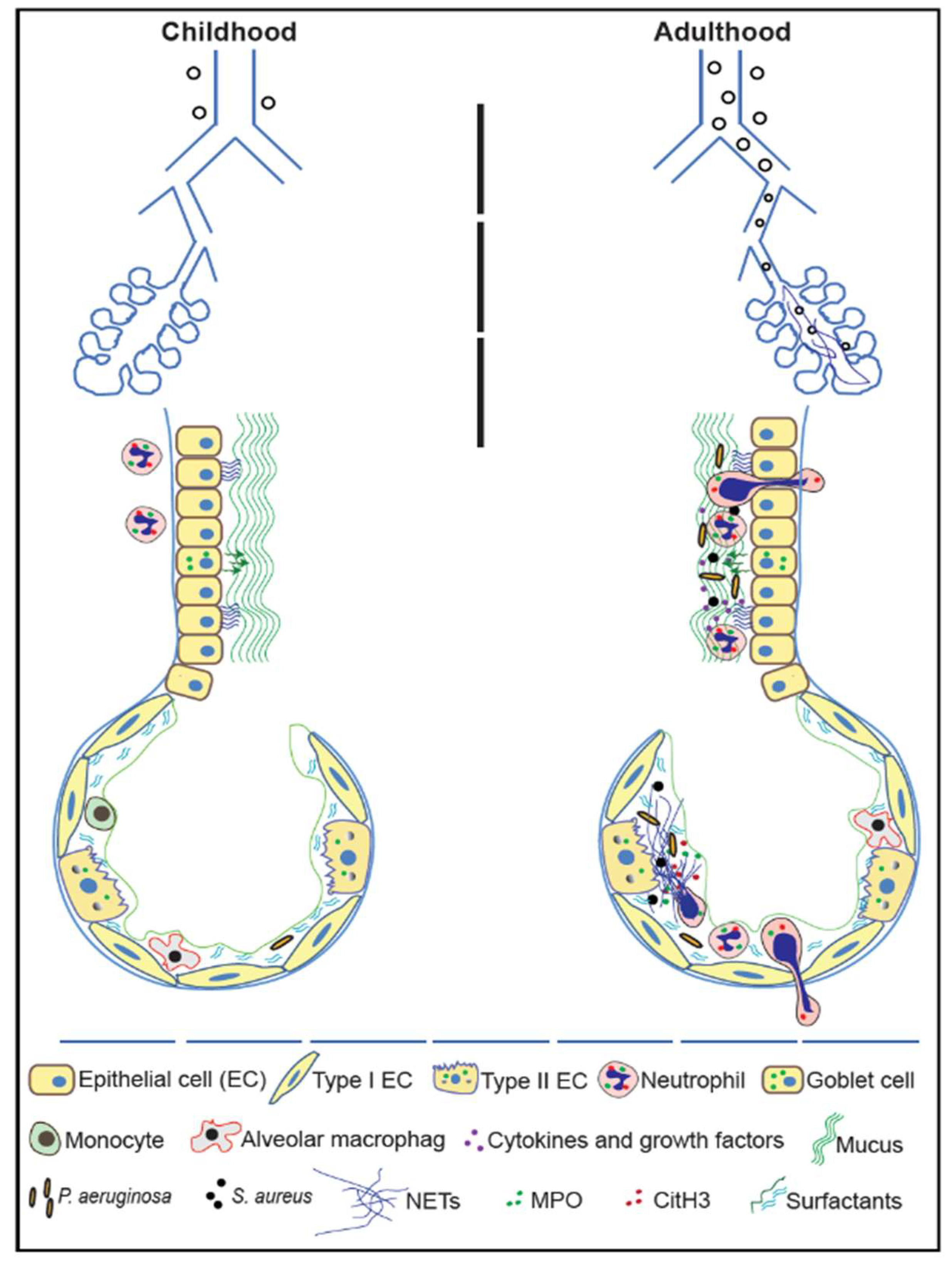

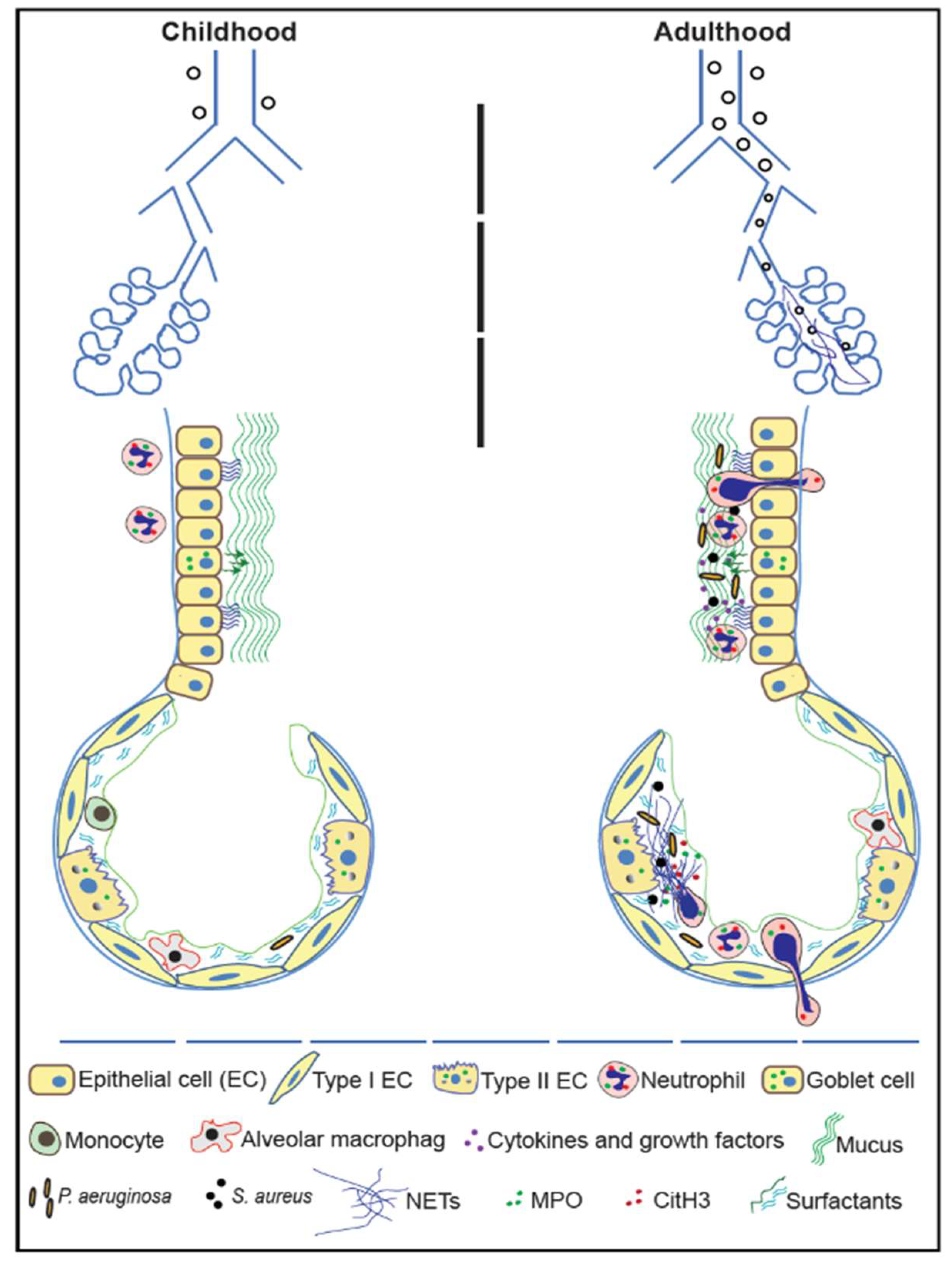{kind=link}
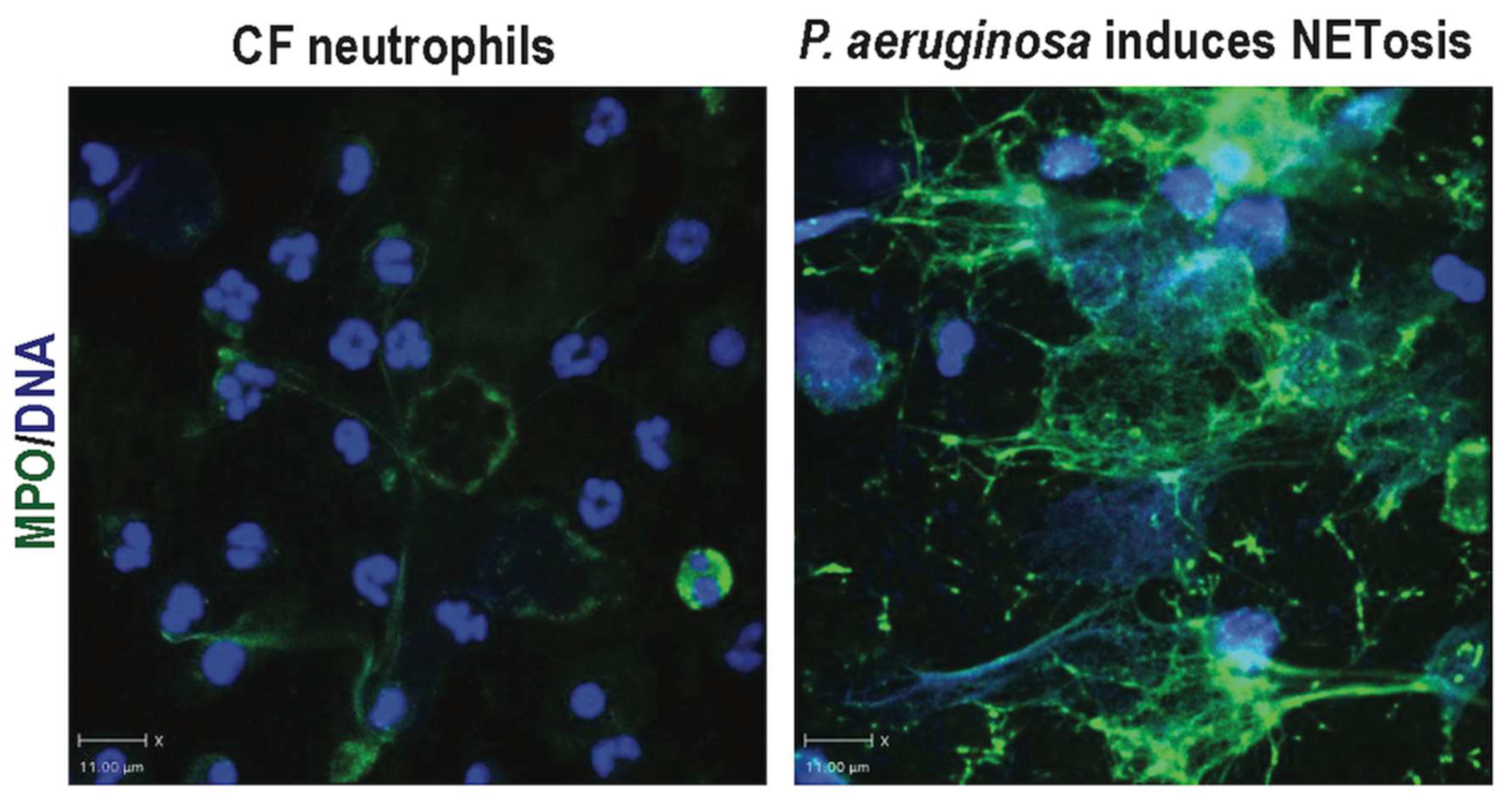
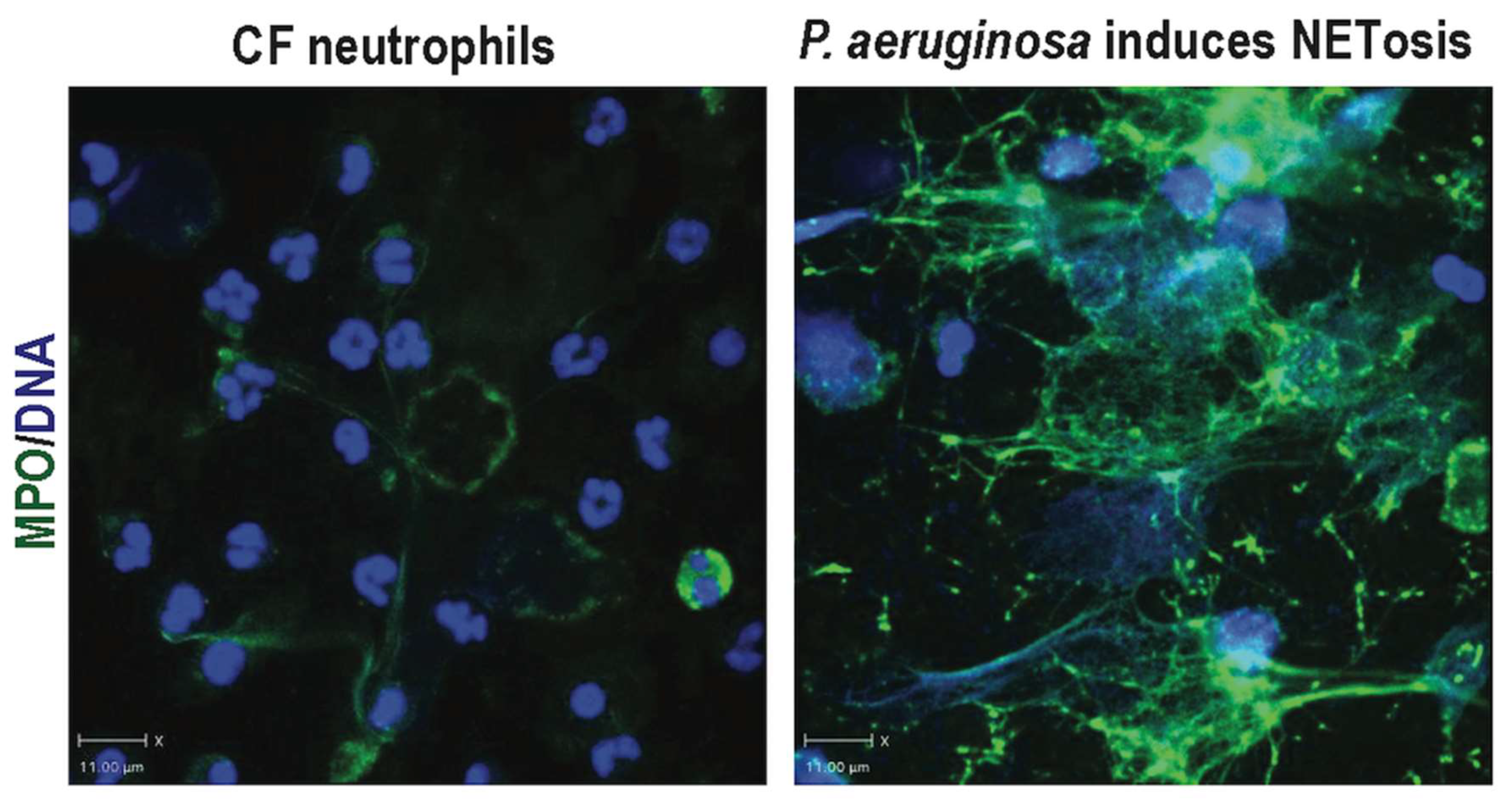{kind=link}
| Class | Molecular Mechanism | Mutation Examples |
|---|---|---|
| 1 | Non-sense mutation: premature stop codon → defective protein synthesis (no CFTR expression) | R553X, G542X [84] |
| 2 | Missense mutation: either (1) Misfolded CFTR protein and, or (2) not transported to the destination (or if so, only in residual amounts) | F508del (Most prevalent), N1303K [69,72] |
| 3 | Missense mutation (AA substitution): reduced of lack of CFTR opening in response to agonists → gating defect | G551D (Ivacaftor corrects), G1244E [69,74] |
| 4 | Missense mutation (AA substitution): restrict Cl− transport across the channel → conductance defect | R117H, R334W [85,86] |
| 5 | Splicing defect: improper processing of CFTR mRNA → less CFTR protein abundance on the cell surface but the proteins are normal | A455E (Least prevalent) [81] |
| 6 | CFTR is functional, but unstable due to rapid removal and degradation | N287Y [87] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.A.; Ali, Z.S.; Sweezey, N.; Grasemann, H.; Palaniyar, N. Progression of Cystic Fibrosis Lung Disease from Childhood to Adulthood: Neutrophils, Neutrophil Extracellular Trap (NET) Formation, and NET Degradation. Genes 2019, 10, 183. https://doi.org/10.3390/genes10030183
Khan MA, Ali ZS, Sweezey N, Grasemann H, Palaniyar N. Progression of Cystic Fibrosis Lung Disease from Childhood to Adulthood: Neutrophils, Neutrophil Extracellular Trap (NET) Formation, and NET Degradation. Genes. 2019; 10(3):183. https://doi.org/10.3390/genes10030183
Chicago/Turabian StyleKhan, Meraj A., Zubair Sabz Ali, Neil Sweezey, Hartmut Grasemann, and Nades Palaniyar. 2019. "Progression of Cystic Fibrosis Lung Disease from Childhood to Adulthood: Neutrophils, Neutrophil Extracellular Trap (NET) Formation, and NET Degradation" Genes 10, no. 3: 183. https://doi.org/10.3390/genes10030183
APA StyleKhan, M. A., Ali, Z. S., Sweezey, N., Grasemann, H., & Palaniyar, N. (2019). Progression of Cystic Fibrosis Lung Disease from Childhood to Adulthood: Neutrophils, Neutrophil Extracellular Trap (NET) Formation, and NET Degradation. Genes, 10(3), 183. https://doi.org/10.3390/genes10030183