Complete Genome Sequence Analysis and Characterization of Selected Iron Regulation Genes of Pasteurella Multocida Serotype A Strain PMTB2.1

Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strain and DNA Extraction
2.2. Genome Sequencing and Assembly
2.3. Genome Annotation and Genomic Organization of PMTB2.1
2.4. Genome Sequence Analysis
2.5. Comparative Genomics of PMTB2.1
2.6. Expression Profiling of Iron-Regulating Genes of PMTB2.1
2.7. Data Availability
3. Results
3.1. Genome Assembly
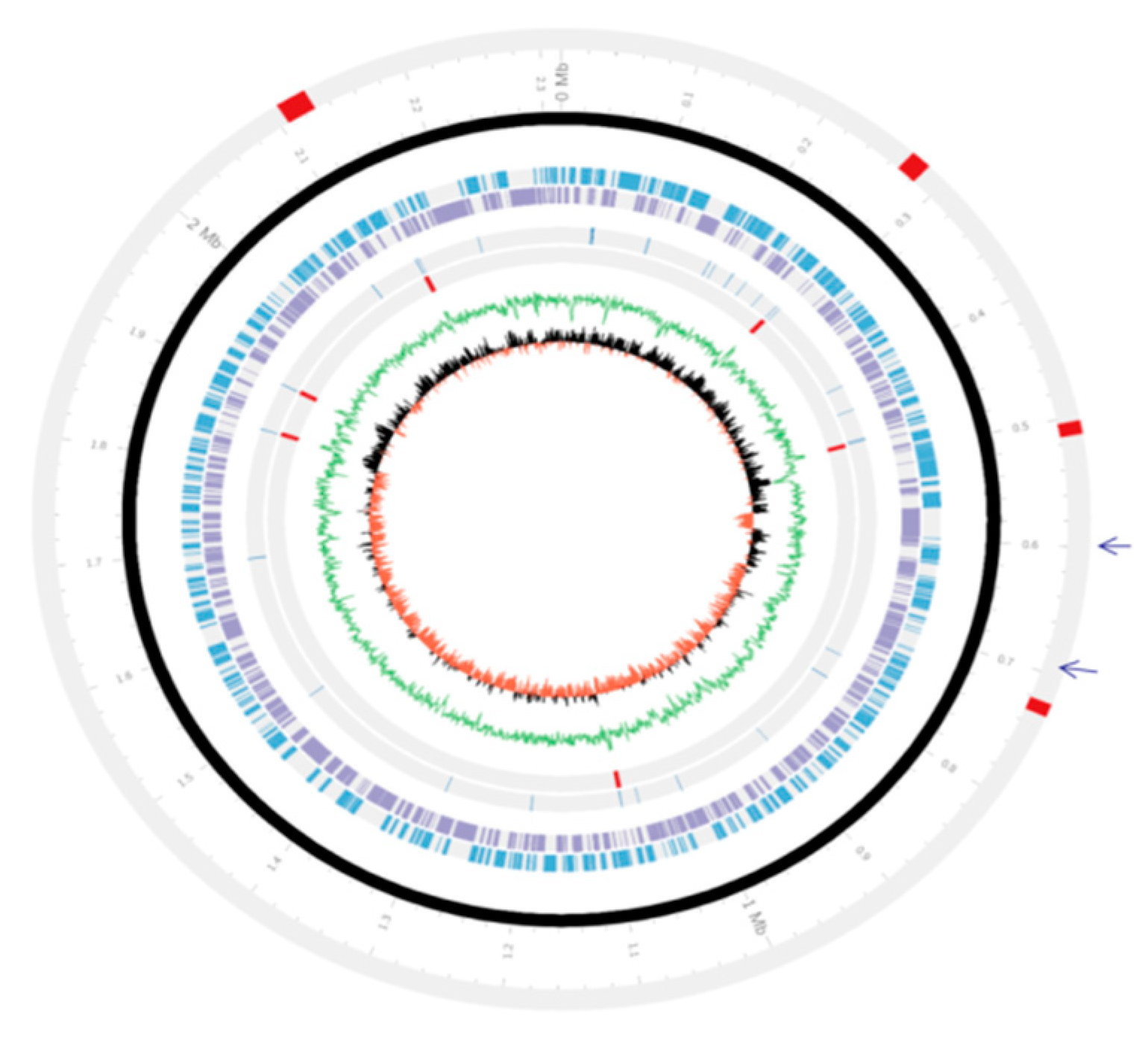
3.2. Genetic Organizations of the PMTB2.1 Complete Genome Sequence
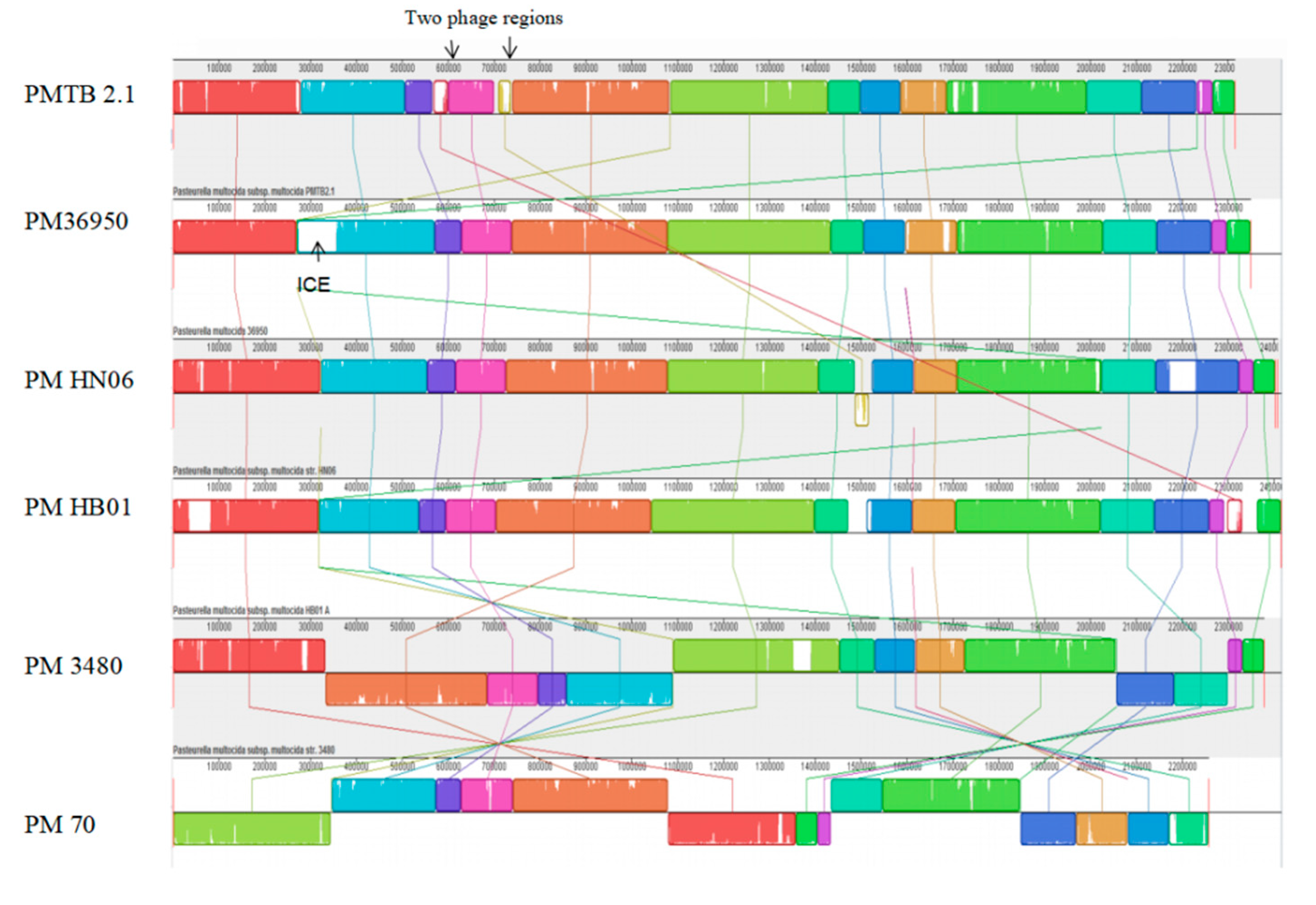
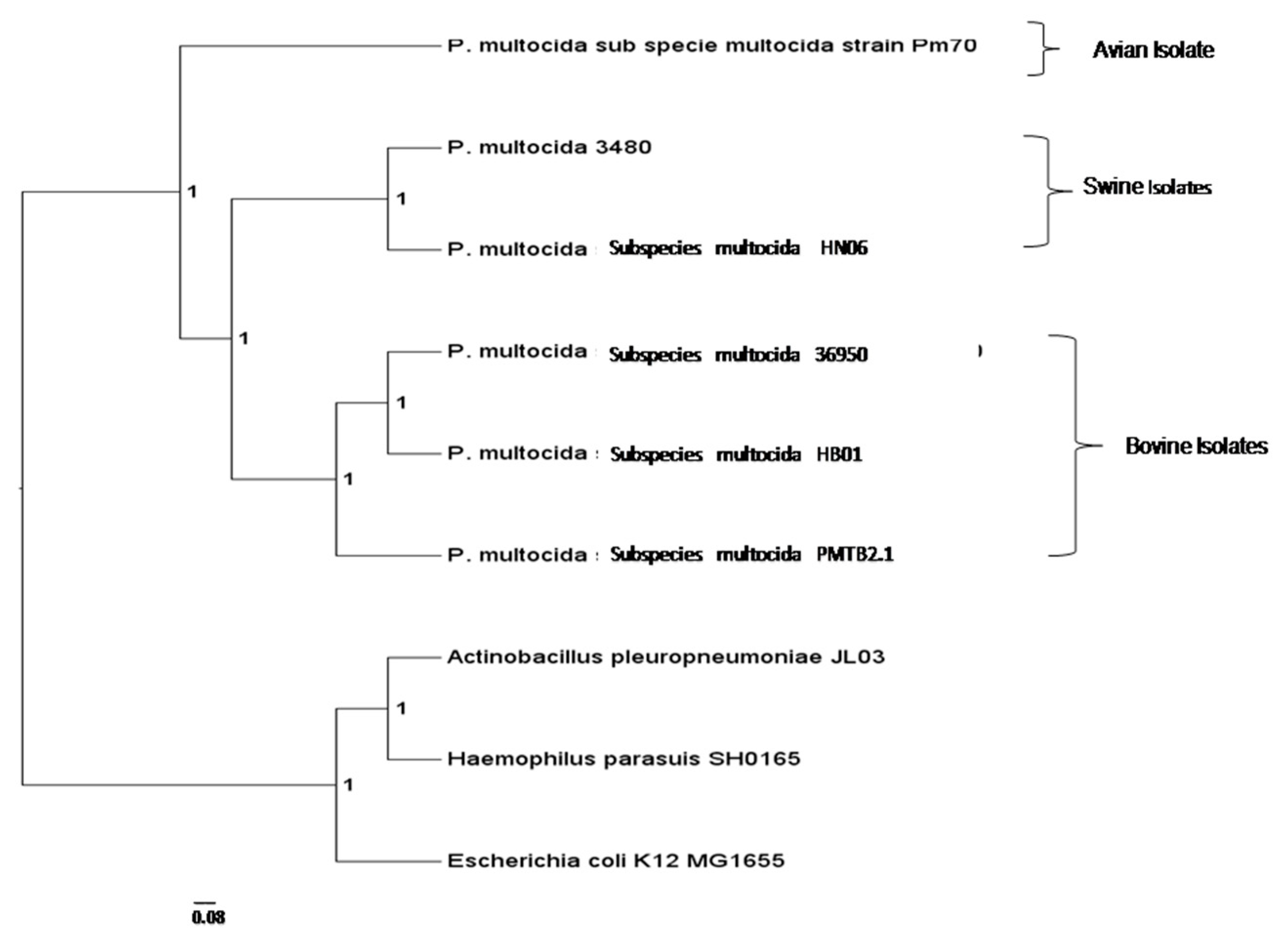
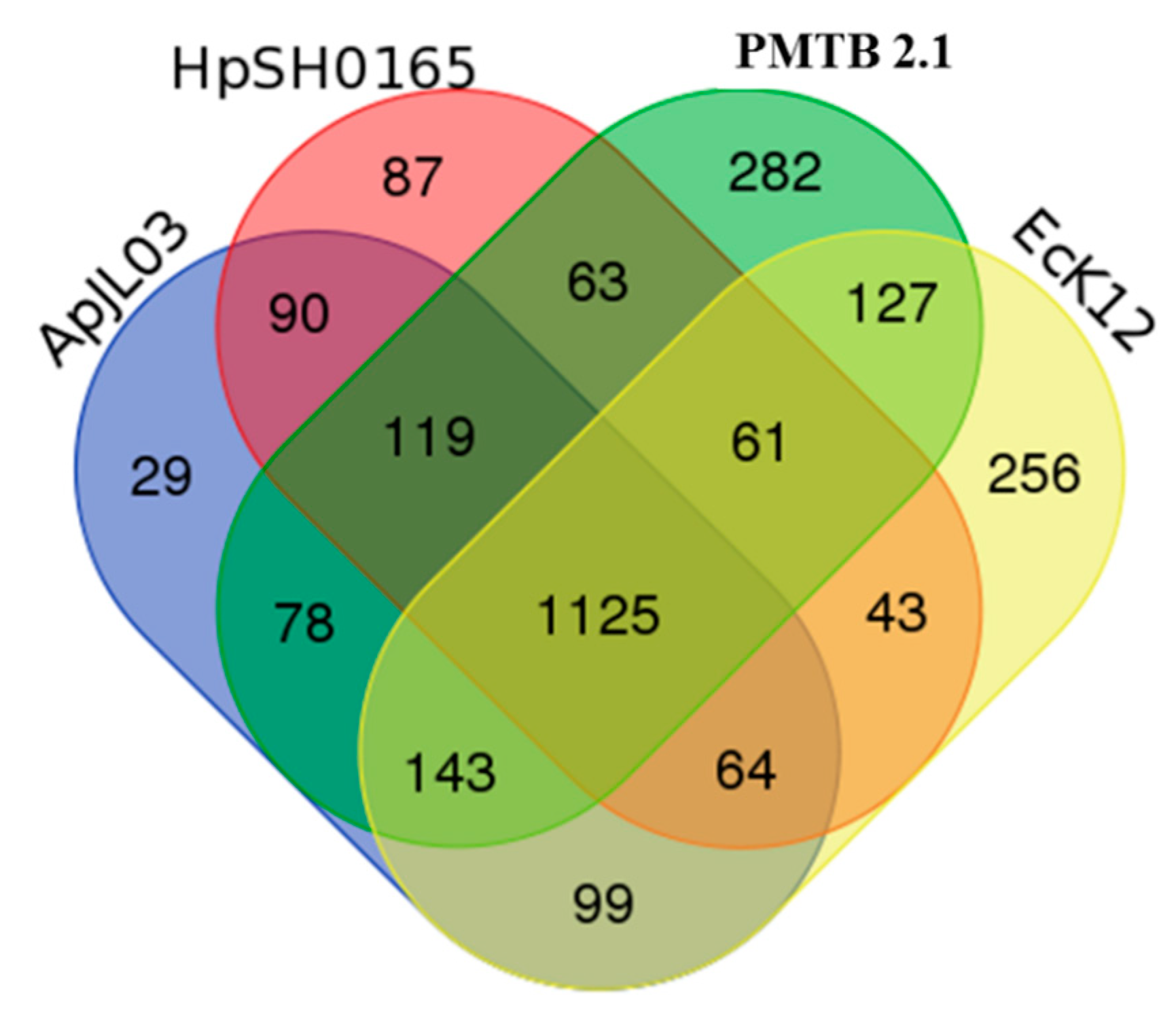
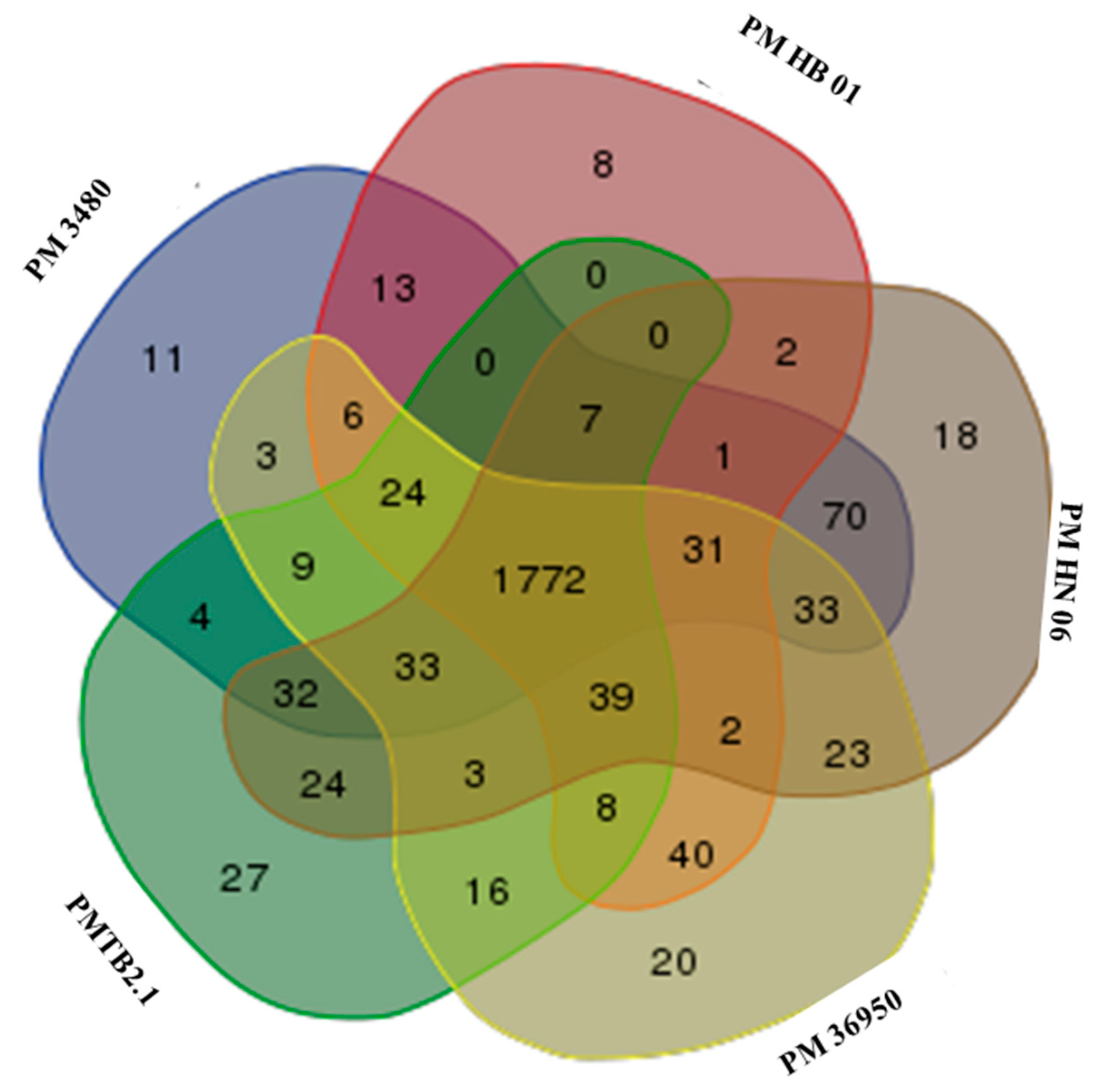
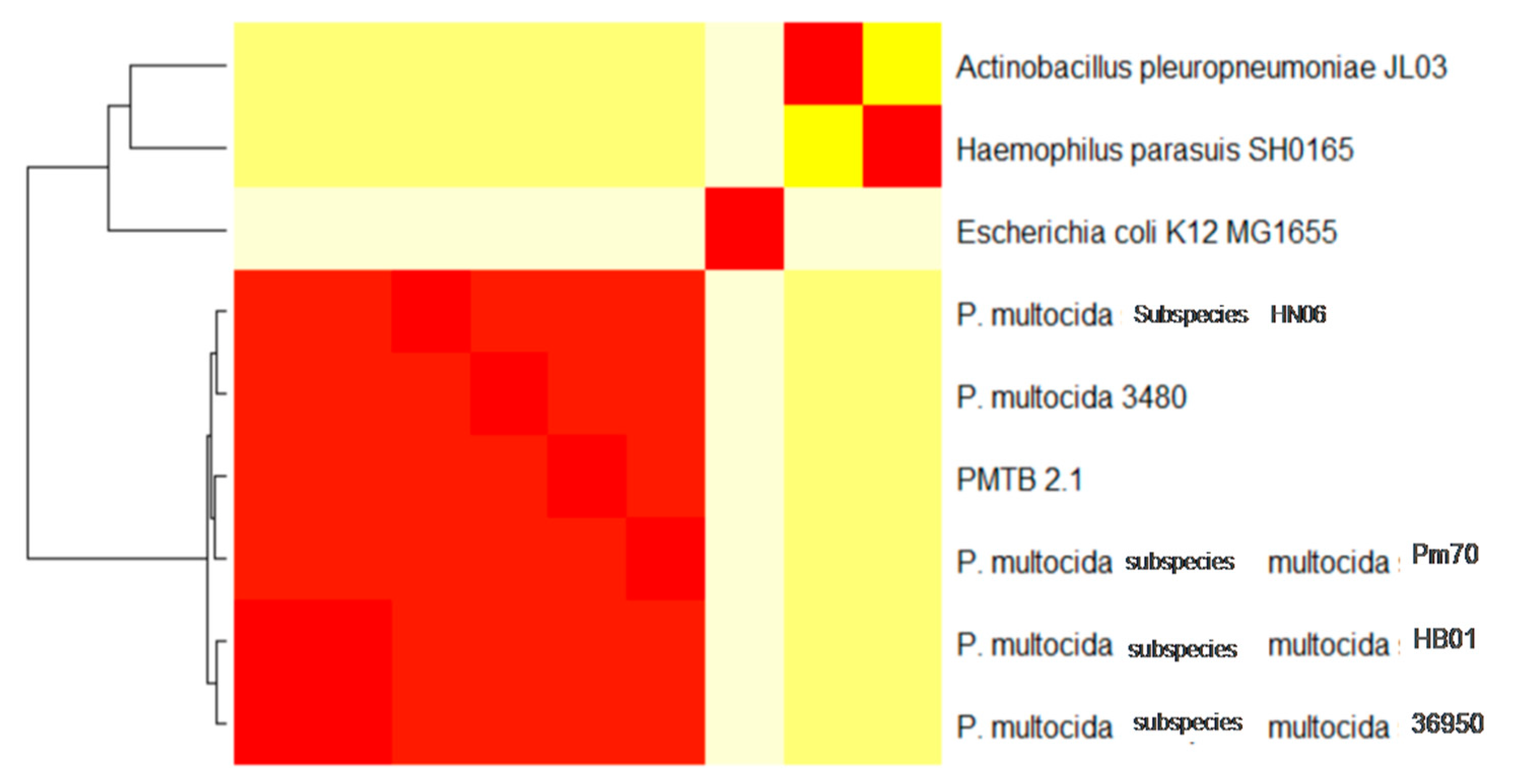
3.3. Comparative Genomic Analysis and Genotyping of PMTB2.1
3.4. Virulence Genes and Antibiotic Resistance Genes of PMTB2.1
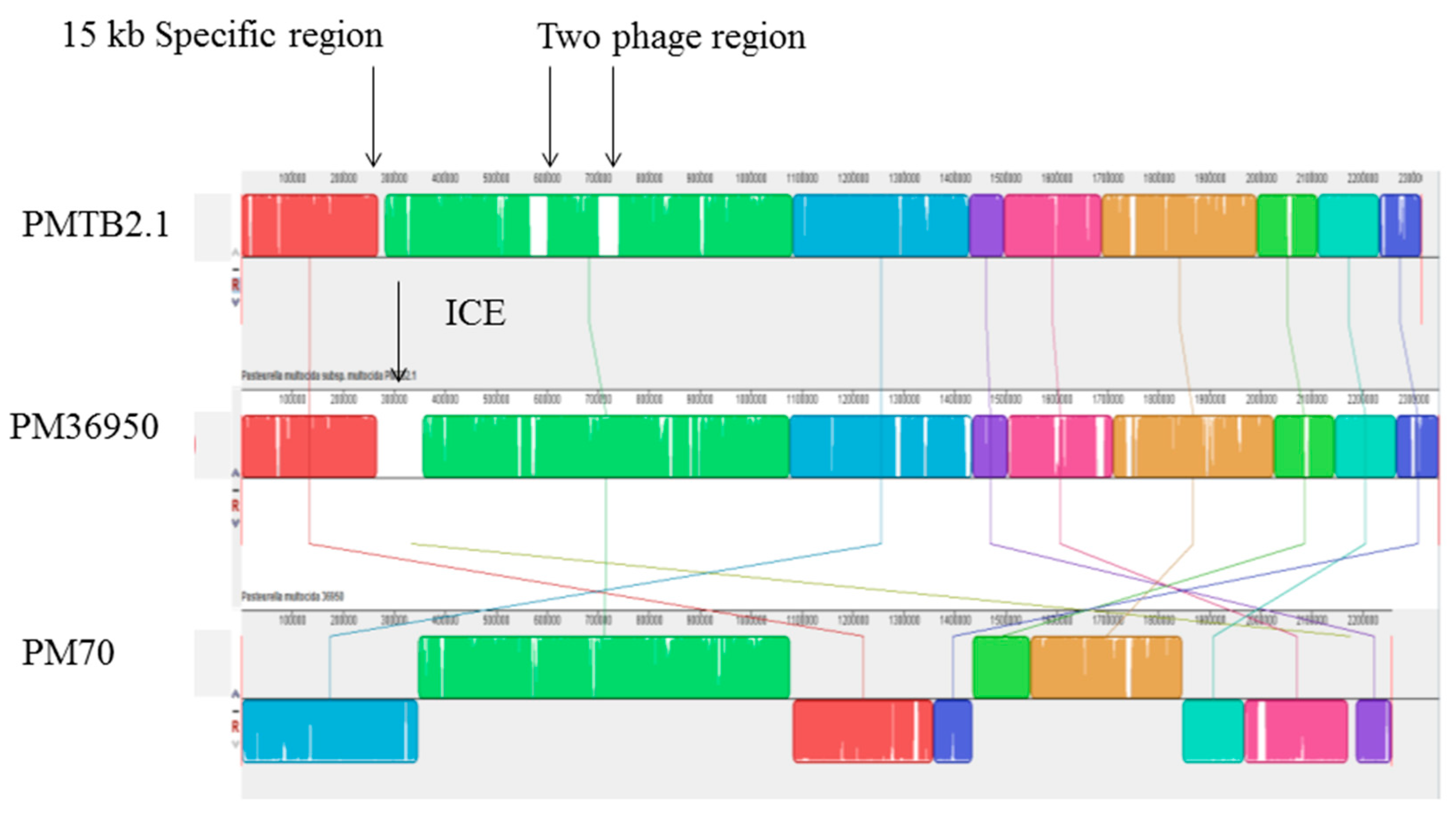
3.5. Phage and Plasmid Sequences of PMTB2.1
3.6. Iron Regulating Genes and Expression Profiling of Selected Iron-Regulating Genes in PMTB2.1
4. Discussion
4.1. Complete Genome Sequencing
4.2. Pathogenomics Analysis
4.3. Phylogenomics Analysis
4.4. Antibiotic Resistance Genes
4.5. Tad Locus Genes of PMTB2.1
4.6. The Capsular and LPS Genes of PMTB2.1
4.7. Bacteriophages in Pasteurella and Prophage Sequence in PMTB 2.1
4.8. Iron Regulating Genes and Expression Profiling of Selected Iron-Regulating Genes in PMTB2.1
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kuhnert, P.; Christensen, H. Pasteurellaceae: Biology, Genomics and Molecular Aspects; Academic Press: Caister, UK, 2008; pp. 34–39. [Google Scholar]
- Carter, G.R. A haemagglutination test for the identification of serological types. Am. J. Vet. Res. 1955, 16, 481–484. [Google Scholar] [PubMed]
- Heddleston, K.L.; Gallagher, J.E.; Rebers, P.A. Fowl cholera: Gel diffusion precipitin test for serotyping Pasteruella multocida from avian species. Avian Dis. 1972, 16, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.A.; Ho, M. Pasteurella multocida: From zoonosis to cellular microbiology. Clin. Microbiol. Rev. 2013, 26, 631–655. [Google Scholar] [CrossRef] [PubMed]
- Boyce, J.D.; Adler, B. The capsule is a virulence determinant in pathogenesis of Pasteurella multocida M1404 (B:2). Infect. Immun. 2000, 68, 3463–3468. [Google Scholar] [CrossRef]
- De Alwis, M.C.L. Hemorrhagic Septicemia, Aciar Monograph Series; ACIAR: Canberra, Australia, 1999. [Google Scholar]
- OIE. Haemorrhagic Septicaemia. In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals; OIE: Paris, France, 2012; pp. 1–13. Available online: www.oie.int/en/international-standard-setting/terrestrial-manual/ (accessed on 11 December 2014).
- Dabo, S.M.; Taylor, J.D.; Confer, A.W. Pasteurella multocida and bovine respiratory disease. Anim. Health Res. Rev. 2007, 8, 129–150. [Google Scholar] [CrossRef]
- Praveena, P.E.; Periasamy, S.; Kumar, A.A.; Singh, N. Pathology of experimental infection by Pasteurella multocida serotype A:1. Vet. Pathol. 2014, 51, 1109–1112. [Google Scholar] [CrossRef]
- Shivachandra, S.B.; Viswas, K.N.; Kumar, A.A. A review of hemorrhagic septicemia in cattle and buffalo. Anim. Health Res. Rev. 2011, 12, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Zamri-Saad, M.; Annas, S. Vaccination against hemorrhagic septicemia of bovines: A review. Pak. Vet. J. 2015, 36, 1–5. [Google Scholar] [CrossRef]
- May, B.J.; Zhang, Q.; Li, L.L.; Paustian, M.L.; Whittam, T.S.; Kapur, V. Complete genomic sequence of Pasteurella multocida, Pm70. Proc. Natl. Acad. Sci. USA 2001, 98, 3460–3465. [Google Scholar] [CrossRef]
- Michael, G.B.; Kadlec, K.; Sweeney, M.T.; Brzuszkiewicz, E.; Liesegang, H.; Daniel, R.; Murray, R.W.; Watts, J.L.; Schwarz, S. ICEPmu1, an integrative conjugative element (ICE) of Pasteurella multocida: Structure and transfer. J. Antimicrob. Chemother. 2012, 67, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Liang, W.; Liu, W.; Wu, B.; Tang, B.; Tan, C.; Zhou, R.; Chen, H. Genomic characterization of Pasteurella multocida HB01, a serotype A bovine isolate from China. Gene 2016, 581, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yang, M.; Xu, Z.; Zheng, H.; Liang, W.; Zhou, R.; Wu, B.; Chen, H. Complete genome sequence of Pasteurella multocida HN06, a toxigenic strain of serogroup D. J. Bacteriol. 2012, 194, 3292–3293. [Google Scholar] [CrossRef] [PubMed]
- Boyce, J.D.; Seemann, T.; Adler, B.; Harper, M. Pathogenomics of Pasteurella multocida. Curr. Top. Microbiol. Immunol. 2012, 361, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Paustian, M.L.; May, B.J.; Kapur, V. Transcriptional response of Pasteurella multocida to nutrient limitation. J. Bacteriol. 2002, 184, 3734–3739. [Google Scholar] [CrossRef] [PubMed]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 2012, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- Townsend, K.M.; Frost, A.J.; Lee, C.W.; John, M.; Dawkins, H.J.S.; Papadimitriou, J.M. Development of PCR assays for species- and type-specific identification of Pasteurella multocida isolates. J. Clin. Microbiol. 1998, 36, 1096–1100. [Google Scholar] [PubMed]
- Jabeen, S.; Yap, H.Y.; Jesse Abdullah, F.F.; Zakaria, Z.; Mat Isa, N.; Tan, Y.C.; Hoh, C.C.; Yee, W.Y.; Omar, A.R. Complete genome sequence of Pasteurella multocida serotype A strain PMTB2.1 isolated from buffaloes died of septicemia in Malaysia. Genome Announc. 2017, 5, e01190-17. [Google Scholar] [CrossRef] [PubMed]
- English, A.C.; Richards, S.; Han, Y.; Wang, M.; Vee, V.; Qu, J.; Qin, X.; Muzny, D.M.; Reid, J.G.; Worley, K.C.; et al. Mind the gap: Upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 2012, 7, e47768. [Google Scholar] [CrossRef]
- Besemer, J.; Borodovsky, M. GeneMark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Got, S.; Garcia-gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, P.; Zakrzewska-Czerwinska, J.; Zawilak, A.; Dudek, M.R.; Cebrat, S. Where does bacterial replication start? Rules for predicting the oriC region. Nucleic Acids Res. 2004, 32, 3781–3791. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, P.; Li, C.; Han, S.; Lopez-Baltazar, J.; Zhang, X.; Wang, X. Identification of lipoxygenase (LOX) genes from legumes and their responses in wild type and cultivated peanut upon Aspergillus flavus infection. Sci. Rep. 2016, 6, 35245. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, B.K.; Laird, M.R.; Shay, J.A.; Winsor, G.L.; Lo, R.; Nizam, F.; Pereira, S.K.; Waglechner, N.; McArthur, A.G.; Langille, M.G.I.; et al. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 2015, 43, 104–108. [Google Scholar] [CrossRef]
- Winnenburg, R.; Baldwin, T.K.; Urban, M.; Rawlings, C.; Kohler, J.; Hammond-Kosack, K.E. PHI-base: A new database for pathogen host interactions. Nucleic Acids Res. 2006, 34, D459–D464. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB: Hierarchical and refined dataset for big data analysis 10-years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Subaaharan, S.; Blackall, L.L.; Blackall, P.J. Development of a multi-locus sequence typing scheme for avian isolates of Pasteurella multocida. Vet. Microbiol. 2010, 141, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T.; Batzoglou, S.; Zhong, Y. Progressive mauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Creevey, C.J.; Muller, J.; Doerks, T.; Thompson, J.D.; Arendt, D.; Bork, P. Identifying single copy orthologs in metazoa. PLoS Comput. Biol. 2011, 7, e1002269. [Google Scholar] [CrossRef] [PubMed]
- Heydari, M.; Miclotte, G.; Demeester, P.; Van de peer, Y.; Fostier, J. Evaluation of the impact of Illumina error correction tools on de novo genome assembly. BMC Bioinformatics 2017, 18, 374. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Nolan, T.; Pfaff, M.W. Quantitative real-time RT-PCR—A perspective. J. Mol. Endocrinol. 2005, 34, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Figurski, D.H.; Fine, D.H.; Perez-Cheeks, B.A.; Grosso, V.W.; Karin, E.; Kram, J.H.; Xu, K.; Hedhli, J. Targeted mutagenesis in the study of the tight adherence (tad) locus of Aggregatibacter actinomycetemcomitans. In Genetic Manipulation of DNA and Protein—Examples from Current Research; Figurski, D., Ed.; InTech Publisher: London, UK, 2013; pp. 953–978. [Google Scholar] [CrossRef]
- Wootton, M. BSAC Methods for Antimicrobial Susceptibility Testing; Version 12; BSAC: Birmingham, UK, 2013. [Google Scholar]
- Moustafa, A.M.; Seemann, T.; Gladman, S.; Adler, B.; Harper, M.; Boyce, J.D.; Bennett, M.D. Comparative genomic analysis of Asian haemorrhagic septicaemia-associated strains of Pasteurella multocida identifies more than 90 haemorrhagic septicaemia- specific genes. PLoS ONE 2015, 10, e130296. [Google Scholar] [CrossRef] [PubMed]
- Habrun, B.; Kompes, G.; Cvetni, Ž.; Špi, S.; Beni, M.; Mitak, M. Antimicrobial sensitivity of Escherichia coli, Salmonella spp., Pasteurella multocida, Streptococcus suis and Actinobacillus pleuropneumoniae isolated from diagnostic samples from large pig breeding farms in Croatia. Vet. Arh. 2010, 80, 571–583. [Google Scholar]
- Sellyei, B.; Varga, Z.; Szentesi-Samu, K.; Kaszanyitzky, E.; Magyar, T. Antimicrobial susceptibility of Pasteurella multocida isolated from swine and poultry. Acta Vet. Hung. 2009, 57, 357–367. [Google Scholar] [CrossRef]
- Hansen, L.M.; McMurry, L.M.; Levy, S.B.; Hirsh, D.C. A new tetracycline resistance determinant, tet(H), from Pasteurella multocida specifying active efflux of tetracycline. Antimicrob. Agents Chemother. 1993, 37, 2699–2705. [Google Scholar] [CrossRef]
- Hansen, L.M.; Blanchard, P.C.; Hirsh, D.C. Distribution of tet(H) among Pasteurella isolates from the United States and Canada. Antimicrob. Agents Chemother. 1996, 40, 1558–1560. [Google Scholar] [CrossRef]
- Miranda, C.D.; Kehrenberg, C.; Ulep, C.; Schwarz, S.; Roberts, M.C. Diversity of tetracycline resistance genes in bacteria from Chilean salmon farms. Antimicrob. Agents Chemother. 2003, 47, 883–888. [Google Scholar] [CrossRef]
- Falagas, M.E.; Rafailidis, P.I.; Matthaiou, D.K. Resistance to polymyxins: Mechanisms, frequency and treatment options. Drug Resist. Updates 2010, 13, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Clock, S.A.; Planet, P.J.; Perez, B.A.; Figureurski, D.H. Outer membrane components of the Tad (tight adherence) secreton of Aggregatibacter actinomycetemcomitans. J. Bacteriol. 2008, 190, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Tomich, M.; Planet, P.J.; Figurski, D.H. The tad locus: Postcards from the widespread colonization island. Nat. Rev. Microbiol. 2007, 5, 363–375. [Google Scholar] [CrossRef]
- Kachlany, S.C.; Planet, P.J.; Desalle, R.; Fine, D.H.; Figurski, D.H.; Kaplan, J.B. flp-1, the first representative of a new pilin gene subfamily, is required for non-specific adherence of Actinobacillus actinomycetemcomitans. Mol. Microbiol. 2001, 40, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, M.K.; Kachlany, S.C.; Fine, D.H.; Figurski, D.H. Nonspecific adherence and fibril biogenesis by Actinobacillus actinomycetemcomitans: TadA protein is an ATPase. J. Bacteriol. 2001, 20, 5927–5936. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Zhang, Y.; Adler, B. The capsule biosynthetic locus of Pasteurella multocida A:1. FEMS Microbiol. Lett. 1998, 166, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Faez, F.J.A.; Lawan, A.; Abdinasir, Y.O.; Zunita, Z.; Rasedee, A. Clinicopathological responses of calves associated with infection of Pasteurella multocida type B and the bacterial lipopolysaccharide and outer membrane protein immunogens. Int. J. Anim. Vet. Adv. 2013, 5, 190–198. [Google Scholar]
- Xu, Z.; Yue, M.; Zhou, R.; Jin, Q.; Fan, Y.; Bei, W.; Chen, H. Genomic characterization of Haemophilus parasuis SH0165, a highly virulent strain of serovar 5 prevalent in China. PLoS ONE 2011, 6, e19631. [Google Scholar] [CrossRef] [PubMed]
- Opiyo, S.O.; Pardy, R.L.; Moriyama, H.; Moriyama, E.N. Evolution of the Kdo2-lipid A biosynthesis in bacteria. BMC Evol. Biol. 2010, 10, 362. [Google Scholar] [CrossRef] [PubMed]
- Campoy, S.; Aranda, J.; Alvarez, G.; Barbe, J.; Llagostera, M. Isolation and sequencing of a temperate transducing phage for Pasteurella multocida. Appl. Environ. Microbiol. 2006, 72, 3154–3160. [Google Scholar] [CrossRef] [PubMed]
- Pullinger, G.D.; Bevir, T.; Lax, A.J. The Pasteurella multocida toxin is encoded within a lysogenic bacteriophage. Mol. Microbiol. 2004, 51, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Jakhetia, R.; Verma, N.K. Identification and molecular characterisation of a novel Mu-like bacteriophage, SfMu, of Shigella flexneri. PLoS ONE 2015, 10, e0124053. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.; Boyce, J.D.; Adler, B. Pasteurella multocida pathogenesis: 125 years after Pasteur. FEMS Microbiol. Lett. 2006, 265, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.; Henestrosa, A.R.; Badiola, I.; Saco, M.; Perez Rozas, D.A.M.; Campoy Frawley, E.R.; Fang, F.C. The ins and outs of bacterial iron metabolism. Mol. Microbiol. 2014, 93, 609–616. [Google Scholar] [CrossRef]
- Hatfaludi, T.; Al-Hasani, K.; Boyce, J.D.; Adler, B. Outer membrane proteins of Pasteurella multocida. Vet. Microbiol. 2010, 144, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.C.; Robinson, A.K.; Rodríguez-Quiñones, F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237. [Google Scholar] [CrossRef]
- Adhikari, P.; Berish, S.A.; Nowalk, A.J.; Veraldi, K.L.; Morse, S.A.; Mietzner, T.A. The fbpABC locus of Neisseria gonorrhoeae functions in the periplasm-to-cytosol transport of iron. J. Bacteriol. 1996, 178, 2145–2149. [Google Scholar] [CrossRef]
- Bearden, S.W.; Perry, R.D. The Yfe system of Yersinia pestis transports iron and manganese and is required for full virulence of plague. Mol. Microbiol. 1999, 32, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, S.; Mizan, S.; Quist, C.; Schroder, P.; Juneau, M.; Dawe, D.; Ritchie, B.; Lee, M.D. Serological response to Pasteurella multocida NanH sialidase in persistently colonized rabbits. Clin. Diagn. Lab. Immunol. 2004, 11, 825–834. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer & Probe | Sequence (5’–3’) | Product Size (bp) | Annealing Temperature (°C) |
|---|---|---|---|
| fbpB F | GCTGCCATTGCAGGCTTAGG | 209 | 61 |
| fbpB R | GGTTGCCACGCGTGTATCTG | ||
| fbpB probe | CGCTCAATGACCACGGTCAGCGCA | ||
| fecE F | GGCTGCGGAAAATCCACGTT | 152 | 49 |
| fecE R | TGGTACCAGGTGTTGTTGTGGT | ||
| fecE probe | GCGCTTGCACGTTTGCTGAAACCCA | ||
| fur F | AGCGGGGTTGAAAATCACCG | 267 | 49 |
| fur R | CACTTTACCGCAGTCCACGC | ||
| fur probe | ACTTGCCCCAACTGAACACCACGA | ||
| yfeA F | ATTACCAAACCGGGTGCGGA | 426 | 61 |
| yfeA R | AGGCGCCTTCACTGGTTACT | ||
| yfeA probe | ACCGGCTGTCGTTGTGACAGAAGG | ||
| nanA F | TGGTACAGGTCCTGGTGTTGG | 204 | 49 |
| nanA R | ACGCTGATTTGTTCGAAGCGT | ||
| nanA probe | TGGGATTTGGGCGCTAGCCCT | ||
| gapdh F | GCTGCCATTGCAGGCTTAGG | 209 | 61 |
| gapdh R | GGTTGCCACGCGTGTATCTG | ||
| gapdh probe | CGCTCAATGACCACGGTCAGCGCA | ||
| gyrB F | GGCTGCGGAAAATCCACGTT | 152 | 61 |
| gyrB R | CACTTTACCGCAGTCCACGC | ||
| gyrB probe | ACTTGCCCCAACTGAACACCACGA |
| Feature of the Sequence of PMTB2.1 | Description |
|---|---|
| Genome size | 2,315,13 |
| Genome GC content | 40.32% |
| oriC length | 415 nucleotides |
| Putative oriC location | 1,837,028–1,837,442 |
| Total CDs | 2097 |
| RNA gene | 79 |
| 16S rRNAs | 6 |
| 23S rRNAs | 6 |
| 5S rRNAs | 7 |
| tRNAs | 56 |
| ncNAs | 4 |
| Bacteria Strain Name | PMTB2.1 | K12 MG1655 | PM 70 | JL03 | SH0165 | PM 36950 | PM HN06 | PM 3480 | PM HB01 |
|---|---|---|---|---|---|---|---|---|---|
| P. multocida PMTB2.1 | 0.00 | 0.80 | 0.08 | 0.72 | 0.72 | 0.10 | 0.11 | 0.09 | 0.12 |
| Escherichia coli K-12 MG1655 | 0.80 | 0.00 | 0.81 | 0.80 | 0.82 | 0.81 | 0.81 | 0.80 | 0.81 |
| P. multocida PM70 | 0.08 | 0.81 | 0.00 | 0.72 | 0.72 | 0.09 | 0.10 | 0.08 | 0.12 |
| A. pleuropneumoniae JL03 | 0.72 | 0.80 | 0.72 | 0.00 | 0.68 | 0.72 | 0.72 | 0.72 | 0.73 |
| H. parasuis SH0165 | 0.72 | 0.82 | 0.72 | 0.68 | 0.00 | 0.72 | 0.72 | 0.72 | 0.73 |
| P. multocida 36950 | 0.10 | 0.81 | 0.09 | 0.72 | 0.72 | 0.00 | 0.12 | 0.10 | 0.06 |
| P. multocida HN06 | 0.11 | 0.81 | 0.10 | 0.72 | 0.72 | 0.12 | 0.00 | 0.07 | 0.13 |
| P. multocida 3480 | 0.09 | 0.80 | 0.08 | 0.72 | 0.72 | 0.10 | 0.07 | 0.00 | 0.12 |
| P. multocida HB01 | 0.12 | 0.81 | 0.12 | 0.73 | 0.73 | 0.06 | 0.13 | 0.12 | 0.00 |
| Pasteurella Species | GenBank Accession No. | Host and Serotype | Genome Size (bp) | GC Content (%) | Reference | Compared with PMTB2.1 |
|---|---|---|---|---|---|---|
| P. multocida strain PM70 | AE004439.1 | Avian (F) | 2,257,487 | 40.4 | May et al., 2001 | 57,651 bp less |
| P. multocida strain 36950 | CP003022.1 | Bovine (A) | 2,349,518 | 40.4 | Michael et al., 2011 | 34,380 bp more |
| P. multocida strain 3480 | CP001409.1 | Swine (A) | 2,378,127 | 40.3 | Unpublished | 62,989 bp more |
| P. multocida strain HN06 | CP003313.1 | Swine (D) toxigenic | 2,402,218 | 40.2 | Liu et al., 2012 | 87,080 bp more |
| P. multocida strain HB01 | CP006976 | Bovine (A) | 2,416,068 | 40.3 | Peng et al., 2016 | 100,930 bp more |
| P. multocida strain PMTB2.1 | CP007205.1 | Buffalo (A) | 2,315,138 | 40.4 | This study | - |
| No | Gene ID | Annotation | Gene Name |
|---|---|---|---|
| 1 | AW43_04840 | pilus assembly protein TadG | tadG |
| 2 | AW43_04845 | protein Tad F | tadF |
| 3 | AW43_04850 | protein TadE | tadE |
| 4 | AW43_04855 | NrfG protein | tadD |
| 5 | AW43_04860 | pilus assembly protein TadC | tadC |
| 6 | AW43_04865 | MaoC protein | tadB |
| 7 | AW43_04870 | pilus assembly protein CpaF | tadA |
| 8 | AW43_04875 | pilus assembly protein Protein: (Flp pilus assembly protein, ATPase CpaE) | tadZ |
| 9 | AW43_04880 | protein RcpB | rcpB |
| 10 | AW43_04885 | Secretin | rcpA |
| 11 | AW43_04890 | flp operon protein C | rcpC |
| 12 | AW43_04895 | flp operon protein B | tadV |
| 13 | AW43_04900 | pilus assembly protein | flp2 |
| 14 | AW43_04905 | fimbrial protein | flp1 |
| Time Points | Relative Fold Changes of Iron-Related Genes and nanA Gene in Iron-Limited Environment | ||||
|---|---|---|---|---|---|
| fbpB | fecE | fur | yfeA | nanA | |
| 30 | 3.38 b ± 0.19 | 281.24 b ± 1.23 | 4.80 b ± 0.24 | 26.02 b ± 0.11 | 1.41 ± 0.31 |
| 60 | 25.69 b ± 1.50 | 4.70 a ± 0.36 | 2.88 a ± 0.07 | 12.11 a ± 0.10 | 1.09 ± 0.31 |
| 120 | −1.13 a ± 0.50 | −1.39 b ± 0.32 | 4.82 b ± 0.29 | 42.11 b ± 0.42 | −1.67 ± 0.40 |
| 180 | −4.95 b ± 0.59 | −1.58 b ± 1.09 | 5.46 a ± 0.05 | 11.65 b ± 0.12 | 1.04 ± 1.50 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabeen, S.; Yap, H.Y.; Abdullah, F.F.J.; Zakaria, Z.; Isa, N.M.; Tan, Y.C.; Joo, Y.S.; Satharasinghe, D.A.; Omar, A.R. Complete Genome Sequence Analysis and Characterization of Selected Iron Regulation Genes of Pasteurella Multocida Serotype A Strain PMTB2.1. Genes 2019, 10, 81. https://doi.org/10.3390/genes10020081
Jabeen S, Yap HY, Abdullah FFJ, Zakaria Z, Isa NM, Tan YC, Joo YS, Satharasinghe DA, Omar AR. Complete Genome Sequence Analysis and Characterization of Selected Iron Regulation Genes of Pasteurella Multocida Serotype A Strain PMTB2.1. Genes. 2019; 10(2):81. https://doi.org/10.3390/genes10020081
Chicago/Turabian StyleJabeen, Shagufta, Huan Y. Yap, Faez Firdaus J. Abdullah, Zunita Zakaria, Nurulfiza M. Isa, Yung C. Tan, Yap S. Joo, Dilan A. Satharasinghe, and Abdul R. Omar. 2019. "Complete Genome Sequence Analysis and Characterization of Selected Iron Regulation Genes of Pasteurella Multocida Serotype A Strain PMTB2.1" Genes 10, no. 2: 81. https://doi.org/10.3390/genes10020081
APA StyleJabeen, S., Yap, H. Y., Abdullah, F. F. J., Zakaria, Z., Isa, N. M., Tan, Y. C., Joo, Y. S., Satharasinghe, D. A., & Omar, A. R. (2019). Complete Genome Sequence Analysis and Characterization of Selected Iron Regulation Genes of Pasteurella Multocida Serotype A Strain PMTB2.1. Genes, 10(2), 81. https://doi.org/10.3390/genes10020081