N6-Methyladenosine Landscape of Glioma Stem-Like Cells: METTL3 Is Essential for the Expression of Actively Transcribed Genes and Sustenance of the Oncogenic Signaling

Abstract
:1. Introduction
2. Materials and Methods
2.1. Neurosphere Cell Culturing
2.2. RNA Isolation, Reverse Transcription and qPCR Analysis
2.3. RNA Preparation for RIP-Sequencing
2.4. m6A RNA Immunoprecipitation Enrichment
2.5. m6A RNA Immunoprecipitation Sequencing
2.6. Transfection of Cells with Plasmids and siRNAs
2.7. RNA Stability Assay
2.8. Western Blot
2.9. RNA Immunoprecipitation of METTL3
2.10. Luciferase Reporter Assay
2.11. RNA Isolation, cDNA Synthesis and qRT-PCR
2.12. Lentivirus Preparation
2.13. RNA Editing
2.14. Differential Alternative Splicing
2.15. Alignment of RNA-Sequences
2.16. Gene Set Enrichment Analysis (GSEA)
- Genes were grouped as activated (H3K4me3) or repressed (H3K27me3, H3K27me3 + H3K4me3) in MGG8 GSC compared to NSC.
- Genes were grouped as activated (H3K4me3) or repressed (H3K27me3, H3K27me3 + H3K4me3) in MGG8 GSC compared to MGG8 DGC.
- Genes were grouped as activated (H3K27Ac) in MGG8 GSC compared to MGG8DGC.
- The activated and upregulated/repressed and downregulated genes in GSC compared to NSC based on H3K27me3/H3K4me3marks.
- The activated and upregulated/repressed and downregulated genes in GSC compared to DGC based on H3K27me3/H3K4me3marks.
- The activated and upregulated genes in GSC compared to DGC based onH3K27Ac
2.17. Pathway Analysis
2.18. RNA Immunoprecipitation Sequencing Analysis
2.18.1. Alignment of m6A RNA Immunoprecipitation Peaks
2.18.2. Peak Calling for m6A Samples
2.18.3. Peak Visualization
2.18.4. Distribution of m6A Peak Regions in 5′UTR, exon and 3′UTR
2.19. Cumulative Frequency Distribution
2.20. miRNA Target Prediction
2.21. Motif Finding
3. Results
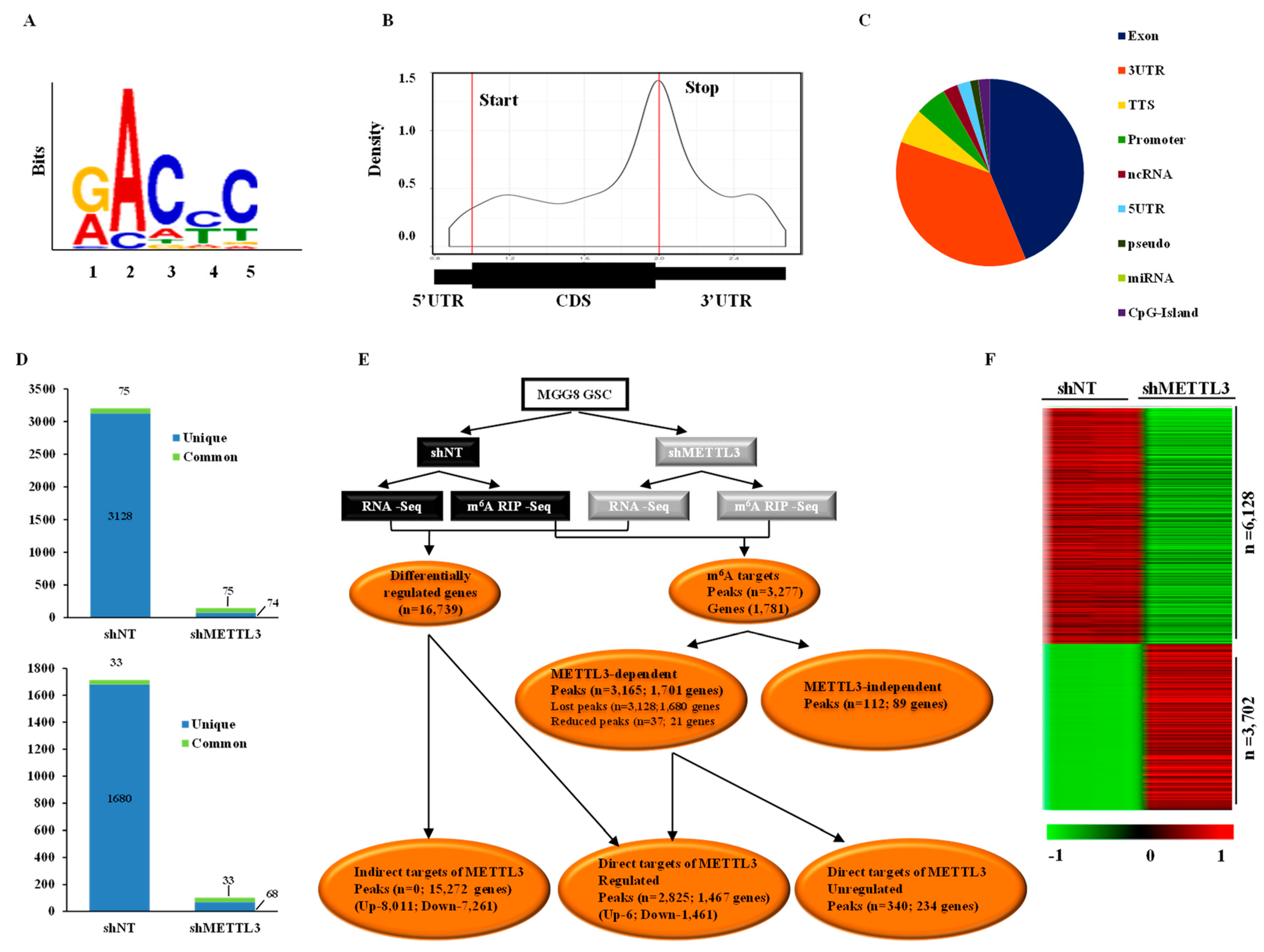
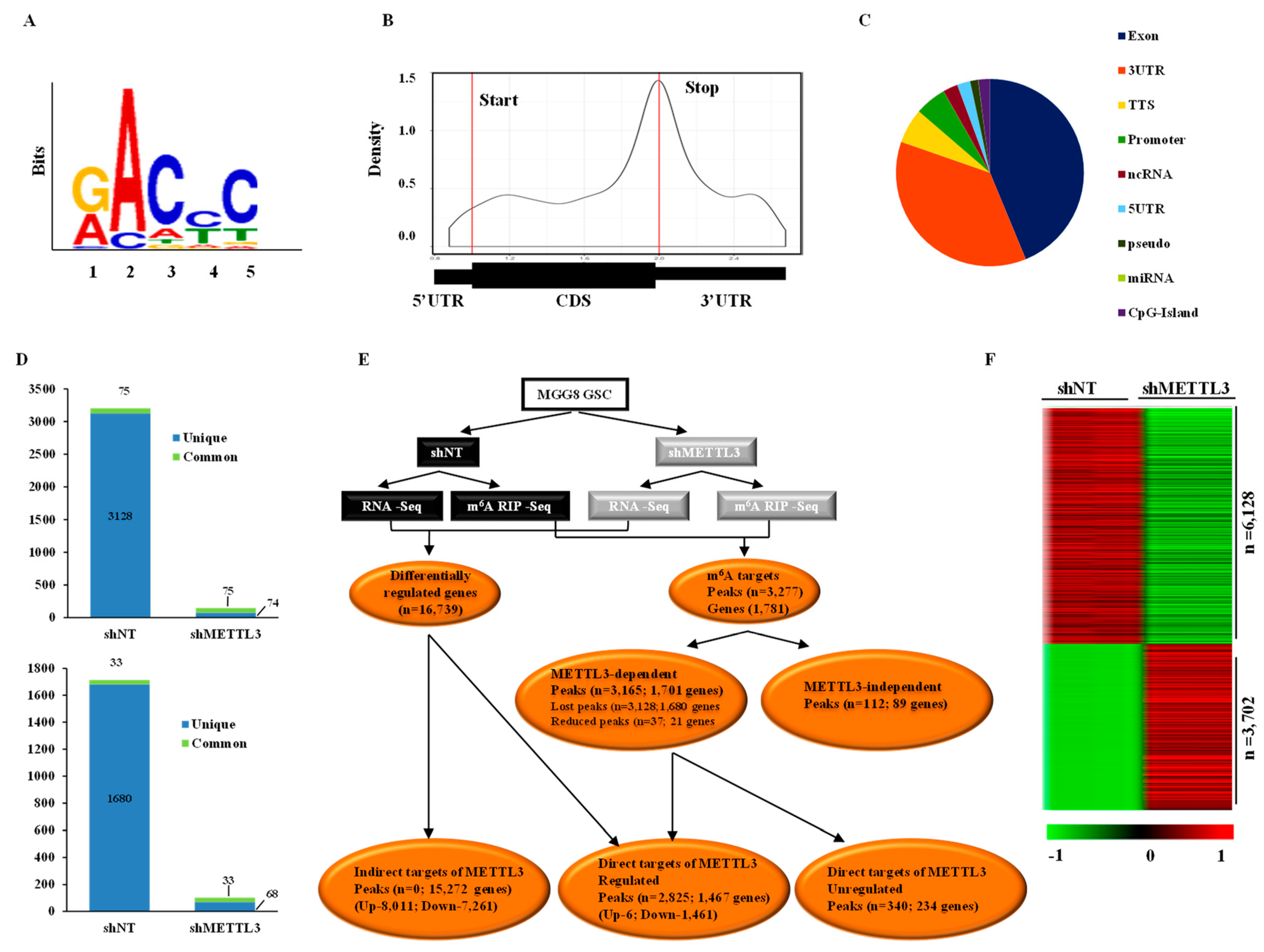
3.1. Transcriptome-Wide Mapping of m6A Modification Landscape in Glioma Stem-Like Cells
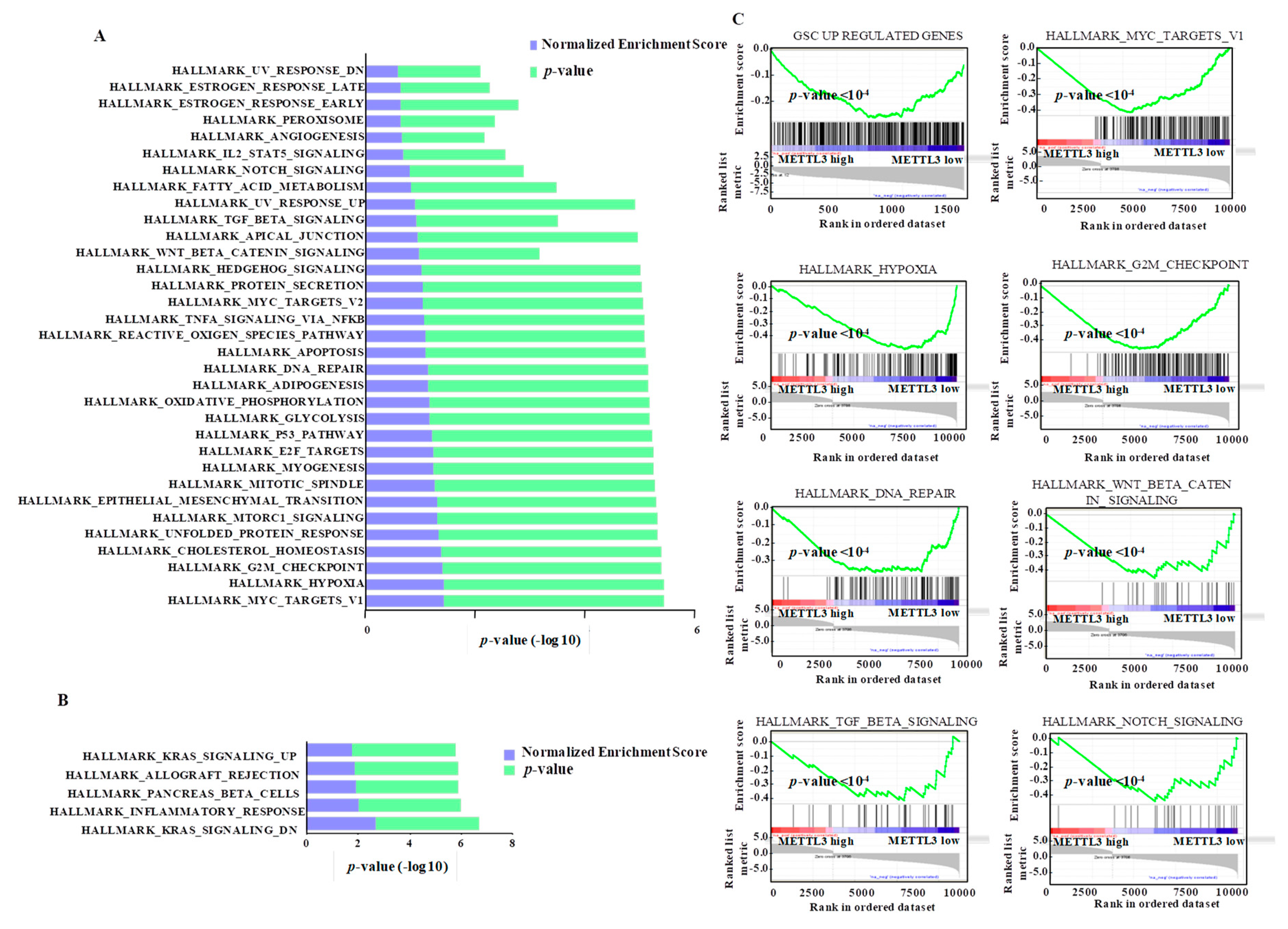
3.2. Impact of METTL3-Mediated m6A Modification on the Glioma Stem-Like Cells Transcriptome
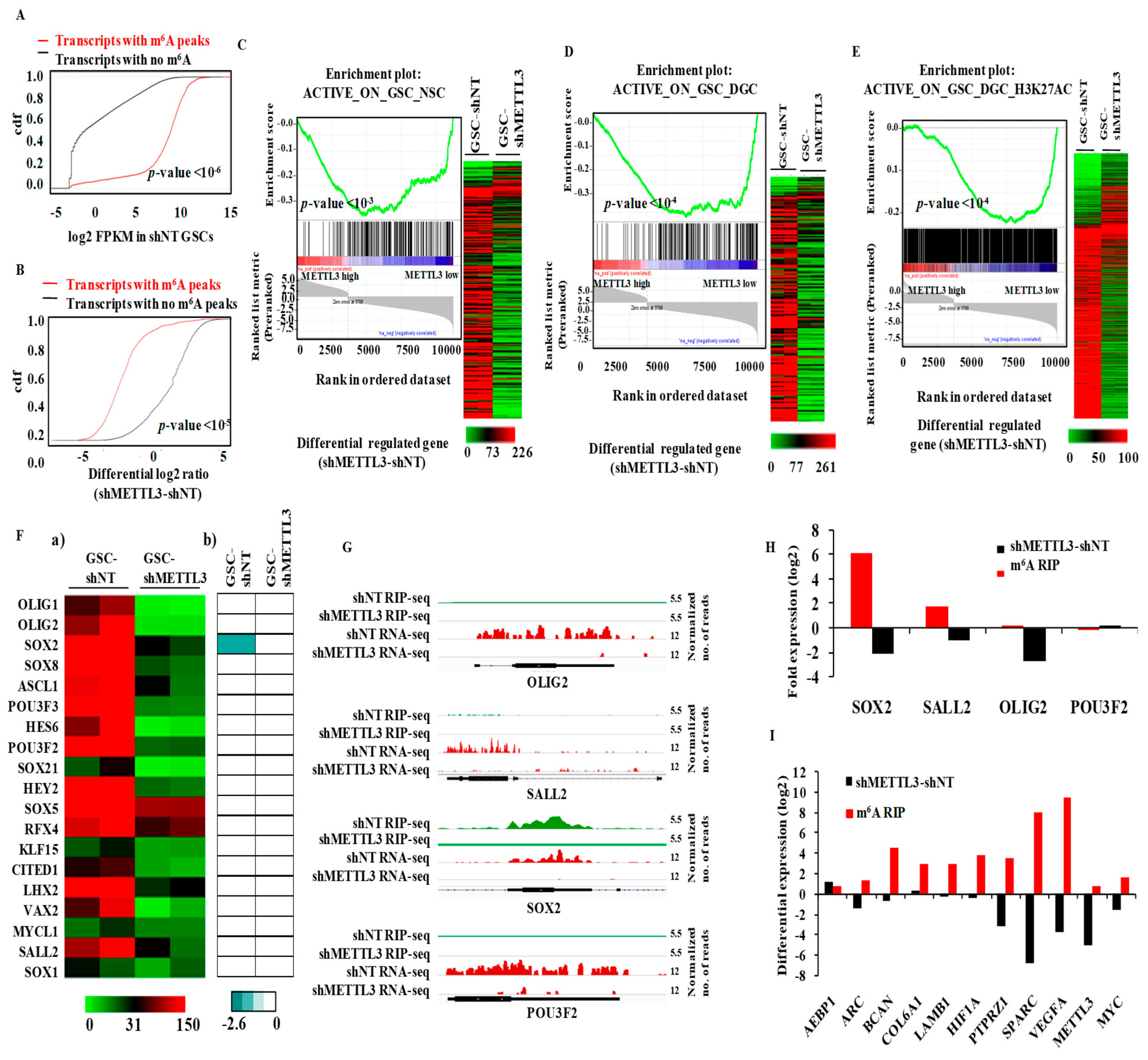
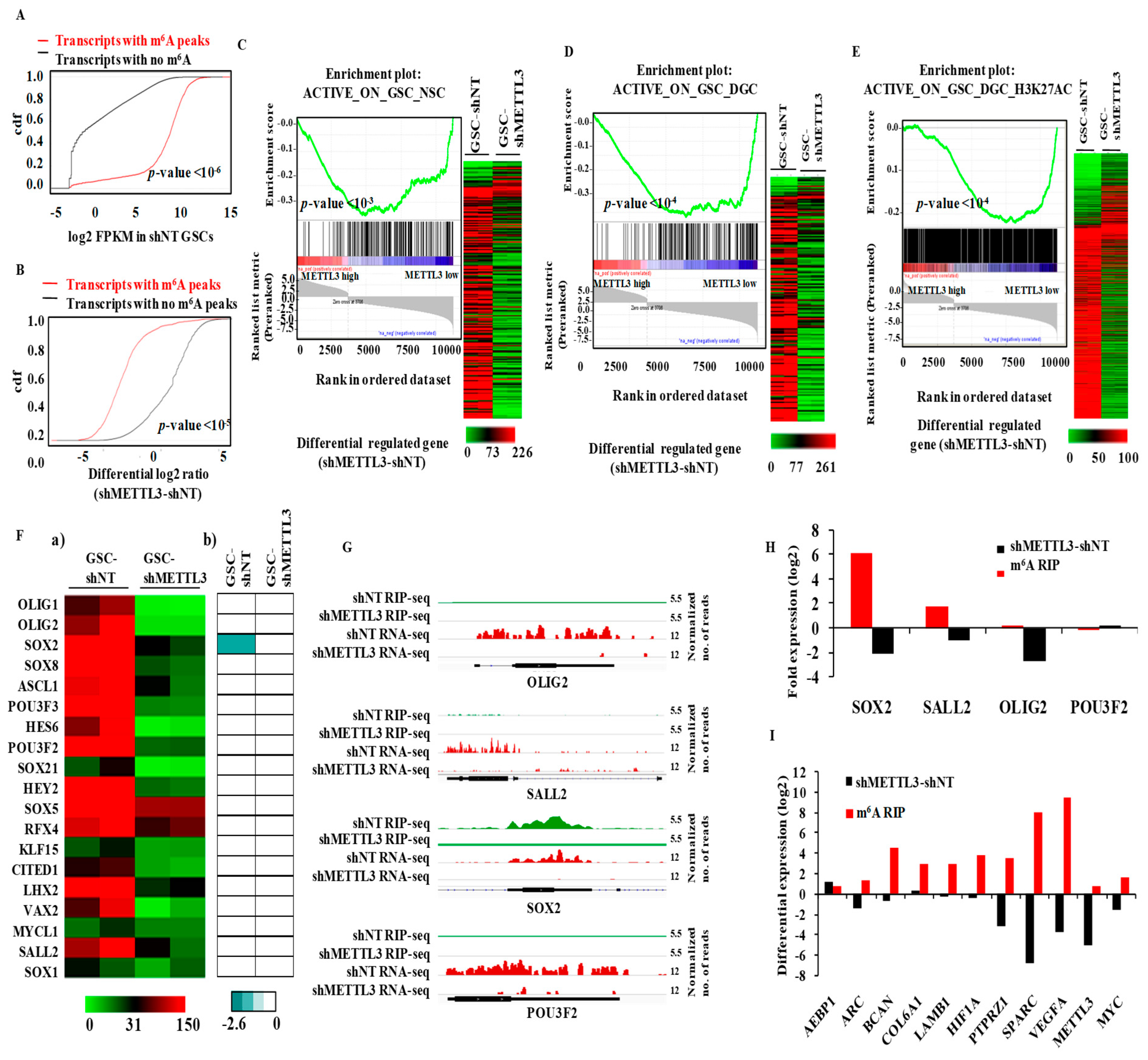
3.3. METTL3 Is Essential for the Expression of Epigenetically Activated Genes in Glioma Stem-Like Cells
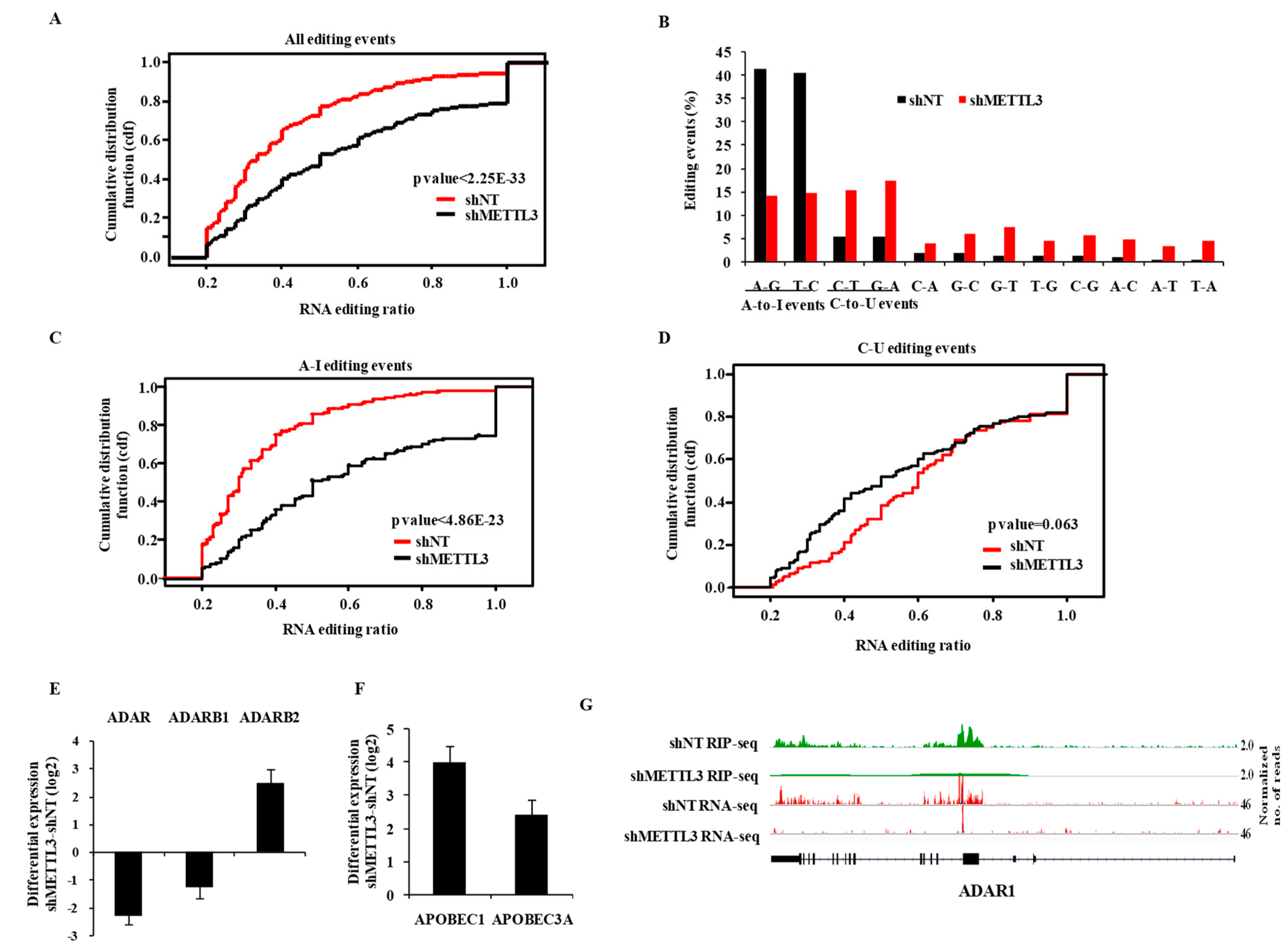
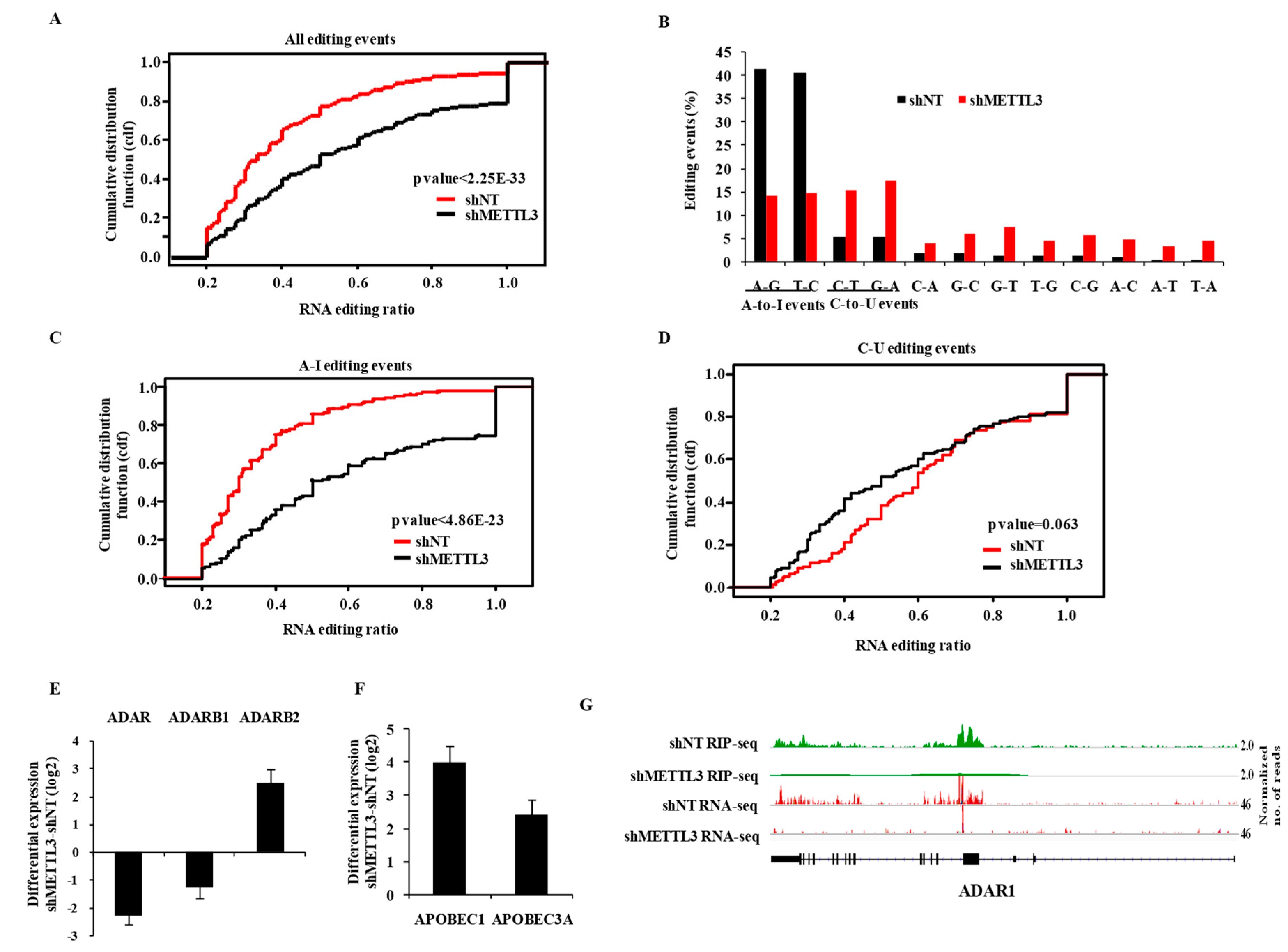
3.4. m6A Modification Regulates RNA Editing
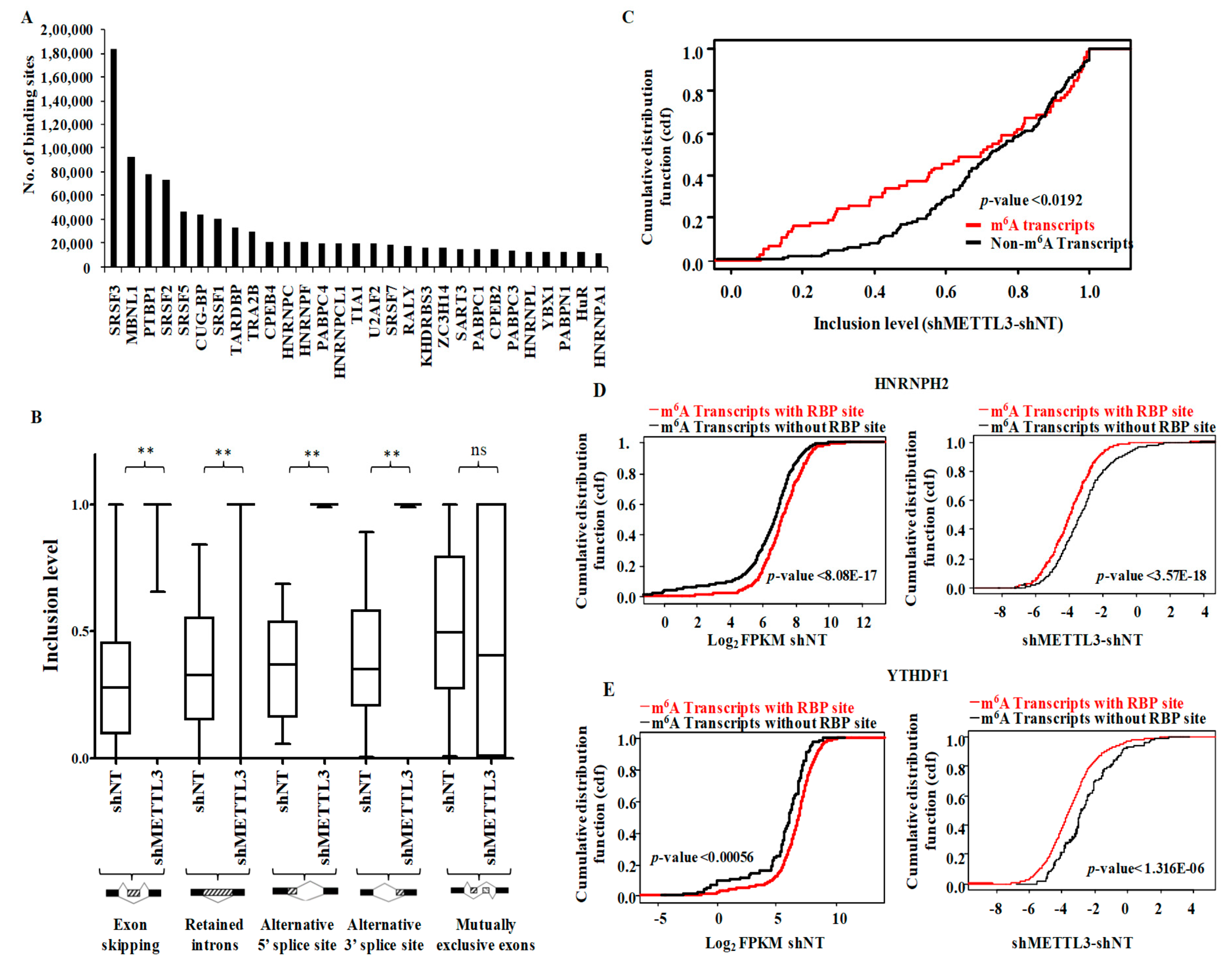
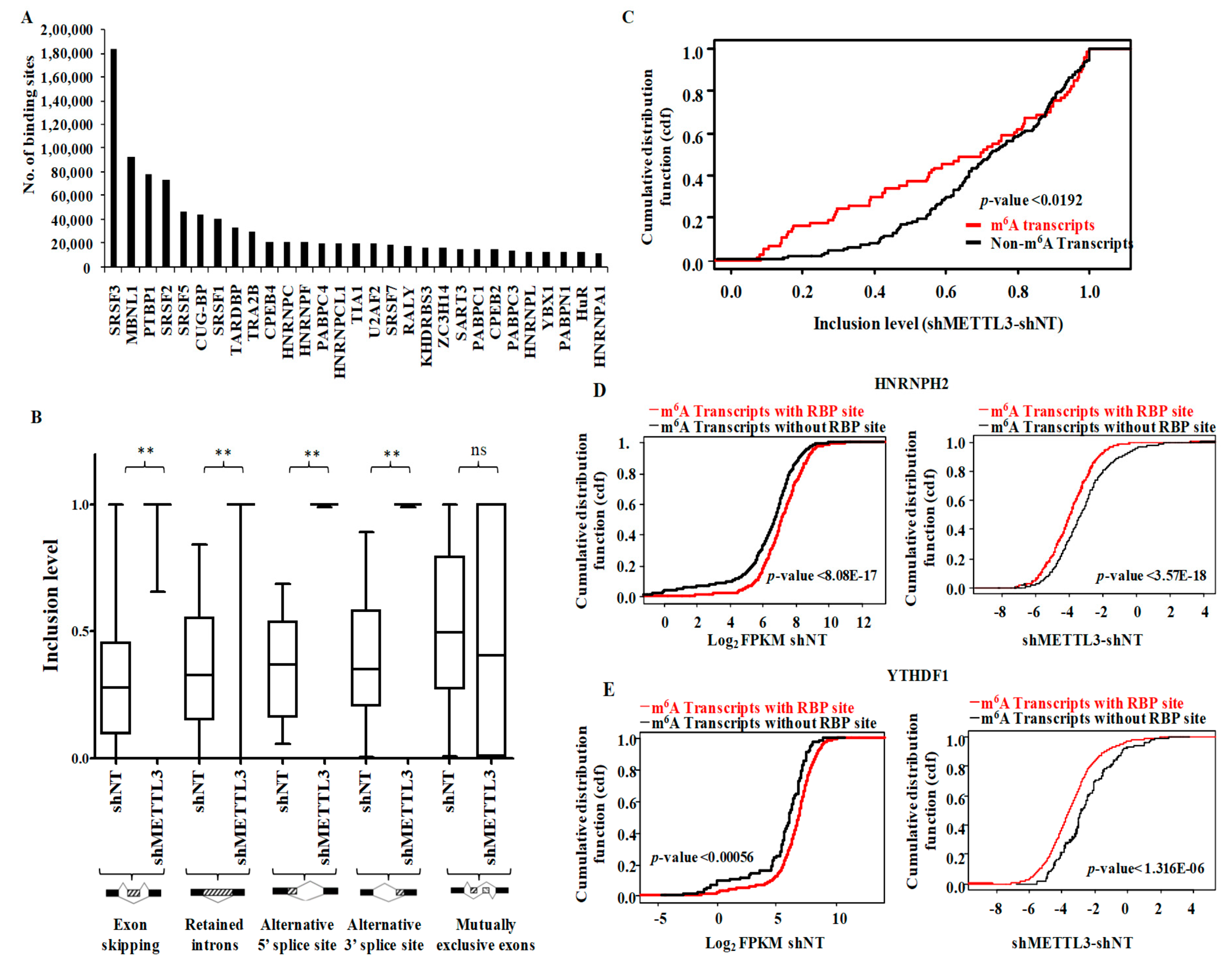
3.5. Functional Association of RNA Processing Factors with m6A Modification
3.6. Regulation of Long Non-Coding RNAs and miRNAs by m6A-Methylome
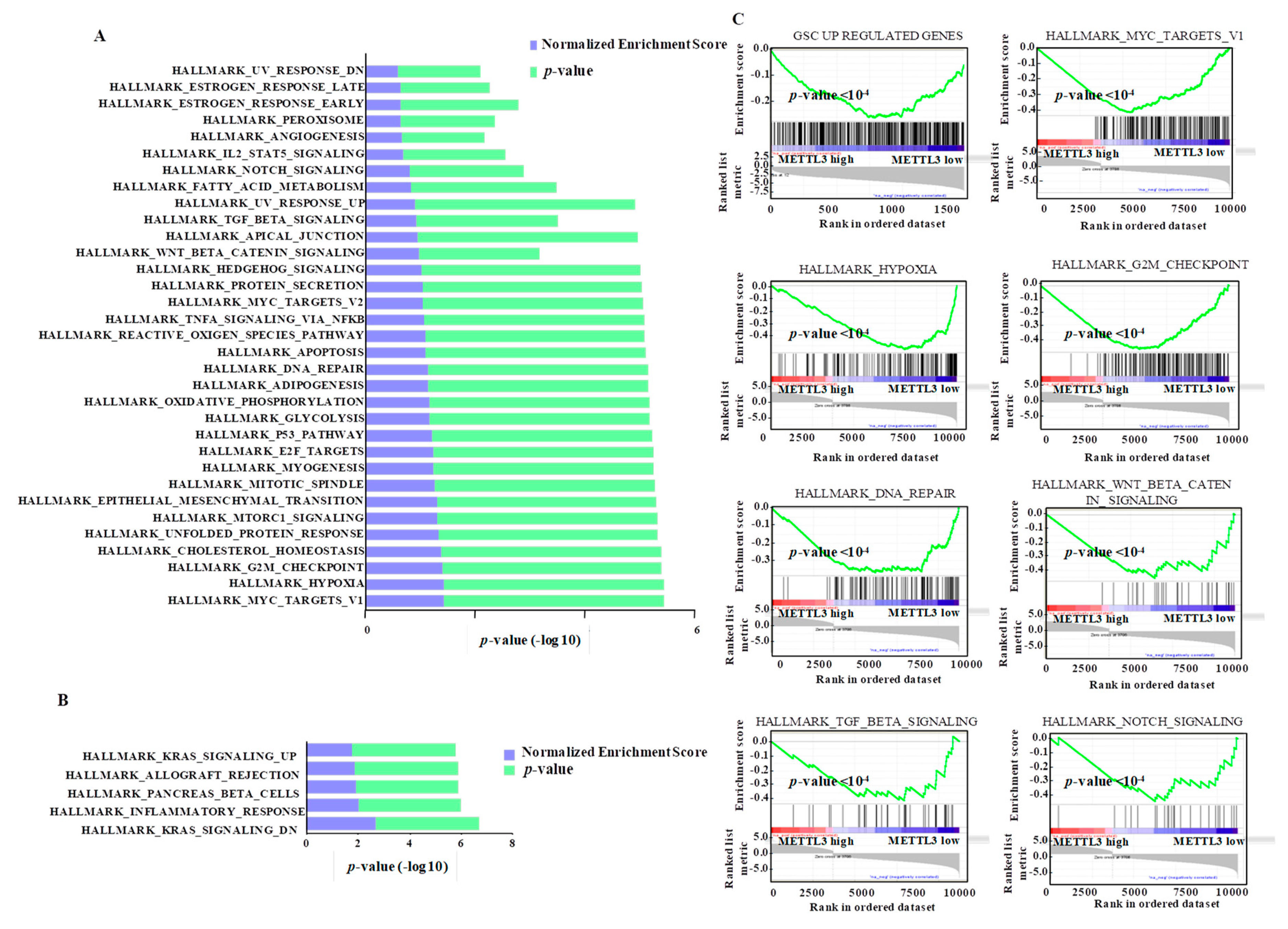
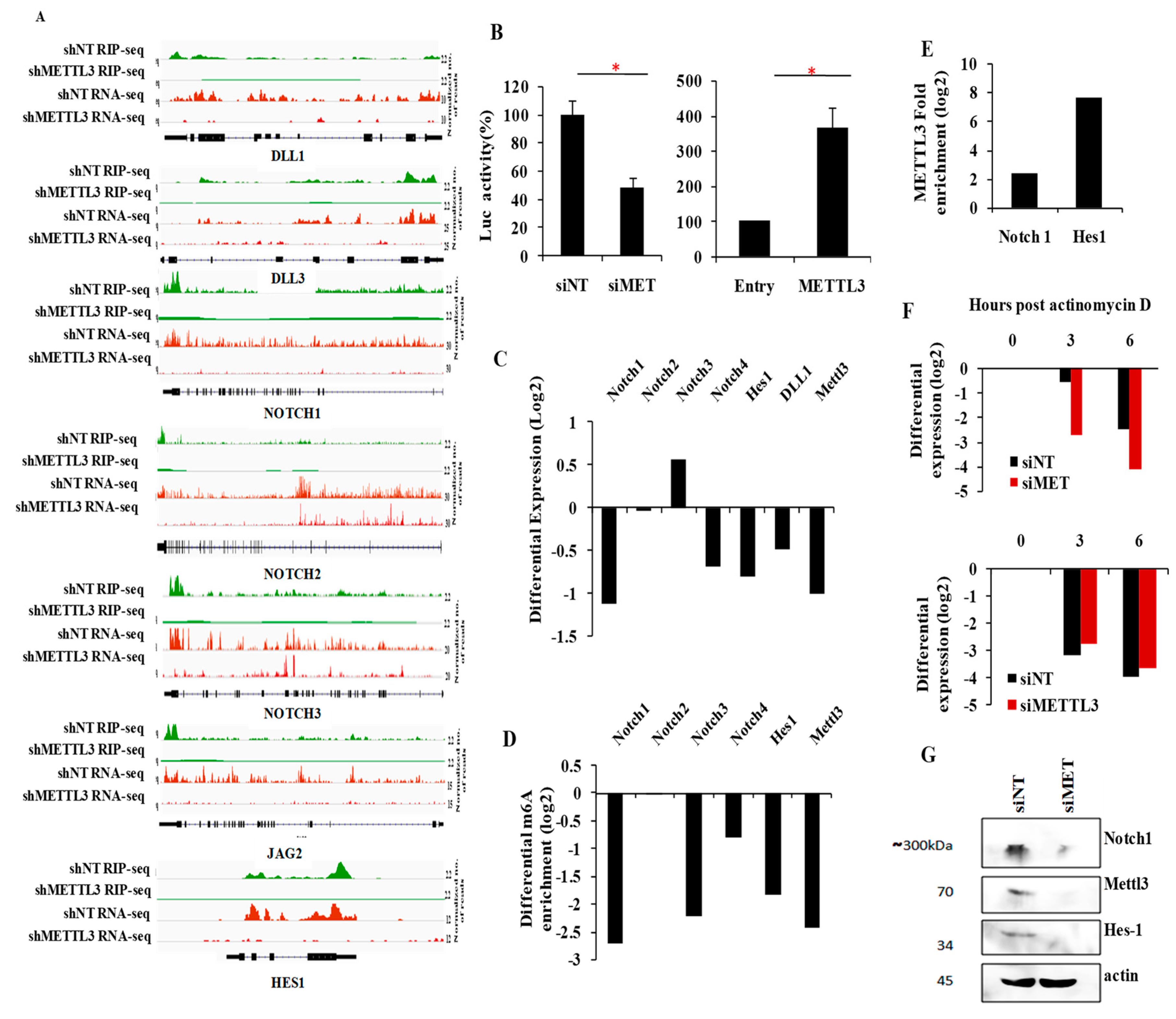
3.7. Integrated Pathway Analysis of Regulated Transcriptome Implies an Oncogenic Role for METTL3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
List of Abbreviations
| m6A | N6-methyladenosine |
| METTL3 | methyltransferase-like3 |
| METTL14 | methyl transferase-like 14 |
| FTO | fat mass and obesity-associated protein |
| ALKBH5 | alkB homolog 5 |
| GSC | glioma stem-like cell |
| DGC | differentiated glioma cell |
| NSC | normal neural stem cell |
References
- Wang, J.; Ma, Y.; Cooper, M.K. Cancer stem cells in glioma: Challenges and opportunities. Transl. Cancer Res. 2013, 2, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vagbo, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, A.; Patil, V.; Arora, A.; Hegde, A.S.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene 2018, 37, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Doxtader, K.A.; Nam, Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol. Cell 2016, 63, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, G.; Zhang, R.; Piskol, R.; Keegan, L.P.; Deng, P.; O’Connell, M.A.; Li, J.B. Identifying RNA editing sites using RNA sequencing data alone. Nat. Methods 2013, 10, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Paz, I.; Kosti, I.; Ares, M., Jr.; Cline, M.; Mandel-Gutfreund, Y. RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014, 42, W361–W367. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Rheinbay, E.; Suva, M.L.; Gillespie, S.M.; Wakimoto, H.; Patel, A.P.; Shahid, M.; Oksuz, O.; Rabkin, S.D.; Martuza, R.L.; Rivera, M.N.; et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013, 3, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Oakes, E.; Anderson, A.; Cohen-Gadol, A.; Hundley, H.A. Adenosine deaminase that acts on RNA 3 (ADAR3) binding to glutamate receptor subunit B pre-mRNA inhibits RNA editing in glioblastoma. J. Biol. Chem. 2017, 292, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, A.; Somasundaram, K. mRNA Traffic Control Reviewed: N6-Methyladenosine (m6A) Takes the Driver’s Seat. BioEssays 2018, 40. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Blue, S.M.; Dominguez, D.; Cody, N.A.L.; Olson, S.; Sundararaman, B.; et al. A Large-scale binding and functional map of human RNA binding proteins. bioRxiv 2018. [Google Scholar] [CrossRef]
- Alkan, S.A.; Martincic, K.; Milcarek, C. The hnRNPs F and H2 bind to similar sequences to influence gene expression. Biochem. J. 2006, 393, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Yugami, M.; Kabe, Y.; Yamaguchi, Y.; Wada, T.; Handa, H. hnRNP-U enhances the expression of specific genes by stabilizing mRNA. FEBS Lett. 2007, 581, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, V.N.; Lingjaerde, O.C.; Russnes, H.G.; Vollan, H.K.; Frigessi, A.; Borresen-Dale, A.L. Principles and methods of integrative genomic analyses in cancer. Nat. Rev. Cancer 2014, 14, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Tan, F.; Hess, K.R.; Yung, W.K. The expression of PAX6, PTEN, vascular endothelial growth factor, and epidermal growth factor receptor in gliomas: Relationship to tumor grade and survival. Clin. Cancer Res. 2003, 9, 3369–3375. [Google Scholar] [PubMed]
- Kim, D.S.; Hubbard, S.L.; Peraud, A.; Salhia, B.; Sakai, K.; Rutka, J.T. Analysis of mammalian septin expression in human malignant brain tumors. Neoplasia 2004, 6, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Liu, L.; Fan, N.; Zhang, Q.; Wang, W.; Zheng, M.; Ma, L.; Li, Y.; Shi, L. The requirement of SEPT2 and SEPT7 for migration and invasion in human breast cancer via MEK/ERK activation. Oncotarget 2016, 7, 61587–61600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.A.; Santosh, V.; Somasundaram, K. Genome-wide expression profiling identifies deregulated miRNAs in malignant astrocytoma. Mod. Pathol. 2010, 23, 1404–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, B.C.; Lefort, K.; Mandinova, A.; Antonini, D.; Devgan, V.; Della Gatta, G.; Koster, M.I.; Zhang, Z.; Wang, J.; Tommasi di Vignano, A.; et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006, 20, 1028–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, M.E.; Hess, S.; Meyer, K.D.; Verhagen, L.A.; Koch, L.; Bronneke, H.S.; Dietrich, M.O.; Jordan, S.D.; Saletore, Y.; Elemento, O.; et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat. Neurosci. 2013, 16, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Lence, T.; Akhtar, J.; Bayer, M.; Schmid, K.; Spindler, L.; Ho, C.H.; Kreim, N.; Andrade-Navarro, M.A.; Poeck, B.; Helm, M.; et al. m6A modulates neuronal functions and sex determination in Drosophila. Nature 2016, 540, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Molinie, B.; Wang, J.; Lim, K.S.; Hillebrand, R.; Lu, Z.X.; Van Wittenberghe, N.; Howard, B.D.; Daneshvar, K.; Mullen, A.C.; Dedon, P.; et al. m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome. Nat. Methods 2016, 13, 692–698. [Google Scholar] [CrossRef]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m6A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef]
- Du, H.; Zhao, Y.; He, J.; Zhang, Y.; Xi, H.; Liu, M.; Ma, J.; Wu, L. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 2016, 7, 12626. [Google Scholar] [CrossRef]
- Ivanova, I.; Much, C.; Di Giacomo, M.; Azzi, C.; Morgan, M.; Moreira, P.N.; Monahan, J.; Carrieri, C.; Enright, A.J.; O’Carroll, D. The RNA m6A reader YTHDF2 is essential for the post-transcriptional regulation of the maternal transcriptome and oocyte competence. Mol. Cell 2017, 67, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m6A Methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.G.; et al. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bogler, O.; et al. m6A Demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 2017, 31, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Xue, Y.; Zheng, J.; Liu, X.; Ma, J.; Liu, Y. WTAP expression predicts poor prognosis in malignant glioma patients. J. Mol. Neurosci. 2016, 60, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Salmon-Divon, M.; Amariglio, N.; Rechavi, G. Transcriptome-wide mapping of N6-methyladenosine by m6A-seq based on immunocapturing and massively parallel sequencing. Nat. Protocols 2013, 8, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Bansal, H.; Yihua, Q.; Iyer, S.P.; Ganapathy, S.; Proia, D.A.; Penalva, L.O.; Uren, P.J.; Suresh, U.; Carew, J.S.; Karnad, A.B.; et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 2014, 28, 1171–1174. [Google Scholar] [CrossRef] [Green Version]
- Yi, C.; Pan, T. Cellular dynamics of RNA modification. Acc. Chem. Res. 2011, 44, 1380–1388. [Google Scholar] [CrossRef]
- Kwok, C.T.; Marshall, A.D.; Rasko, J.E.; Wong, J.J. Genetic alterations of m6A regulators predict poorer survival in acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 39. [Google Scholar] [CrossRef]
- Xiang, Y.; Laurent, B.; Hsu, C.H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef]
- Hongay, C.F.; Orr-Weaver, T.L. Drosophila Inducer of MEiosis 4 (IME4) is required for Notch signaling during oogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 14855–14860. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.; Shen, J.; Cheng, C.L.; Tsang, L.H.; Ho, D.W.; Chiu, D.K.; Lee, J.M.; et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef] [PubMed]
- Spitale, R.C.; Flynn, R.A.; Zhang, Q.C.; Crisalli, P.; Lee, B.; Jung, J.W.; Kuchelmeister, H.Y.; Batista, P.J.; Torre, E.A.; Kool, E.T.; et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 2015, 519, 486–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017, 45, 6051–6063. [Google Scholar] [CrossRef] [PubMed]
- Lareau, L.F.; Brenner, S.E. Regulation of splicing factors by alternative splicing and NMD is conserved between kingdoms yet evolutionarily flexible. Mol. Biol. Evol. 2015, 32, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Bartel, F.; Taubert, H.; Harris, L.C. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell 2002, 2, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Bohlman, S.; Manfredi, J.J. p53-independent effects of Mdm2. Sub-Cell. Biochem. 2014, 85, 235–246. [Google Scholar] [CrossRef]
- Steinman, H.A.; Burstein, E.; Lengner, C.; Gosselin, J.; Pihan, G.; Duckett, C.S.; Jones, S.N. An alternative splice form of Mdm2 induces p53-independent cell growth and tumorigenesis. J. Biol. Chem. 2004, 279, 4877–4886. [Google Scholar] [CrossRef] [PubMed]
- Fry, N.J.; Law, B.A.; Ilkayeva, O.R.; Holley, C.L.; Mansfield, K.D. N6-methyladenosine is required for the hypoxic stabilization of specific mRNAs. RNA 2017, 23, 1444–1455. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, X.; Lu, Z.; Zhao, B.S.; Ma, H.; Hsu, P.J.; Liu, C.; He, C. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017, 27, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Arhin, G.K.; Boots, M.; Bagga, P.S.; Milcarek, C.; Wilusz, J. Downstream sequence elements with different affinities for the hnRNP H/H’ protein influence the processing efficiency of mammalian polyadenylation signals. Nucleic Acids Res. 2002, 30, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chopp, M.; Lu, Y.; Buller, B.; Jiang, F. MiR-15b and miR-152 reduce glioma cell invasion and angiogenesis via NRP-2 and MMP-3. Cancer Lett. 2013, 329, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.J.; Choi, Y.E.; Kim, E.S.; Han, Y.H.; Park, M.J.; Bae, I.H. miR-29b attenuates tumorigenicity and stemness maintenance in human glioblastoma multiforme by directly targeting BCL2L2. Oncotarget 2015, 6, 18429–18444. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primers (5′-3′) |
|---|---|
| METTL3-FP | ACCTATGCTGACCATTACCAAG |
| METTL3-RP | CTGTTGGTTCAGAAGGCTCTC |
| SOX2-FP | AACCCCAAGATGCACAACTC |
| SOX2-RP | GCTTAGCCTCGTCGATGAAC |
| OLIG2-FP | CCAGAGCCCGATGACCTTTT |
| OLIG2-RP | AGGACGACTTGAAGCCACTG |
| POU3F2-FP | TGACGATCTCCACGCAGTAG |
| POU3F2-RP | GGCAGAAAGCTGTCCAAGTC |
| SALL2-FP | TAATCTCGGACTGCGAAGGT |
| SALL2-RP | TAGAACATGCGTTCTGGTGG |
| ATP5G-FP | CCAGACGGGAGTTCCAGAC |
| ATP5G-RP | GACGGGTTCCTGGCATAGC |
| DLL1-FP | GCAGCTCTTCACCCTGTTCT |
| DLL1-RP | GGTGCAGGAGAAGTCGTTCA |
| NOTCH1-FP | GAGGCCTGGCAGACTATGC |
| NOTCH1-RP | CTTGTACTCCGTCAGCGTGA |
| HES1-FP | AGTGAAGCACCTCCGGAAC |
| HES1-RP | TCACCTCGTTCATGCACTC |
| NOTCH2-fp | CTGTGAGTGTCTGAAGGGTTATG |
| NOTCH2-rp | GGCACTGGAAACGATTGACTTT |
| NOTCH3-fp | CGTGGCTTCTTTCTACTGTGC |
| NOTCH3-rp | CGTTCACCGGATTTGTGTCAC |
| NOTCH4-fp | CTGGGTGTCAATGGAGAGGGA |
| NOTCH4-rp | GGGTGAGACGTGCCAGTTTC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Visvanathan, A.; Patil, V.; Abdulla, S.; Hoheisel, J.D.; Somasundaram, K. N6-Methyladenosine Landscape of Glioma Stem-Like Cells: METTL3 Is Essential for the Expression of Actively Transcribed Genes and Sustenance of the Oncogenic Signaling. Genes 2019, 10, 141. https://doi.org/10.3390/genes10020141
Visvanathan A, Patil V, Abdulla S, Hoheisel JD, Somasundaram K. N6-Methyladenosine Landscape of Glioma Stem-Like Cells: METTL3 Is Essential for the Expression of Actively Transcribed Genes and Sustenance of the Oncogenic Signaling. Genes. 2019; 10(2):141. https://doi.org/10.3390/genes10020141
Chicago/Turabian StyleVisvanathan, Abhirami, Vikas Patil, Shibla Abdulla, Jörg D. Hoheisel, and Kumaravel Somasundaram. 2019. "N6-Methyladenosine Landscape of Glioma Stem-Like Cells: METTL3 Is Essential for the Expression of Actively Transcribed Genes and Sustenance of the Oncogenic Signaling" Genes 10, no. 2: 141. https://doi.org/10.3390/genes10020141
APA StyleVisvanathan, A., Patil, V., Abdulla, S., Hoheisel, J. D., & Somasundaram, K. (2019). N6-Methyladenosine Landscape of Glioma Stem-Like Cells: METTL3 Is Essential for the Expression of Actively Transcribed Genes and Sustenance of the Oncogenic Signaling. Genes, 10(2), 141. https://doi.org/10.3390/genes10020141