The Mitochondrial Genomes of Neuropteridan Insects and Implications for the Phylogeny of Neuroptera


Abstract
:1. Introduction
2. Materials and Methods
2.1. Taxonomic Sampling
2.2. Molecular Data Collection
2.3. Sequence Alignment
2.4. DNA Analyses
2.5. Phylogenetic Analyses
3. Results
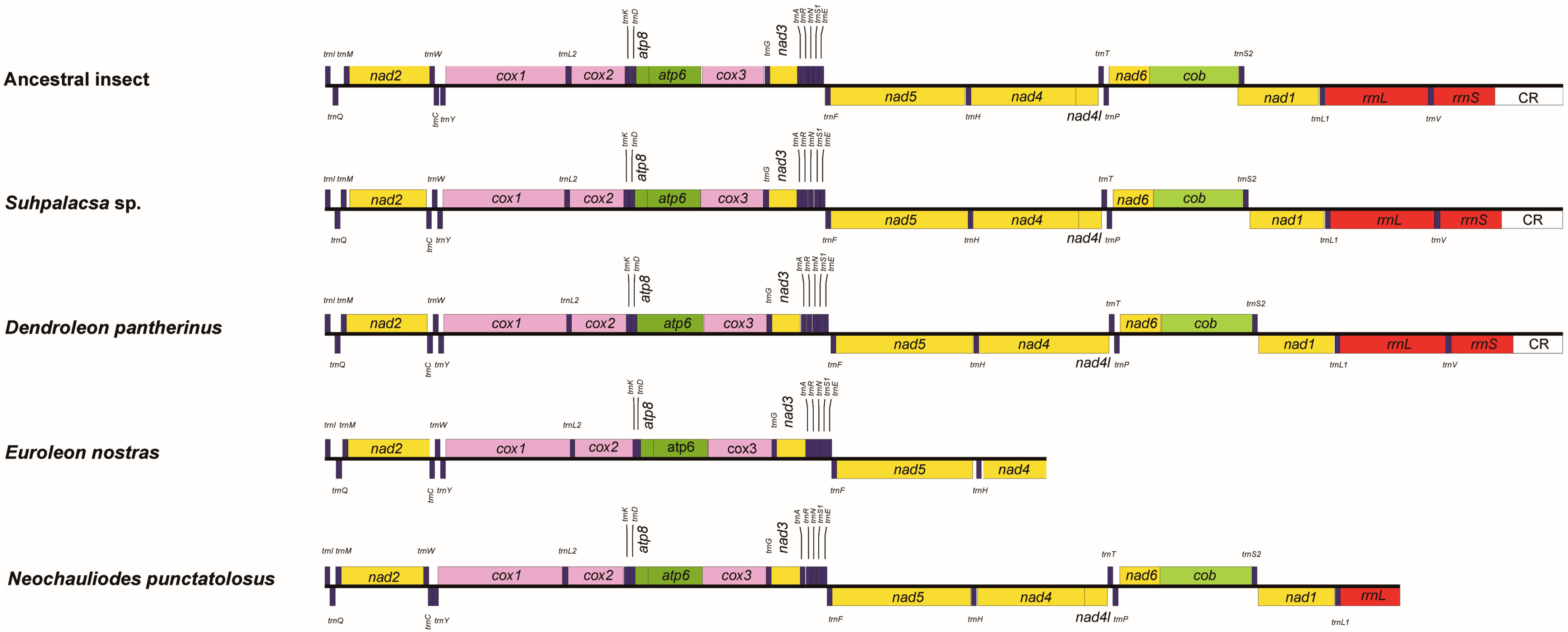
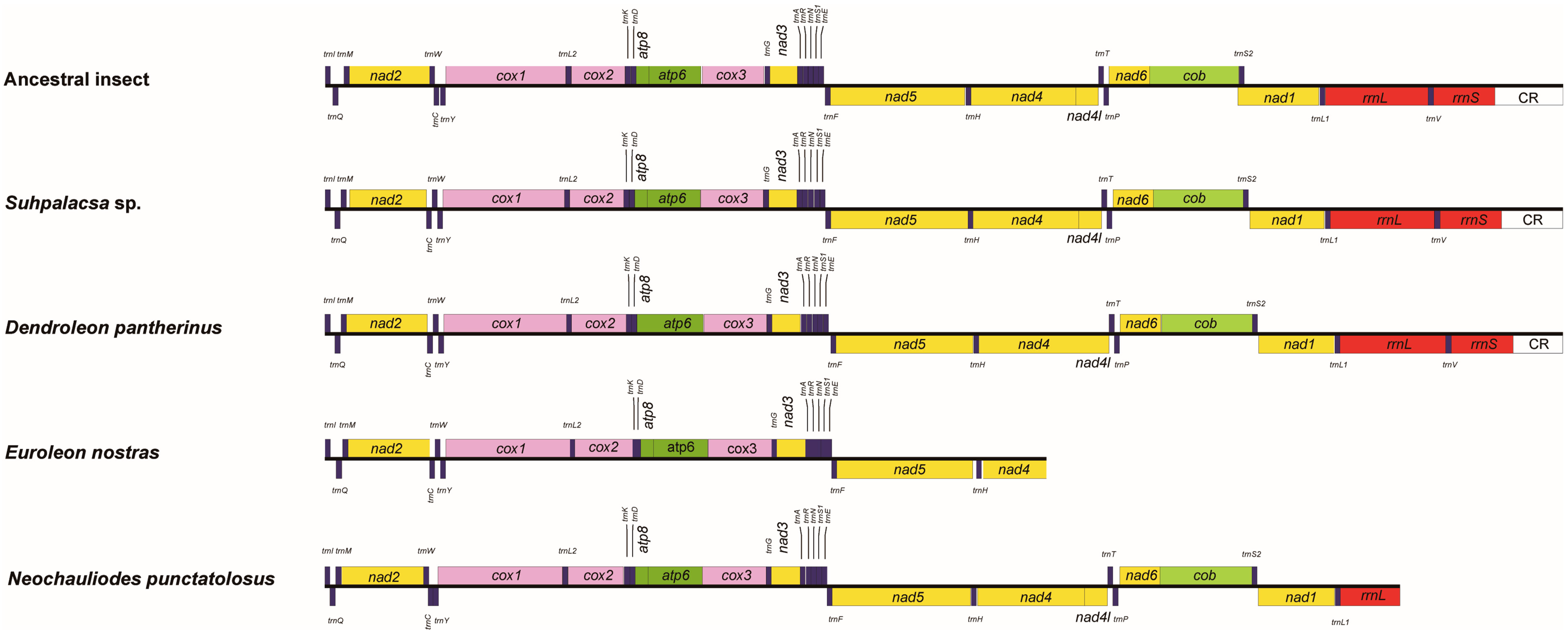
3.1. Reconstruction of Mitochondrial Genomes
3.2. Genome Structure
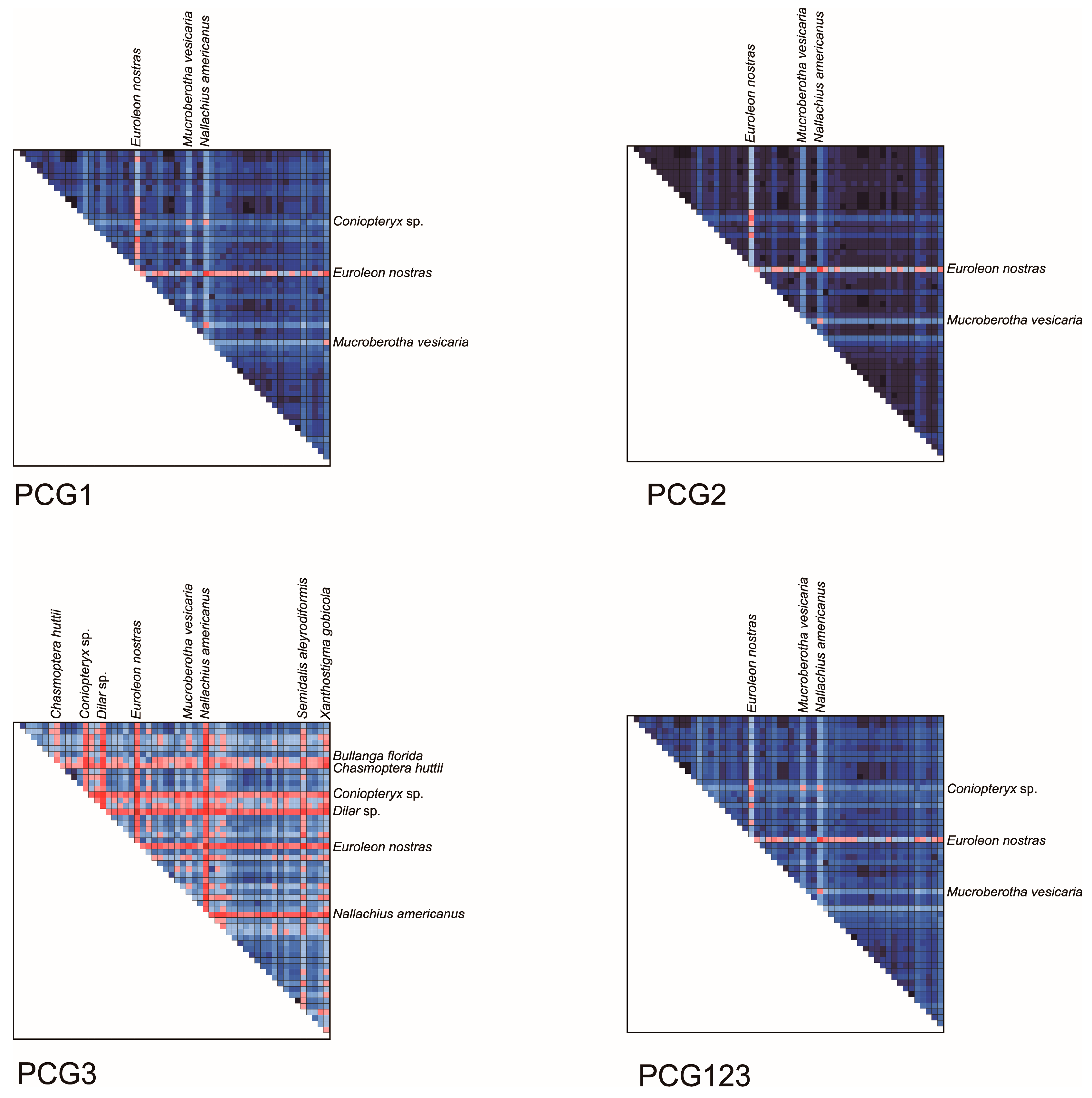
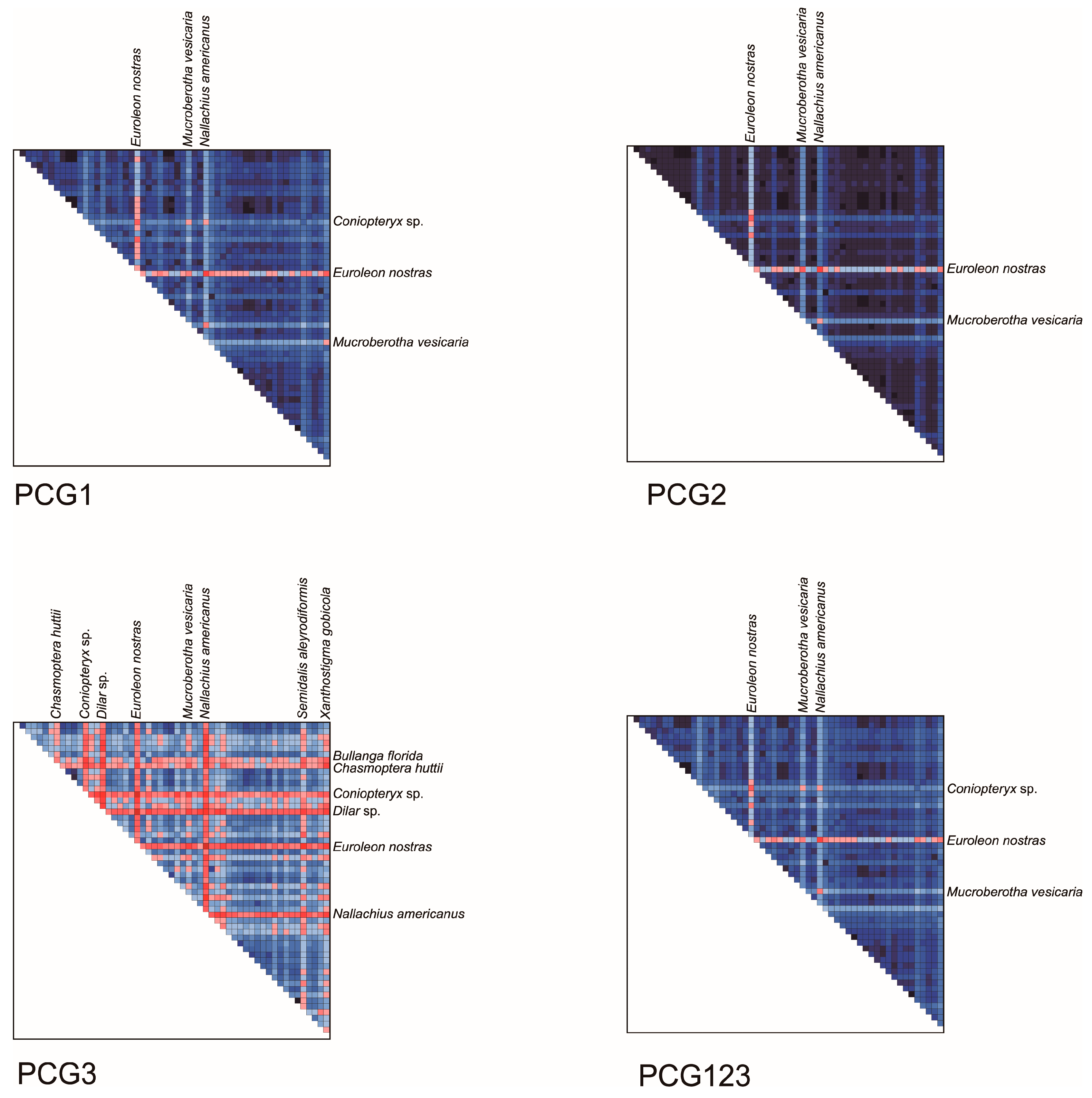
3.3. Characteristics of Data Matrix
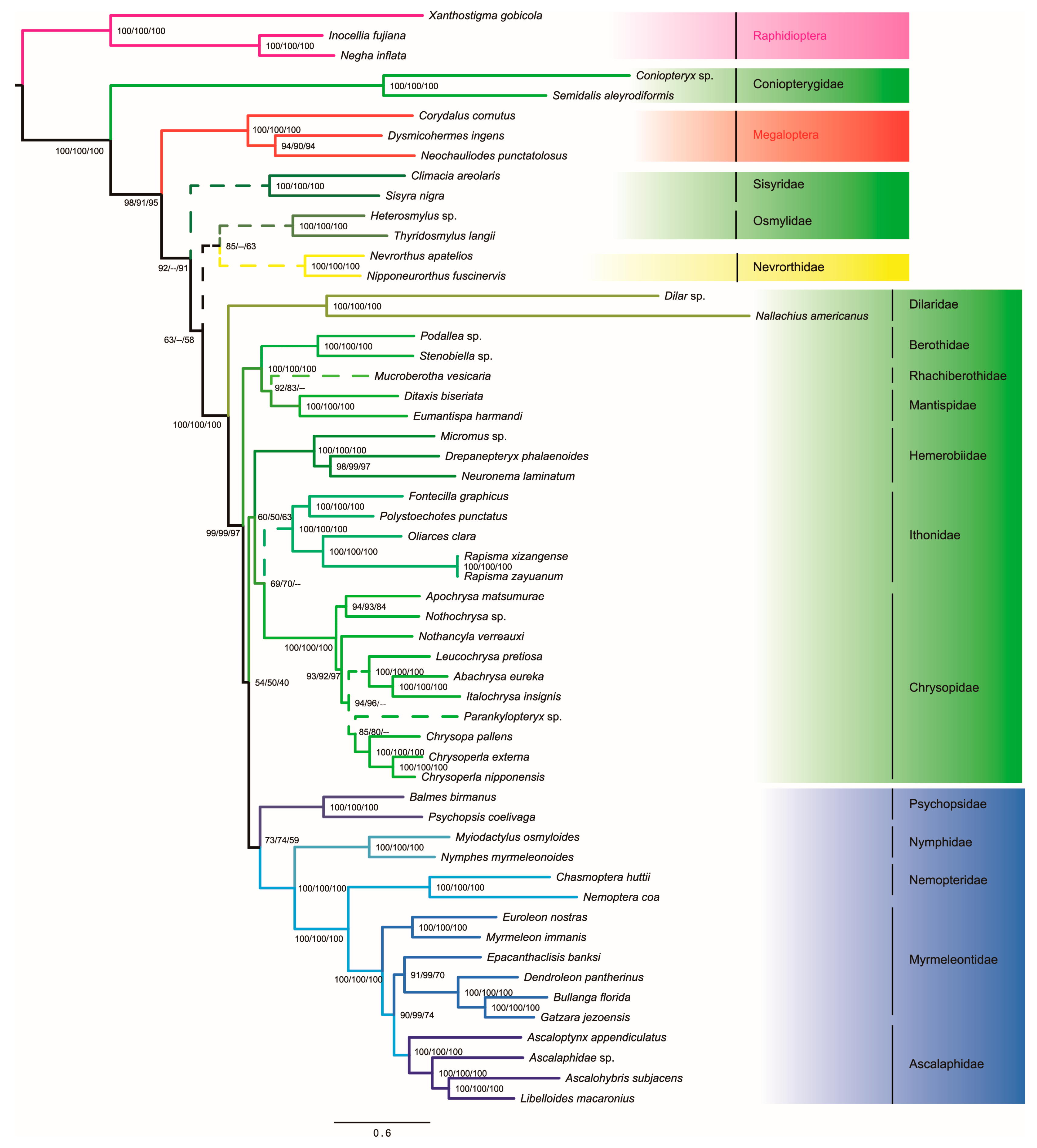
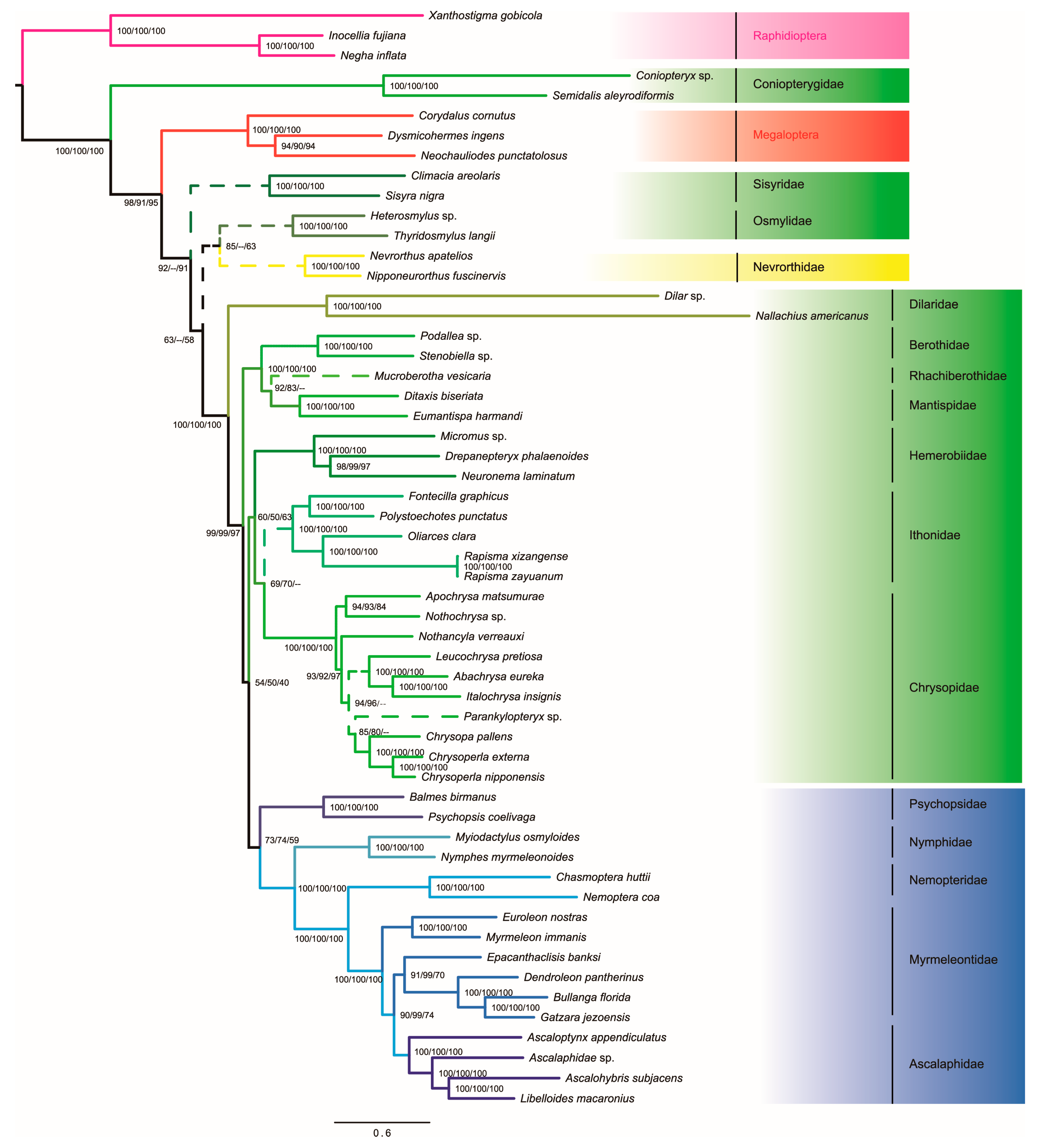
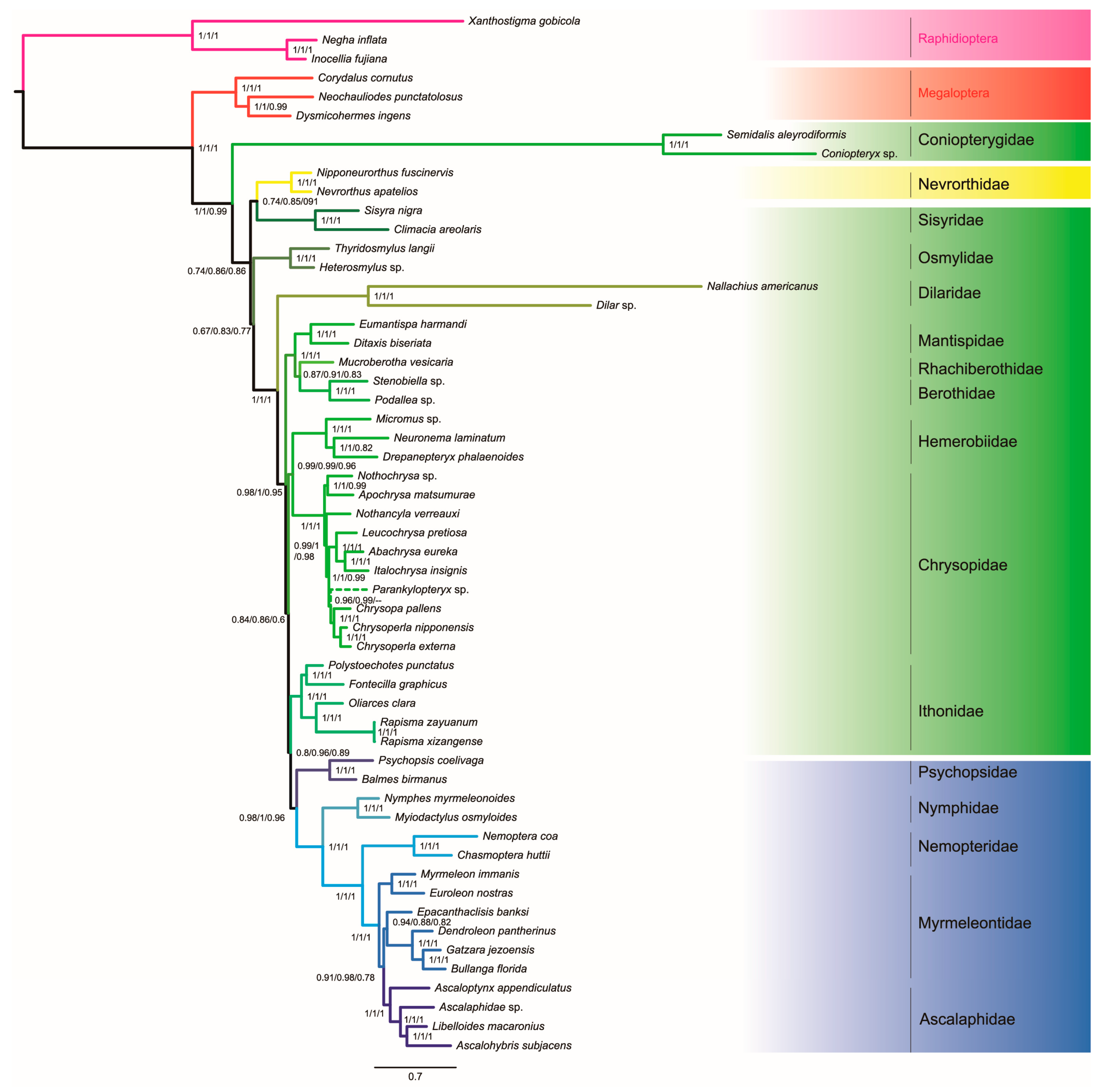
3.4. Phylogenetic Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Oswald, J.D. Lacewing Digital Library. Available online: http://lacewing.tamu.edu/ (accessed on 12 September 2015).
- Aspöck, U.; Plant, J.D.; Nemeschkal, H.L. Cladistic analysis of Neuroptera and their systematic position within Neuropterida (Insecta: Holometabola: Neuropterida: Neuroptera). Syst. Entomol. 2001, 26, 73–86. [Google Scholar] [CrossRef]
- Winterton, S.L.; Makarkin, V.N. Phylogeny of moth lacewings and giant lacewings (Neuroptera: Ithonidae, Polystoechotidae) using DNA sequence data, morphology, and fossils. Ann. Entomol. Soc. Am. 2010, 103, 511–522. [Google Scholar] [CrossRef]
- Winterton, S.L.; Lemmon, A.R.; Gillung, J.P.; Garzon, I.J.; Badano, D.; Bakkes, D.K.; Breitkreuz, L.C.V.; Engel, M.S.; Lemmon, E.M.; Liu, X.Y.; et al. Evolution of lacewings and allied orders using anchored phylogenomics (Neuroptera, Megaloptera, Raphidioptera). Syst. Entomol. 2018, 43, 330–354. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Garzón-Orduña, I.J.; Winterton, S.L.; Yan, Y.; Aspöck, U.; Aspöck, H.; Yang, D. Mitochondrial phylogenomics illuminates the evolutionary history of Neuropterida. Cladistics 2017, 33, 617–636. [Google Scholar] [CrossRef]
- Aspöck, U.; Haring, E.; Aspöck, H. The phylogeny of the Neuropterida: Long lasting and current controversies and challenges (Insecta: Endopterygota). Arthropod. Syst. Phyl. 2012, 70, 119–129. [Google Scholar]
- Beutel, R.G.; Friedrich, F.; Aspöck, U. The larval head of Nevrorthidae and the phylogeny of Neuroptera (Insecta). Zool. J. Linn. Soc-Lond. 2010, 158, 533–562. [Google Scholar] [CrossRef]
- Stange, L.A. Systematic catalog, bibliography, and classification of the world antlions (Insecta: Neuroptera: Myrmeleontidae). Am. Entomol. Inst. 2004, 74, 565. [Google Scholar]
- Badano, D.; Pantaleoni, R.A. The larvae of European Myrmeleontidae (Neuroptera). Zootaxa 2014, 3762, 1–71. [Google Scholar] [CrossRef]
- Michel, B.; Clamens, A.L.; Béthoux, O.; Kergoat, G.J.; Condamine, F.L. A first higher-level time-calibrated phylogeny of antlions (Neuroptera: Myrmeleontidae). Mol. Phylogenet. Evol. 2017, 107, 103–116. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, Y.; Liu, X.; Winterton, S.L.; Yang, D. The first mitochondrial genomes of antlion (Neuroptera: Myrmeleontidae) and split-footed lacewing (Neuroptera: Nymphidae), with phylogenetic implications of Myrmeleontiformia. Int. J. Biol. Sci. 2014, 10, 895. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, J. The mitochondrial genome of Gatzara jezoensis (Neuroptera: Myrmeleontidae) and phylogenetic analysis of Neuroptera. Biochem. Syst. Ecol. 2017, 71, 230–235. [Google Scholar] [CrossRef]
- Song, N.; Lin, A.; Zhao, X. Insight into higher-level phylogeny of Neuropterida: Evidence from secondary structures of mitochondrial rRNA genes and mitogenomic data. PLoS ONE 2018, 13, e0191826. [Google Scholar] [CrossRef] [PubMed]
- Winterton, S.L.; Hardy, N.B.; Wiegmann, B.M. On wings of lace: Phylogeny and Bayesian divergence time estimates of Neuropterida (Insecta) based on morphological and molecular data. Syst. Entomol. 2010, 35, 349–378. [Google Scholar] [CrossRef]
- Crampton-Platt, A.; Timmermans, M.J.; Gimmel, M.L.; Kutty, S.N.; Cockerill, T.D.; Vun Khen, C.; Vogler, A.P. Soup to Tree: The phylogeny of beetles inferred by mitochondrial metagenomics of a Bornean rainforest sample. Mol. Biol. Evol. 2015, 32, 2302–2316. [Google Scholar] [CrossRef]
- Gillett, C.P.; Crampton-Platt, A.; Timmermans, M.J.; Jordal, B.H.; Emerson, B.C.; Vogler, A.P. Bulk de novo mitogenome assembly from pooled total DNA elucidates the phylogeny of weevils (Coleoptera: Curculionoidea). Mol. Biol. Evol. 2014, 31, 2223–2237. [Google Scholar] [CrossRef]
- Li, H.; Leavengood, J.M., Jr.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.; Zhou, X.; Cai, W. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. Biol. Sci. 2017, 284. [Google Scholar] [CrossRef]
- Li, H.; Shao, R.; Song, N.; Song, F.; Jiang, P.; Li, Z.; Cai, W. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 2015, 5, 8527. [Google Scholar] [CrossRef]
- Liu, Y.; Song, F.; Jiang, P.; Wilson, J.J.; Cai, W.; Li, H. Compositional heterogeneity in true bug mitochondrial phylogenomics. Mol. Phylogenet. Evol. 2018, 118, 135–144. [Google Scholar] [CrossRef]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef]
- Simon, S.; Hadrys, H. A comparative analysis of complete mitochondrial genomes among Hexapoda. Mol. Phylogenet. Evol. 2013, 69, 393–403. [Google Scholar] [CrossRef]
- Song, N.; Li, H.; Song, F.; Cai, W.Z. Molecular phylogeny of Polyneoptera (Insecta) inferred from expanded mitogenomic data. Sci. Rep. 2016, 6, 36175. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, M.J.; Barton, C.; Haran, J.; Ahrens, D.; Culverwell, C.L.; Ollikainen, A.; Dodsworth, S.; Foster, P.G.; Bocak, L.; Vogler, A.P. Family-Level Sampling of Mitochondrial Genomes in Coleoptera: Compositional Heterogeneity and Phylogenetics. Genome Biol. Evol. 2015, 8, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Lepage, T.; Blanquart, S. PhyloBayes 3: A Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 2009, 25, 2286–2288. [Google Scholar] [CrossRef]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J. PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar] [CrossRef]
- Lartillot, N.; Brinkmann, H.; Philippe, H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 2007, 7, S4. [Google Scholar] [CrossRef] [PubMed]
- Boussau, B.; Walton, Z.; Delgado, J.A.; Collantes, F.; Beani, L.; Stewart, I.J.; Cameron, S.A.; Whitfield, J.B.; Johnston, J.S.; Holland, P.W.H.; et al. Strepsiptera, phylogenomics and the long branch attraction problem. PLoS ONE 2014, 9, e107709. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.; Liu, J.; Sun, C.; Vogler, A.P.; Cai, W. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multisample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic. Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Kuck, P.; Meusemann, K. FASconCAT: Convenient handling of data matrices. Mol. Phylogenet. Evol. 2010, 56, 1115–1118. [Google Scholar] [CrossRef]
- Kuck, P.; Meid, S.A.; Gross, C.; Wagele, J.W.; Misof, B. AliGROOVE—Visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinform. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Xia, X.; Lemey, P. Assessing substitution saturation with DAMBE. In The phylogenetic handbook: A practical approach to DNA and protein phylogeny, 2nd ed.; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Xia, X.H. DAMBE5: A Comprehensive Software Package for Data Analysis in molecular biological evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gateway Comput. Environ. Workshop (GCE) 2010, 14, 1–8. [Google Scholar]
- Aspöck, U. Phylogeny of the Neuropterida (Insecta: Holometabola). Zool. Scr. 2002, 31, 51–55. [Google Scholar] [CrossRef]
- Beutel, R.G.; Zimmermann, D.; Kraus, M.; Randolf, S.; Wipfler, B. Head morphology of Osmylus fulvicephalus (Osmylidae, Neuroptera) and its phylogenetic implications. Org. Divers. Evol. 2010, 10, 311–329. [Google Scholar] [CrossRef]
- Haring, E.; Aspöck, U. Phylogeny of the Neuropterida: A first molecular approach. Syst. Entomol. 2004, 29, 415–430. [Google Scholar] [CrossRef]
- Engel, M.S.; Winterton, S.L.; Breitkreuz, L.C.V. Phylogeny and Evolution of Neuropterida: Where Have Wings of Lace Taken Us? Annu. Rev. Entomol. 2018, 63, 531–551. [Google Scholar] [CrossRef] [PubMed]
- Zwick, P. Beschreibung der aquatischen Larve von Neurorthus [sic] fallax (Rambur) und Errichtung der neuen Planipennierfamilie Neurorthidae [sic] fam. nov. Gewässer Abwässer 1967, 44, 65–86. [Google Scholar]
- Randolf, S.; Zimmermann, D.; Aspöck, U. Head anatomy of adult Sisyra terminalis (Insecta: Neuroptera: Sisyridae) - functional adaptations and phylogenetic implications. Arthropod. Struct. Dev. 2013, 42, 565–582. [Google Scholar] [CrossRef] [PubMed]
- Randolf, S.; Zimmermann, D.; Aspöck, U. Head anatomy of adult Nevrorthus apatelios and basal splitting events in Neuroptera (Neuroptera: Nevrorthidae). Arthropod Syst. Phyl. 2014, 72, 111–136. [Google Scholar]
- Winterton, S.L.; Zhao, J.; Garzon-Orduna, I.J.; Wang, Y.J.; Liu, Z.Q. The phylogeny of lance lacewings (Neuroptera: Osmylidae). Syst. Entomol. 2017, 42, 555–574. [Google Scholar] [CrossRef]
- Aspöck, U.; Aspöck, H. Phylogenetic relevance of the genital sclerites of Neuropterida (Insecta: Holometabola). Syst. Entomol. 2008, 33, 97–127. [Google Scholar] [CrossRef]
- Winterton, S.L. Molecular phylogeny of Neuropterida with emphasis on the lacewings (Neuroptera). Entomol. Abh 2003, 61, 158–160. [Google Scholar]
- Zimmermann, D.; Klepal, W.; Aspöck, U. The first holistic SEM study of Coniopterygidae (Neuroptera) - structural evidence and phylogenetic implications. Eur. J. Entomol. 2009, 106, 651–662. [Google Scholar] [CrossRef]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef]
- Randolf, S.; Zimmermann, D.; Aspöck, U. Head anatomy of adult Coniopteryx pygmaea Enderlein 1906: Effects of miniaturization and the systematic position of Coniopterygidae (Insecta: Neuroptera). Arthropod. Struct. Dev. 2017, 46, 304–322. [Google Scholar] [CrossRef]
- Zimmermann, D.; Randolf, S.; Metscher, B.; Aspöck, U. The function and phylogenetic implications of the tentorium in adult Neuroptera (Insecta). Arthropod Struct. Dev. 2011, 40, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Willmann, R. The phylogenetic position of the Rhachiberothinae and the basal sister-group relationships within the Mantispidae (Neuroptera). Syst. Entomol. 1990, 15, 253–265. [Google Scholar] [CrossRef]
- Aspöck, U.; Mansell, M. Arevision of the family Rhachiberothidae Tjeder, 1959, stat. n. (Neuroptera). Syst. Entomol. 1994, 19, 181–206. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Order | Suborder | Family | Species | Accession |
|---|---|---|---|---|---|
| number | |||||
| Ingroup | Neuroptera | Myrmeleontiformia | Ascalaphidae | Ascalohybris subjacens | KC758703 |
| Neuroptera | Myrmeleontiformia | Ascalaphidae | Ascaloptynx appendiculatus | FJ171324 | |
| Neuroptera | Myrmeleontiformia | Ascalaphidae | Libelloides macaronius | NC_015609 | |
| Neuroptera | Myrmeleontiformia | Ascalaphidae | Suhpalacsa sp. | MK301247 | |
| Neuroptera | Hemerobiiformia | Rhachiberothidae | Mucroberotha vesicaria | KT425092 | |
| Neuroptera | Hemerobiiformia | Berothidae | Podallea sp. | KT425091 | |
| Neuroptera | Hemerobiiformia | Berothidae | Stenobiella sp. | KT425081 | |
| Neuroptera | Hemerobiiformia | Chrysopidae | Abachrysa eureka | KY587199 | |
| Neuroptera | Hemerobiiformia | Chrysopidae | Apochrysa matsumurae | NC_015095 | |
| Neuroptera | Hemerobiiformia | Chrysopidae | Chrysopa pallens | NC_019618 | |
| Neuroptera | Hemerobiiformia | Chrysopidae | Chrysoperla externa | KU877169 | |
| Neuroptera | Hemerobiiformia | Chrysopidae | Chrysoperla nipponensis | NC_015093 | |
| Neuroptera | Hemerobiiformia | Chrysopidae | Italochrysa insignis | KY587200 | |
| Neuroptera | Hemerobiiformia | Chrysopidae | Leucochrysa pretiosa | KY587201 | |
| Neuroptera | Hemerobiiformia | Chrysopidae | Nothancyla verreauxi | KP264629 | |
| Neuroptera | Hemerobiiformia | Chrysopidae | Nothochrysa sp. | KP264630 | |
| Neuroptera | Hemerobiiformia | Chrysopidae | Parankylopteryx sp. | KY587202 | |
| Neuroptera | Hemerobiiformia | Coniopterygidae | Coniopteryx sp. | KT425078 | |
| Neuroptera | Hemerobiiformia | Coniopterygidae | Semidalis aleyrodiformis | KT425067 | |
| Neuroptera | Hemerobiiformia | Dilaridae | Dilar sp. | KT425073 | |
| Neuroptera | Hemerobiiformia | Dilaridae | Nallachius americanus | KT425071 | |
| Neuroptera | Hemerobiiformia | Hemerobiidae | Drepanepteryx phalaenoides | KT425087 | |
| Neuroptera | Hemerobiiformia | Hemerobiidae | Micromus sp. | KT425075 | |
| Neuroptera | Hemerobiiformia | Hemerobiidae | Neuronema laminatum | KR078257 | |
| Neuroptera | Hemerobiiformia | Ithonidae | Fontecilla graphicus | KT425072 | |
| Neuroptera | Hemerobiiformia | Ithonidae | Oliarces clara | KT425090 | |
| Neuroptera | Hemerobiiformia | Ithonidae | Polystoechotes punctatus | FJ171325 | |
| Neuroptera | Hemerobiiformia | Ithonidae | Rapisma xizangense | KF626446 | |
| Neuroptera | Hemerobiiformia | Ithonidae | Rapisma zayuanum | KF626447 | |
| Neuroptera | Hemerobiiformia | Mantispidae | Ditaxis biseriata | FJ859906 | |
| Neuroptera | Hemerobiiformia | Mantispidae | Eumantispa harmandi | KT425080 | |
| Neuroptera | Myrmeleontiformia | Myrmeleontidae | Dendroleon pantherinus | MK301246 | |
| Neuroptera | Myrmeleontiformia | Myrmeleontidae | Euroleon nostras | MK301248 | |
| Neuroptera | Myrmeleontiformia | Myrmeleontidae | Bullanga florida | KX369241 | |
| Neuroptera | Myrmeleontiformia | Myrmeleontidae | Epacanthaclisis banksi | KF701327 | |
| Neuroptera | Myrmeleontiformia | Myrmeleontidae | Gatzara jezoensis | KY364372 | |
| Neuroptera | Myrmeleontiformia | Myrmeleontidae | Myrmeleon immanis | KJ461323 | |
| Neuroptera | Myrmeleontiformia | Nemopteridae | Chasmoptera huttii | KT425069 | |
| Neuroptera | Myrmeleontiformia | Nemopteridae | Nemoptera coa | KT425079 | |
| Neuroptera | Nevrorthiformia | Nevrorthidae | Nevrorthus apatelios | KT425074 | |
| Neuroptera | Nevrorthiformia | Nevrorthidae | Nipponeurorthus fuscinervis | KT425076 | |
| Neuroptera | Myrmeleontiformia | Nymphidae | Myiodactylus osmyloides | KT425089 | |
| Neuroptera | Myrmeleontiformia | Nymphidae | Nymphes myrmeleonoides | KJ461322 | |
| Neuroptera | Myrmeleontiformia | Osmylidae | Heterosmylus sp. | KT425077 | |
| Neuroptera | Myrmeleontiformia | Osmylidae | Thyridosmylus langii | KC515397 | |
| Neuroptera | Myrmeleontiformia | Psychopsidae | Balmes birmanus | KT425083 | |
| Neuroptera | Myrmeleontiformia | Psychopsidae | Psychopsis coelivaga | KT425082 | |
| Neuroptera | Hemerobiiformia | Sisyridae | Climacia areolaris | KT425088 | |
| Neuroptera | Hemerobiiformia | Sisyridae | Sisyra nigra | KT425070 | |
| Outgroup | Raphidioptera | Inocelliidae | Inocellia fujiana | KT425085 | |
| Raphidioptera | Inocelliidae | Negha inflata | KT425086 | ||
| Raphidioptera | Raphidiidae | Xanthostigma gobicola | KT425093 | ||
| Megaloptera | Corydalidae | Neochauliodes punctatolosus | MK301249 | ||
| Megaloptera | Sialidae | Dysmicohermes ingens | KJ806318 | ||
| Megaloptera | Corydalidae | Corydalus cornutus | FJ171323 | ||
| Species | Contig Length | Mapped Bases | Mean Coverage | Total Number of Reads | Mapped Paired Reads |
|---|---|---|---|---|---|
| Suhpalacsa sp. | 15,540 | 8,365,370 | 538 | 163,544,336 | 55,770/0.03% |
| Dendroleon pantherinus | 15,416 | 5,518,285 | 357 | 179,658,614 | 36,789/0.02% |
| Euroleon nostras | 9095 | 2,222,880 | 244 | 128,598,236 | 14,820/0.01% |
| Neochauliodes punctatolosus | - | - | - | - | - |
| N. punctatolosus contig-1 | 7013 | 340,174 | 49 | 128,598,236 | 2268/0.00% |
| N. punctatolosus contig-2 | 6391 | 607,749 | 95 | 128,598,236 | 4052/0.00% |
| Gene partitions | NumOTU | Issa | Iss.cSymb | Psymc | Iss.cAsymd | Pasyme |
|---|---|---|---|---|---|---|
| PCG1 | 32 | 0.349 | 0.808 | 0.000 | 0.554 | 0.000 |
| PCG2 | 32 | 0.240 | 0.808 | 0.000 | 0.554 | 0.000 |
| PCG3 | 32 | 0.736 | 0.808 | 0.000 | 0.554 | 0.000 |
| PCG123 | 32 | 0.406 | 0.818 | 0.000 | 0.572 | 0.000 |
| tRNA+rRNA | 32 | 0.452 | 0.807 | 0.000 | 0.549 | 0.000 |
| tRNA | 32 | 0.361 | 0.773 | 0.000 | 0.488 | 0.000 |
| rRNA | 32 | 0.511 | 0.787 | 0.000 | 0.515 | 0.798 |
| Species | dN | dS | dN/dS |
|---|---|---|---|
| Abachrysa eureka | 0.101 | 4.517 | 0.022 |
| Apochrysa matsumurae | 0.100 | 4.614 | 0.022 |
| Ascalohybris subjacens | 0.130 | 4.758 | 0.027 |
| Ascaloptynx appendiculatus | 0.109 | 4.770 | 0.023 |
| Balmes birmanus | 0.099 | 4.621 | 0.021 |
| Bullanga florida | 0.113 | 4.808 | 0.023 |
| Chasmoptera huttii | 0.126 | 4.839 | 0.026 |
| Chrysopa pallens | 0.097 | 4.601 | 0.021 |
| Chrysoperla externa | 0.097 | 4.580 | 0.021 |
| Chrysoperla nipponensis | 0.097 | 4.691 | 0.021 |
| Climacia areolaris | 0.131 | 4.647 | 0.028 |
| Coniopteryx sp. | 0.202 | 4.835 | 0.042 |
| Corydalus cornutus | 0.145 | 4.789 | 0.030 |
| Dendroleon pantherinus | 0.111 | 4.780 | 0.023 |
| Dilar sp. | 0.162 | 4.949 | 0.033 |
| Ditaxis biseriata | 0.105 | 4.648 | 0.023 |
| Drepanepteryx phalaenoides | 0.114 | 4.688 | 0.024 |
| Dysmicohermes ingens | 0.132 | 4.614 | 0.029 |
| Epacanthaclisis banksi | 0.106 | 4.795 | 0.022 |
| Eumantispa harmandi | 0.103 | 4.544 | 0.023 |
| Euroleon nostras | 0.110 | 4.743 | 0.023 |
| Fontecilla graphicus | 0.102 | 4.574 | 0.022 |
| Gatzara jezoensis | 0.114 | 4.704 | 0.024 |
| Heterosmylus sp. | 0.125 | 4.598 | 0.027 |
| Inocellia fujiana | 0.173 | 4.584 | 0.038 |
| Italochrysa insignis | 0.106 | 4.594 | 0.023 |
| Leucochrysa pretiosa | 0.098 | 4.554 | 0.022 |
| Libelloides macaronius | 0.111 | 4.835 | 0.023 |
| Micromus sp. | 0.113 | 4.626 | 0.024 |
| Mucroberotha vesicaria | 0.100 | 4.626 | 0.022 |
| Myiodactylus osmyloides | 0.108 | 4.669 | 0.023 |
| Myrmeleon immanis | 0.105 | 4.733 | 0.022 |
| Nallachius americanus | 0.175 | 4.860 | 0.036 |
| Negha inflata | 0.174 | 4.570 | 0.038 |
| Nemoptera coa | 0.140 | 4.768 | 0.029 |
| Neochauliodes punctatolosus | 0.136 | 4.779 | 0.028 |
| Neuronema laminatum | 0.118 | 4.667 | 0.025 |
| Nevrorthus apatelios | 0.119 | 4.654 | 0.025 |
| Nipponeurorthus fuscinervis | 0.118 | 4.596 | 0.026 |
| Nothancyla verreauxi | 0.099 | 4.545 | 0.022 |
| Nothochrysa sp. | 0.097 | 4.583 | 0.021 |
| Nymphes myrmeleonoides | 0.107 | 4.536 | 0.024 |
| Oliarces clara | 0.106 | 4.624 | 0.023 |
| Parankylopteryx sp. | 0.100 | 4.544 | 0.022 |
| Podallea sp. | 0.113 | 4.696 | 0.024 |
| Polystoechotes punctatus | 0.098 | 4.586 | 0.021 |
| Psychopsis coelivaga | 0.102 | 4.704 | 0.022 |
| Rapisma xizangense | 0.120 | 4.452 | 0.027 |
| Rapisma zayuanum | 0.120 | 4.450 | 0.027 |
| Semidalis aleyrodiformis | 0.185 | 4.728 | 0.039 |
| Sisyra nigra | 0.131 | 4.600 | 0.028 |
| Stenobiella sp. | 0.108 | 4.626 | 0.023 |
| Suhpalacsa sp. | 0.109 | 4.798 | 0.023 |
| Thyridosmylus langii | 0.134 | 4.725 | 0.028 |
| Xanthostigma gobicola | 0.185 | 4.691 | 0.039 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, N.; Li, X.-X.; Zhai, Q.; Bozdoğan, H.; Yin, X.-M. The Mitochondrial Genomes of Neuropteridan Insects and Implications for the Phylogeny of Neuroptera. Genes 2019, 10, 108. https://doi.org/10.3390/genes10020108
Song N, Li X-X, Zhai Q, Bozdoğan H, Yin X-M. The Mitochondrial Genomes of Neuropteridan Insects and Implications for the Phylogeny of Neuroptera. Genes. 2019; 10(2):108. https://doi.org/10.3390/genes10020108
Chicago/Turabian StyleSong, Nan, Xin-Xin Li, Qing Zhai, Hakan Bozdoğan, and Xin-Ming Yin. 2019. "The Mitochondrial Genomes of Neuropteridan Insects and Implications for the Phylogeny of Neuroptera" Genes 10, no. 2: 108. https://doi.org/10.3390/genes10020108
APA StyleSong, N., Li, X.-X., Zhai, Q., Bozdoğan, H., & Yin, X.-M. (2019). The Mitochondrial Genomes of Neuropteridan Insects and Implications for the Phylogeny of Neuroptera. Genes, 10(2), 108. https://doi.org/10.3390/genes10020108