D-loop Mutations in Renal Cell Carcinoma Improve Predictive Accuracy for Cancer-Related Death by Integrating with Mutations in the NADH Dehydrogenase Subunit 1 Gene

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Populations and Tissues
2.2. RCC Tissue Sampling
2.3. DNA Extraction, Polymerase Chain Reaction (PCR), and Sanger Sequencing
2.4. Somatic Mutations in the D-loop Region
2.5. Somatic Mutations in the MT-ND1 Gene
2.6. Statistical Analysis
3. Results
3.1. Demographic and Pathological Features of the Study Population
3.2. Mitochondrial D-loop Mutations
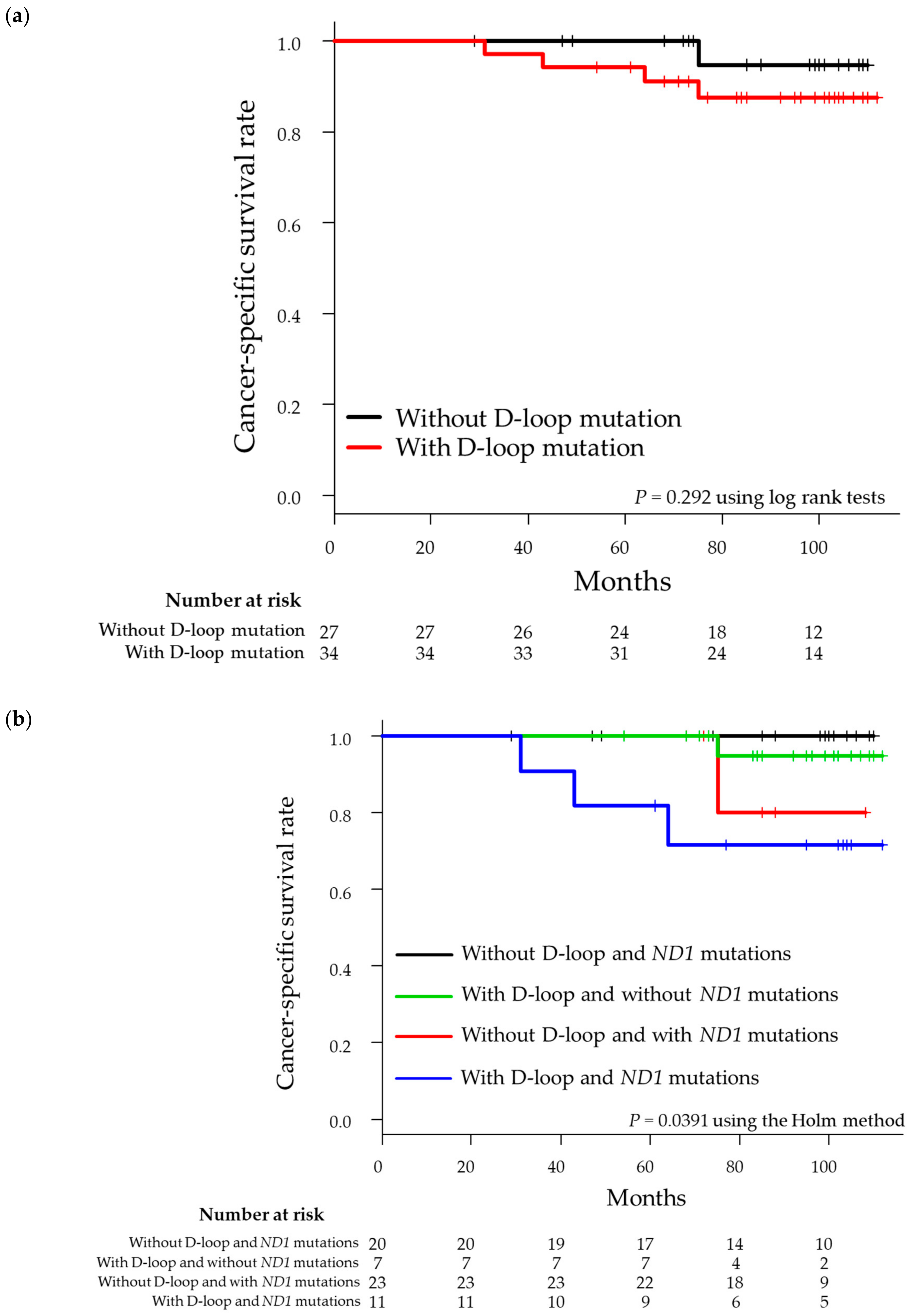
3.3. Survival Analysis for Integration of Mutations in the MT-ND1 Gene and D-loop Region
3.4. Relationship Between Mutation Site and Other Cancers Based on Database Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Inamura, K. Renal cell tumors: Understanding their molecular pathological epidemiology and the 2016 WHO classification. Int. J. Mol. Sci. 2017, 18, 2195. [Google Scholar] [CrossRef] [PubMed]
- Schraml, P.; Struckmann, K.; Hatz, F.; Sonnet, S.; Kully, C.; Gasser, T.; Sauter, G.; Mihatsch, M.J.; Moch, H. VHL mutations and their correlation with tumour cell proliferation, microvessel density, and patient prognosis in clear cell renal cell carcinoma. J. Pathol. 2002, 196, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Humphrey, P.A.; Ulbright, T.M.; Reuter, V.E. WHO classification of tumours of the urinary system and male genital organs. In WHO Classification of Tumours, 4th ed.; WHO Press: Geneva, Switzerland, 2016; Volume 8. [Google Scholar]
- Speicher, M.R.; Schoell, B.; du Manoir, S.; Schrock, E.; Ried, T.; Cremer, T.; Störkel, S.; Kovacs, A.; Kovacs, G. Specific loss of chromosomes 1, 2, 6, 10, 13, 17, and 21 in chromophobe renal cell carcinomas revealed by comparative genomic hybridization. Am. J. Pathol. 1994, 145, 356–364. [Google Scholar] [PubMed]
- Brunelli, M.; Eble, J.N.; Zhang, S.; Martignoni, G.; Delahunt, B.; Cheng, L. Eosinophilic and classic chromophobe renal cell carcinomas have similar frequent losses of multiple chromosomes from among chromosomes 1, 2, 6, 10, and 17, and this pattern of genetic abnormality is not present in renal oncocytoma. Mod. Pathol. 2005, 18, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef]
- Larman, T.C.; De Palma, S.R.; Hadjipanayis, A.G.; The Cancer Genome Atlas Research Network; Protopopov, A.; Zhang, J.; Gabriel, S.B.; Chin, L.; Seidman, C.E.; Kucherlapati, R.; et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc. Natl. Acad. Sci. USA. 2012, 109, 14087–14091. [Google Scholar] [CrossRef]
- Lata, S.; Neeru, S.; Neelam, P.; Sameer, B.; Seema, S.; Tapas, C.N.; Seema, K. Mutational analysis of the mitochondrial DNA displacement-loop region in human retinoblastoma with patient outcome. Pathol. Oncol. Res. 2018, 25, 503–512. [Google Scholar]
- Lian, L.; Rui, X.; Jiantao, C.; Wenmei, L.; Youyong, L. Investigation of frequent somatic mutations of MTND5 gene in gastric cancer cell lines and tissues. Mitochondrial DNA B 2018, 3, 1002–1008. [Google Scholar]
- Kim, H.; Komiyama, T.; Inomoto, C.; Kamiguchi, H.; Kajiwara, H.; Kobayashi, H.; Nakamura, N.; Terachi, T. Mutations in the mitochondrial ND1 gene are associated with postoperative prognosis of localized renal cell carcinoma. Int. J. Mol. Sci. 2016, 17, 2049. [Google Scholar] [CrossRef] [PubMed]
- Felix, A.U.; Felipe, M.; Alenka, L.; Cesar, C. The mitochondrial complex(I)ty of cancer. Front. Oncol. 2017, 7. [Google Scholar]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Sanchez-Cespedes, M.; Parrella, P.; Nomoto, S.; Cohen, D.; Xiao, Y.; Esteller, M.; Jeronimo, C.; Jordan, R.C.; Nicol, T.; Koch, W.M.; et al. Identification of a mononucleotide repeat as a major target for mitochondrial DNA alterations in human tumors. Cancer Res. 2001, 61, 7015–7019. [Google Scholar] [PubMed]
- Yoneyama, H.; Hara, T.; Kato, Y.; Yamori, T.; Matsuura, E.T.; Koike, K. Nucleotide sequence variation is frequent in the mitochondrial DNA displacement loop region of individual human tumor cells. Mol. Cancer Res. 2005, 3, 14–20. [Google Scholar] [PubMed]
- Nishikawa, M.; Nishiguchi, S.; Shiomi, S.; Tamori, A.; Koh, N.; Takeda, T.; Kubo, S.; Hirohashi, K.; Kinoshita, H.; Sato, E.; et al. Somatic mutation of mitochondrial DNA in cancerous and noncancerous liver tissue in individuals with hepatocellular carcinoma. Cancer Res. 2001, 61, 1843–1845. [Google Scholar]
- Boaventura, P.; Pereira, D.; Mendes, A.; Batista, R.; da Silva, A.F.; Guimaraes, I.; Honavar, M.; Teixeira-Gomes, J.; Lopes, J.M.; Maximo, V.; et al. Mitochondrial D310 D-loop instability and histological subtypes in radiation-induced cutaneous basal cell carcinomas. J. Dermatol. Sci. 2014, 73, 31–39. [Google Scholar] [CrossRef]
- Duberow, D.P.; Brait, M.; Hoque, M.O.; Theodorescu, D.; Sidransky, D.; Dasgupta, S.; Mathies, R.A. High-performance detection of somatic D-loop mutation in urothelial cell carcinoma patients by polymorphism ratio sequencing. J. Mol. Med. 2016, 94, 1015–1024. [Google Scholar] [CrossRef]
- Akouchekian, M.; Houshmand, M.; Hemati, S.; Ansaripour, M.; Shafa, M. High rate of mutation in mitochondrial DNA displacement loop region in human colorectal cancer. Dis. Colon Rectum 2009, 52, 526–530. [Google Scholar] [CrossRef]
- Bai, Y.; Guo, Z.; Xu, J.; Liu, S.; Zhang, J.; Cui, L.; Zhang, H.; Zhang, S. Single nucleotide polymorphisms in the D-loop region of mitochondrial DNA is associated with renal cell carcinoma outcome. Mitochondrial DNA 2015, 26, 224–226. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y. Investigation of the freely available easy-to-use software “EZR” for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Uno, H.; Cai, T.; Pencina, M.J.; D’Agostino, R.B.; Wei, L.J. On the C-statistics for evaluating overall adequacy of risk prediction procedures with censored survival data. Stat. Med. 2011, 30, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 2017, 8, 656. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Chang, S.C.; Ou, L.H.; Chen, C.M.; Hsieh, S.S.; Chung, Y.P.; King, K.L.; Lin, S.L.; Wei, Y.H. Mitochondrial DNA alterations correlate with the pathological status and the immunological ER, PR, HER-2/neu, p53 and Ki-67 expression in breast invasive ductal carcinoma. Oncol. Rep. 2015, 33, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J. NCCN Clinical Practice Guidelines in Oncology. In NCCN Guidelines®; NCCN: Pennsylvania, PA, USA, 2019. [Google Scholar]
- Powles, T.; Albiges, L.; Staehler, M.; Bensalah, K.; Dabestani, S.; Giles, R.H.; Hofmann, F.; Hora, M.; Kuczyk, M.A.; Lam, T.B.; et al. Updated European Association of Urology Guidelines Recommendations for the Treatment of First-line Metastatic Clear Cell Renal Cancer. Eur. Urol. 2018, 73, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Lin, J.C.; Wang, C.C.; Jiang, R.S.; Wang, W.Y.; Liu, S.A. Impact of somatic mutations in the D-loop of mitochondrial DNA on the survival of oral squamous cell carcinoma patients. PLoS ONE 2015, 10, e0124322. [Google Scholar] [CrossRef]
- Lin, C.S.; Chang, S.C.; Wang, L.S.; Chou, T.Y.; Hsu, W.H.; Wu, Y.C.; Wei, Y.H. The role of mitochondrial DNA alterations in esophageal squamous cell carcinomas. J. Thorac. Cardiovasc. Surg. 2010, 139, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Guo, Z.; Xu, J.; Zhang, J.; Cui, L.; Zhang, H.; Zhang, S. The 9-bp deletion at position 8272 in region V of mitochondrial DNA is associated with renal cell carcinoma outcome. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 1973–1975. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Variable | Overall (N = 61) | With D-Loop Mutations (N = 34) | Without D-Loop Mutations (N = 27) | p Value |
|---|---|---|---|---|
| Age (years), median (range) | 62 (39–80) | 67 (39–80) | 60 (39–79) | 0.188 |
| Sex | ||||
| Female | 14 | 10 (16.4) | 4 (6.6) | 0.228 |
| Male | 47 | 24 (39.3) | 23 (37.7) | |
| Histological subtype | ||||
| Clear cell subtype | 54 | 28 (45.9) | 26 (42.6) | 0.121 |
| Non clear subtype | 7 | 6 (9.8) | 1 (1.6) | |
| Tumor diameter (mm), median (range) | 32 (12–105) | 36.5 (13–100) | 28 (12–105) | 0.069 |
| Pathological T stage | 0.156 | |||
| T1a | 39 | 18 | 21 | |
| T1b | 14 | 11 | 3 | |
| T2a | 3 | 1 | 2 | |
| T2b | 0 | 0 | 0 | |
| T3a | 1 | 1 | 0 | |
| T3b | 3 | 2 | 1 | |
| T4 | 1 | 1 | 0 | |
| ISUP grade | 0.156 | |||
| 1 | 2 | 1 | 1 | |
| 2 | 50 | 25 | 25 | |
| 3 | 7 | 6 | 1 | |
| 4 | 2 | 2 | 0 | |
| MVI | ||||
| Absence | 48 | 25 (41.0) | 23 (37.7) | 0.352 |
| Presence | 13 | 9 (14.8) | 4 (6.6) |
| Patient | Base Change in the D-Loop | Number of Mutations | Base Change in MT-ND1 | Number of Mutations | Sum of Mutations |
|---|---|---|---|---|---|
| hk346 | None | 0 | None | 0 | 0 |
| hk347 | None | 0 | C4197Y | 2 | 2 |
| T4248Y | |||||
| hk348 | None | 0 | None | 0 | 0 |
| hk350 | C16260Y | 1 | None | 0 | 1 |
| hk352 | None | 0 | None | 0 | 0 |
| hk354 | C146Y | 2 | None | 0 | 2 |
| T152Y | |||||
| hk355 | A16183C | 4 | C3497T | 1 | 5 |
| T16189Y | |||||
| T16217Y | |||||
| T16223Y | |||||
| hk356 | None | 0 | None | 0 | 0 |
| hk357 | T16189Y | 1 | C4197Y | 1 | 2 |
| hk358 | None | 0 | None | 0 | 0 |
| hk359 | G94R | 1 | None | 0 | 1 |
| hk361 | C146Y | 3 | None | 0 | 3 |
| T152Y | |||||
| C16261Y | |||||
| hk362 | T16209Y | 2 | C3970Y | 1 | 3 |
| C16291Y | |||||
| hk363 | None | 0 | T4248Y | 1 | 1 |
| hk364 | C16527T | 1 | C3497T | 2 | 3 |
| G3635A | |||||
| hk365 | None | 0 | None | 0 | 0 |
| hk366 | None | 0 | None | 0 | 0 |
| hk367 | G16390R | 1 | None | 0 | 1 |
| hk368 | None | 0 | C4197Y | 2 | 2 |
| T4248Y | |||||
| hk369 | C16290Y | 2 | None | 0 | 2 |
| G16319R | |||||
| hk370 | A202R | 8 | None | 0 | 8 |
| 309insCC | |||||
| T16136C | |||||
| A16183C | |||||
| T16217Y | |||||
| T16223Y | |||||
| C16261Y | |||||
| C16527T | |||||
| hk371 | None | 0 | None | 0 | 0 |
| hk372 | T16311Y | 2 | A4200W | 2 | 4 |
| A16316R | T4216Y | ||||
| hk374 | None | 0 | None | 0 | 0 |
| hk375 | None | 0 | None | 0 | 0 |
| hk376 | 8C 303 2C | 2 | None | 0 | 2 |
| T310Y | |||||
| hk377 | T60W | 3 | None | 0 | 3 |
| C16261T | |||||
| T16368C | |||||
| hk379 | C16291T | 1 | None | 0 | 1 |
| hk380 | None | 0 | None | 0 | 0 |
| hk382 | None | 0 | A4200W | 2 | 2 |
| T4216Y | |||||
| hk383 | None | 0 | C3572ins | 1 | 1 |
| hk384 | 249Adel | 3 | None | 0 | 3 |
| A16203G | |||||
| C16291Y | |||||
| hk385 | G68A | 2 | G3496T | 3 | 5 |
| G16390R | C4141Y | ||||
| T4248Y | |||||
| hk387 | None | 0 | C4197Y | 2 | 2 |
| T4248Y | |||||
| hk388 | 249Adel | 3 | None | 0 | 3 |
| T485C | |||||
| C16344Y | |||||
| hk389 | C16245Y | 1 | None | 0 | 1 |
| hk390 | None | 0 | None | 0 | 0 |
| hk391 | None | 0 | None | 0 | 0 |
| hk392 | T16195C | 3 | G4048R | 2 | 5 |
| T16297C | C4071Y | ||||
| T16298C | |||||
| hk393 | T60C | 2 | T3368Y | 1 | 3 |
| G263A | |||||
| hk394 | None | 0 | A4200W | 2 | 2 |
| T4216Y | |||||
| hk395 | None | 0 | None | 0 | 0 |
| hk397 | None | 0 | None | 0 | 0 |
| hk398 | None | 0 | None | 0 | 0 |
| hk400 | T72Y | 2 | None | 0 | 2 |
| T310Y | |||||
| hk401 | A202G | 1 | T4117Y | 1 | 2 |
| hk402 | None | 0 | None | 0 | 0 |
| hk403 | G73R | 4 | C3328Y | 2 | 6 |
| C194Y | C3970T | ||||
| T204C | |||||
| T310Y | |||||
| hk404 | 249Adel | 2 | None | 0 | 2 |
| A16203G | |||||
| hk405 | C16320A | 1 | G4113R | 1 | 2 |
| hk407 | G73A | 8 | None | 0 | 8 |
| T131C | |||||
| C16111Y | |||||
| T16140Y | |||||
| C16234Y | |||||
| T16243Y | |||||
| C16291Y | |||||
| A16463G | |||||
| hk408 | None | 0 | None | 0 | 0 |
| hk410 | C6 568 C8 | 1 | None | 0 | 1 |
| hk411 | G251R | 2 | None | 0 | 2 |
| C194Y | |||||
| hk412 | None | 0 | None | 0 | 0 |
| hk413 | 249Adel | 2 | None | 0 | 2 |
| T16172Y | |||||
| hk414 | None | 0 | None | 0 | 0 |
| hk415 | C16291T | 1 | None | 0 | 1 |
| hk416 | C16291Y | 1 | None | 0 | 1 |
| hk417 | A16254G | 1 | None | 0 | 1 |
| hk418 | 249Adel | 5 | None | 0 | 5 |
| G16129R | |||||
| C16232M | |||||
| T16249Y | |||||
| C16344Y |
| Variables | Overall (N = 61) | Number of D-Loop Mutations Median (IQR) | p Value |
|---|---|---|---|
| Age (years) | 0.012 | ||
| ≤62 | 31 | 0 (0–1) | |
| >62 | 30 | 1.5 (0–2.75) | |
| Sex | 0.311 | ||
| Female | 14 | 1 (0.25–2) | |
| male | 47 | 1 (0–2) | |
| Histological subtype | 0.045 | ||
| Clear cell subtype | 54 | 1 (0–2) | |
| Non-clear cell subtype | 7 | 2 (1–4) | |
| Tumor diameter | 0.029 | ||
| ≤32 | 31 | 0 (0–1.5) | |
| >32 | 30 | 1 (0.25–2.0) | |
| Pathological T stage (pT) | 0.197 | ||
| ≤pT2 | 56 | 1 (0–2) | |
| ≥pT3 | 5 | 2 (1–3) | |
| ISUP grade | 0.029 | ||
| ≤ISUP2 | 52 | 0.5 (0–2) | |
| ≥ISUP3 | 9 | 2.0 (1–3) | |
| MVI | 0.257 | ||
| Absencee | 48 | 1 (0–2) | |
| Presence | 13 | 1 (0–2) |
| Model | C-Index | Lower | Upper |
|---|---|---|---|
| 95% CI | 95% CI | ||
| ND1 | 0.757 | 0.419 | 1.000 |
| ND1 and D-loop | 0.810 | 0.527 | 1.000 |
| Patient | D-Loop | MT-ND1 | Sites of Recurrence | ||||
|---|---|---|---|---|---|---|---|
| Our study Polymorphisms | Cancer Types | MITOMAP Polymorphisms | Our study Polymorphisms | Cancer Types | MITOMAP Polymorphisms | Cancer Types | |
| hk385 | G68A | Esophageal cancer PMID: 14639607 | G68A | T4248Y | Thyroid tumors PMID: 10803467 | T4248C | Liver Lung |
| G16390R | 1. Nasopharyngeal carcinoma PMID: 18376149 | G16390A | None | None | |||
| 2. Breast cancer PMID: 16568452 | G16390 A/G | ||||||
| 3. Ovarian cancer PMID: 18842121 | G16390A | ||||||
| hk392 | T16195C | None | G4048R | 1. Ovarian carcinoma PMID: 11507041 | G4048A | Local Lung | |
| 2. Colon cancer PMID: 29842994 | G4048A | ||||||
| T16297C | Nasopharyngeal carcinoma (PMID: 18376149) | T16297C | C4071Y | Ovarian carcinoma PMID: 11507041 | C4071T | ||
| T16298C | 1. Nasopharyngeal carcinoma PMID: 18376149 | T16298C | None | None | |||
| 2. Ovarian cancer PMID: 18842121 | T16298C | ||||||
| hk394 | None | None | T4216Y | 1. Thyroid tumors PMID: 10803467 | T4216C | Lung | |
| 2. Prostate cancer PMID: 11526508 | T4216C | ||||||
| 3. Colorectal cancer PMID: 19050702 | T4216C | ||||||
| hk400 | T72Y | Ovarian cancer PMID: 18842121 | T72C | None | None | Bone Local Lung | |
| T310Y | Nasopharyngeal carcinoma PMID: 18376149 | T310C | |||||
| hk403 | A73R | 1. Ovarian cancer PMID: 16942794, 18842121 | A73G | C3970T | 1. Ovarian carcinoma PMID: 11507041 | C3970T | Bone |
| 2. Nasopharyngeal carcinoma PMID: 18376149 | A73G | ||||||
| 2. Colorectal cancer PMID: 19050702 | C3970T | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Komiyama, T.; Nitta, M.; Kawamura, Y.; Hasegawa, M.; Shoji, S.; Orihashi, Y.; Inomoto, C.; Kajiwara, H.; Nakamura, N.; et al. D-loop Mutations in Renal Cell Carcinoma Improve Predictive Accuracy for Cancer-Related Death by Integrating with Mutations in the NADH Dehydrogenase Subunit 1 Gene. Genes 2019, 10, 998. https://doi.org/10.3390/genes10120998
Kim H, Komiyama T, Nitta M, Kawamura Y, Hasegawa M, Shoji S, Orihashi Y, Inomoto C, Kajiwara H, Nakamura N, et al. D-loop Mutations in Renal Cell Carcinoma Improve Predictive Accuracy for Cancer-Related Death by Integrating with Mutations in the NADH Dehydrogenase Subunit 1 Gene. Genes. 2019; 10(12):998. https://doi.org/10.3390/genes10120998
Chicago/Turabian StyleKim, Hakushi, Tomoyoshi Komiyama, Masahiro Nitta, Yoshiaki Kawamura, Masanori Hasegawa, Sunao Shoji, Yasushi Orihashi, Chie Inomoto, Hiroshi Kajiwara, Naoya Nakamura, and et al. 2019. "D-loop Mutations in Renal Cell Carcinoma Improve Predictive Accuracy for Cancer-Related Death by Integrating with Mutations in the NADH Dehydrogenase Subunit 1 Gene" Genes 10, no. 12: 998. https://doi.org/10.3390/genes10120998
APA StyleKim, H., Komiyama, T., Nitta, M., Kawamura, Y., Hasegawa, M., Shoji, S., Orihashi, Y., Inomoto, C., Kajiwara, H., Nakamura, N., Kobayashi, H., & Miyajima, A. (2019). D-loop Mutations in Renal Cell Carcinoma Improve Predictive Accuracy for Cancer-Related Death by Integrating with Mutations in the NADH Dehydrogenase Subunit 1 Gene. Genes, 10(12), 998. https://doi.org/10.3390/genes10120998