RNA-Dependent RNA Polymerase Speed and Fidelity are not the Only Determinants of the Mechanism or Efficiency of Recombination


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Plasmids, In Vitro Transcription, Cell Transfection, and Virus Quantification
2.3. Luciferase Assays
2.4. Virus Sequencing
2.5. One-Step Growth Analysis
2.6. Quantitative RT-PCR
2.7. Ribavirin-Sensitivity Assay
2.8. Cell-Based Recombination Assay
2.9. Polymerase Expression and Purification
2.10. Poly(rU) Polymerase Activity Assay
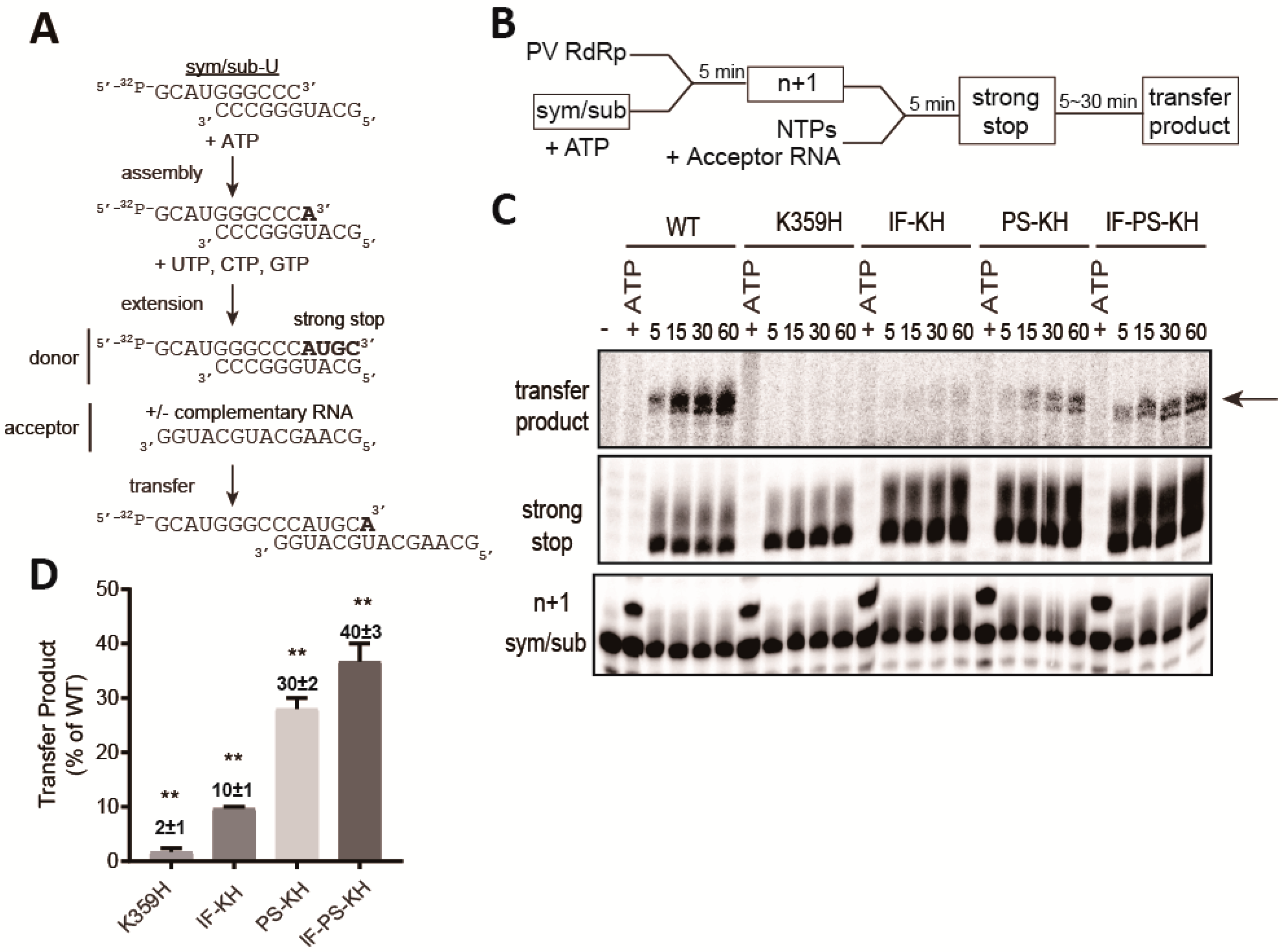
2.11. Sym/Sub-Based Template Switching Assay
3. Results
3.1. K359H PV Requires Two Second-Site Substitutions to Restore a “Wild-Type” Growth Phenotype
3.2. Characterization of the Double- and Triple-Mutant Viruses and Their Polymerases
3.3. Biochemical Properties of the RdRp Other Than Speed and Fidelity Contribute to the Efficiency of Recombination in Cell Culture
3.4. Poly(rU) Polymerase Activity as a Predictor of the Efficiency of Copy-Choice Recombination in Cell Culture
3.5. Polymerase Determinants Supporting Efficient Copy-Choice Recombination do not Overlap Completely With Determinants Supporting Efficient Forced-Copy-Choice Recombination
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- NIAID Emerging Infectious Diseases/Pathogens. Available online: https://www.niaid.nih.gov/research/emerging-infectious-diseases-pathogens (accessed on 12 September 2019).
- Graci, J.D.; Cameron, C.E. Therapeutically targeting RNA viruses via lethal mutagenesis. Future Virol. 2008, 3, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Perales, C.; Domingo, E. Antiviral Strategies Based on Lethal Mutagenesis and Error Threshold. Curr. Top. Microbiol. Immunol. 2016, 392, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, D.F.; Barnard, D.L. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Cameron, C.E.; Moustafa, I.M.; Arnold, J.J. Fidelity of Nucleotide Incorporation by the RNA-Dependent RNA Polymerase from Poliovirus. Enzymes 2016, 39, 293–323. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.A.; August, A.; Arnold, J.J.; Cameron, C.E. Polymerase Mechanism-Based Method of Viral Attenuation. Methods Mol. Biol. 2016, 1349, 83–104. [Google Scholar] [CrossRef]
- Weeks, S.A.; Lee, C.A.; Zhao, Y.; Smidansky, E.D.; August, A.; Arnold, J.J.; Cameron, C.E. A Polymerase mechanism-based strategy for viral attenuation and vaccine development. J. Biol. Chem. 2012, 287, 31618–31622. [Google Scholar] [CrossRef]
- Graham, R.L.; Becker, M.M.; Eckerle, L.D.; Bolles, M.; Denison, M.R.; Baric, R.S. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med. 2012, 18, 1820–1826. [Google Scholar] [CrossRef]
- Vignuzzi, M.; Wendt, E.; Andino, R. Engineering attenuated virus vaccines by controlling replication fidelity. Nat. Med. 2008, 14, 154–161. [Google Scholar] [CrossRef]
- Li, C.; Wang, H.; Yuan, T.; Woodman, A.; Yang, D.; Zhou, G.; Cameron, C.E.; Yu, L. Foot-and-mouth disease virus type O specific mutations determine RNA-dependent RNA polymerase fidelity and virus attenuation. Virology 2018, 518, 87–94. [Google Scholar] [CrossRef]
- Korboukh, V.K.; Lee, C.A.; Acevedo, A.; Vignuzzi, M.; Xiao, Y.; Arnold, J.J.; Hemperly, S.; Graci, J.D.; August, A.; Andino, R.; et al. RNA virus population diversity, an optimum for maximal fitness and virulence. J. Biol. Chem. 2014, 289, 29531–29544. [Google Scholar] [CrossRef]
- Rozen-Gagnon, K.; Stapleford, K.A.; Mongelli, V.; Blanc, H.; Failloux, A.B.; Saleh, M.C.; Vignuzzi, M. Alphavirus mutator variants present host-specific defects and attenuation in mammalian and insect models. PLoS Pathog. 2014, 10, e1003877. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Wang, H.; Xie, X.; Li, C.; Zhou, G.; Yang, D.; Yu, L. Ribavirin-resistant variants of foot-and-mouth disease virus: The effect of restricted quasispecies diversity on viral virulence. J. Virol. 2014, 88, 4008–4020. [Google Scholar] [CrossRef] [PubMed]
- Gnadig, N.F.; Beaucourt, S.; Campagnola, G.; Borderia, A.V.; Sanz-Ramos, M.; Gong, P.; Blanc, H.; Peersen, O.B.; Vignuzzi, M. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, E2294–E2303. [Google Scholar] [CrossRef]
- Coffey, L.L.; Beeharry, Y.; Borderia, A.V.; Blanc, H.; Vignuzzi, M. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proc. Natl. Acad. Sci. USA 2011, 108, 16038–16043. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.J.; Vignuzzi, M.; Stone, J.K.; Andino, R.; Cameron, C.E. Remote site control of an active site fidelity checkpoint in a viral RNA-dependent RNA polymerase. J. Biol. Chem. 2005, 280, 25706–25716. [Google Scholar] [CrossRef]
- Fitzsimmons, W.J.; Woods, R.J.; McCrone, J.T.; Woodman, A.; Arnold, J.J.; Yennawar, M.; Evans, R.; Cameron, C.E.; Lauring, A.S. A speed-fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol. 2018, 16, e2006459. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, H.; Shi, J.; Yang, D.; Zhou, G.; Chang, J.; Cameron, C.E.; Woodman, A.; Yu, L. Senecavirus-Specific Recombination Assays Reveal the Intimate Link between Polymerase Fidelity and RNA Recombination. J. Virol. 2019. [Google Scholar] [CrossRef]
- Woodman, A.; Lee, K.M.; Janissen, R.; Gong, Y.N.; Dekker, N.H.; Shih, S.R.; Cameron, C.E. Predicting Intraserotypic Recombination in Enterovirus 71. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Te Velthuis, A.J.W.; Long, J.C.; Bauer, D.L.V.; Fan, R.L.Y.; Yen, H.L.; Sharps, J.; Siegers, J.Y.; Killip, M.J.; French, H.; Oliva-Martin, M.J.; et al. Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat. Microbiol. 2018, 3, 1234–1242. [Google Scholar] [CrossRef]
- Rawson, J.M.O.; Nikolaitchik, O.A.; Keele, B.F.; Pathak, V.K.; Hu, W.S. Recombination is required for efficient HIV-1 replication and the maintenance of viral genome integrity. Nucleic Acids Res. 2018, 46, 10535–10545. [Google Scholar] [CrossRef] [PubMed]
- Woodman, A.; Arnold, J.J.; Cameron, C.E.; Evans, D.J. Biochemical and genetic analysis of the role of the viral polymerase in enterovirus recombination. Nucleic Acids Res. 2016, 44, 6883–6895. [Google Scholar] [CrossRef] [PubMed]
- Poirier, E.Z.; Mounce, B.C.; Rozen-Gagnon, K.; Hooikaas, P.J.; Stapleford, K.A.; Moratorio, G.; Vignuzzi, M. Low-Fidelity Polymerases of Alphaviruses Recombine at Higher Rates to Overproduce Defective Interfering Particles. J. Virol. 2015, 90, 2446–2454. [Google Scholar] [CrossRef] [PubMed]
- Castro, C.; Smidansky, E.D.; Arnold, J.J.; Maksimchuk, K.R.; Moustafa, I.; Uchida, A.; Gotte, M.; Konigsberg, W.; Cameron, C.E. Nucleic acid polymerases use a general acid for nucleotidyl transfer. Nat. Struct. Mol. Biol. 2009, 16, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Gohara, D.W.; Ha, C.S.; Kumar, S.; Ghosh, B.; Arnold, J.J.; Wisniewski, T.J.; Cameron, C.E. Production of “authentic” poliovirus RNA-dependent RNA polymerase (3D(pol)) by ubiquitin-protease-mediated cleavage in Escherichia coli. Protein Expr. Purif. 1999, 17, 128–138. [Google Scholar] [CrossRef]
- Gohara, D.W.; Crotty, S.; Arnold, J.J.; Yoder, J.D.; Andino, R.; Cameron, C.E. Poliovirus RNA-dependent RNA polymerase (3Dpol): Structural, biochemical, and biological analysis of conserved structural motifs A and B. J. Biol. Chem. 2000, 275, 25523–25532. [Google Scholar] [CrossRef]
- Kumagai, A.; Ando, R.; Miyatake, H.; Greimel, P.; Kobayashi, T.; Hirabayashi, Y.; Shimogori, T.; Miyawaki, A. A bilirubin-inducible fluorescent protein from eel muscle. Cell 2013, 153, 1602–1611. [Google Scholar] [CrossRef]
- Oh, H.S.; Banerjee, S.; Aponte-Diaz, D.; Sharma, S.D.; Aligo, J.; Lodeiro, M.F.; Ning, G.; Sharma, R.; Arnold, J.J.; Cameron, C.E. Multiple poliovirus-induced organelles suggested by comparison of spatiotemporal dynamics of membranous structures and phosphoinositides. PLoS Pathog. 2018, 14, e1007036. [Google Scholar] [CrossRef]
- Kempf, B.J.; Peersen, O.B.; Barton, D.J. Poliovirus Polymerase Leu420 Facilitates RNA Recombination and Ribavirin Resistance. J. Virol. 2016, 90, 8410–8421. [Google Scholar] [CrossRef]
- Lowry, K.; Woodman, A.; Cook, J.; Evans, D.J. Recombination in enteroviruses is a biphasic replicative process involving the generation of greater-than genome length ‘imprecise’ intermediates. PLoS Pathog. 2014, 10, e1004191. [Google Scholar] [CrossRef]
- Arnold, J.J.; Cameron, C.E. Poliovirus RNA-dependent RNA polymerase (3Dpol) is sufficient for template switching in vitro. J. Biol. Chem. 1999, 274, 2706–2716. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.; Hong, Z.; Andino, R.; Cameron, C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000, 6, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 2003, 100, 7289–7294. [Google Scholar] [CrossRef] [PubMed]
- Severson, W.E.; Schmaljohn, C.S.; Javadian, A.; Jonsson, C.B. Ribavirin causes error catastrophe during Hantaan virus replication. J. Virol. 2003, 77, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Andino, R. Ribavirin and lethal mutagenesis of poliovirus: Molecular mechanisms, resistance and biological implications. Virus Res. 2005, 107, 173–181. [Google Scholar] [CrossRef]
- Sierra, M.; Airaksinen, A.; Gonzalez-Lopez, C.; Agudo, R.; Arias, A.; Domingo, E. Foot-and-mouth disease virus mutant with decreased sensitivity to ribavirin: Implications for error catastrophe. J. Virol. 2007, 81, 2012–2024. [Google Scholar] [CrossRef]
- Arias, A.; Arnold, J.J.; Sierra, M.; Smidansky, E.D.; Domingo, E.; Cameron, C.E. Determinants of RNA-dependent RNA polymerase (in)fidelity revealed by kinetic analysis of the polymerase encoded by a foot-and-mouth disease virus mutant with reduced sensitivity to ribavirin. J. Virol. 2008, 82, 12346–12355. [Google Scholar] [CrossRef]
- Pathak, H.B.; Ghosh, S.K.; Roberts, A.W.; Sharma, S.D.; Yoder, J.D.; Arnold, J.J.; Gohara, D.W.; Barton, D.J.; Paul, A.V.; Cameron, C.E. Structure-function relationships of the RNA-dependent RNA polymerase from poliovirus (3Dpol). A surface of the primary oligomerization domain functions in capsid precursor processing and VPg uridylylation. J. Biol. Chem. 2002, 277, 31551–31562. [Google Scholar] [CrossRef]
- Herold, J.; Andino, R. Poliovirus requires a precise 5′ end for efficient positive-strand RNA synthesis. J. Virol. 2000, 74, 6394–6400. [Google Scholar] [CrossRef]
- Andino, R.; Rieckhof, G.E.; Achacoso, P.L.; Baltimore, D. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 1993, 12, 3587–3598. [Google Scholar] [CrossRef]
- Banerjee, S.; Aponte-Diaz, D.; Yeager, C.; Sharma, S.D.; Ning, G.; Oh, H.S.; Han, Q.; Umeda, M.; Hara, Y.; Wang, R.Y.L.; et al. Hijacking of multiple phospholipid biosynthetic pathways and induction of membrane biogenesis by a picornaviral 3CD protein. PLoS Pathog. 2018, 14, e1007086. [Google Scholar] [CrossRef] [PubMed]
- Dulin, D.; Vilfan, I.D.; Berghuis, B.A.; Hage, S.; Bamford, D.H.; Poranen, M.M.; Depken, M.; Dekker, N.H. Elongation-Competent Pauses Govern the Fidelity of a Viral RNA-Dependent RNA Polymerase. Cell Rep. 2015, 10, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, K.; Baltimore, D. The mechanism of RNA recombination in poliovirus. Cell 1986, 47, 433–443. [Google Scholar] [CrossRef]
- Arnold, J.J.; Cameron, C.E. Poliovirus RNA-dependent RNA polymerase (3D(pol)). Assembly of stable, elongation-competent complexes by using a symmetrical primer-template substrate (sym/sub). J. Biol. Chem. 2000, 275, 5329–5336. [Google Scholar] [CrossRef]
- Peliska, J.A.; Benkovic, S.J. Fidelity of in vitro DNA strand transfer reactions catalyzed by HIV-1 reverse transcriptase. Biochemistry 1994, 33, 3890–3895. [Google Scholar] [CrossRef]
- Peliska, J.A.; Benkovic, S.J. Mechanism of DNA strand transfer reactions catalyzed by HIV-1 reverse transcriptase. Science 1992, 258, 1112–1118. [Google Scholar] [CrossRef]
- Peliska, J.A.; Balasubramanian, S.; Giedroc, D.P.; Benkovic, S.J. Recombinant HIV-1 nucleocapsid protein accelerates HIV-1 reverse transcriptase catalyzed DNA strand transfer reactions and modulates RNase H activity. Biochemistry 1994, 33, 13817–13823. [Google Scholar] [CrossRef]
- Castro, C.; Smidansky, E.; Maksimchuk, K.R.; Arnold, J.J.; Korneeva, V.S.; Gotte, M.; Konigsberg, W.; Cameron, C.E. Two proton transfers in the transition state for nucleotidyl transfer catalyzed by RNA- and DNA-dependent RNA and DNA polymerases. Proc. Natl. Acad. Sci. USA 2007, 104, 4267–4272. [Google Scholar] [CrossRef]
- Parsley, T.B.; Cornell, C.T.; Semler, B.L. Modulation of the RNA binding and protein processing activities of poliovirus polypeptide 3CD by the viral RNA polymerase domain. J. Biol. Chem. 1999, 274, 12867–12876. [Google Scholar] [CrossRef]
- Harris, K.S.; Reddigari, S.R.; Nicklin, M.J.; Hammerle, T.; Wimmer, E. Purification and characterization of poliovirus polypeptide 3CD, a proteinase and a precursor for RNA polymerase. J. Virol. 1992, 66, 7481–7489. [Google Scholar]
- Muller, H.J. The Relation of Recombination to Mutational Advance. Mutat. Res. 1964, 106, 2–9. [Google Scholar] [CrossRef]
- Felsenstein, J. The evolutionary advantage of recombination. Genetics 1974, 78, 737–756. [Google Scholar] [PubMed]
- Chao, L. Fitness of RNA virus decreased by Muller’s ratchet. Nature 1990, 348, 454–455. [Google Scholar] [CrossRef] [PubMed]
- Kempf, B.J.; Watkins, C.L.; Peersen, O.B.; Barton, D.J. Picornavirus RNA Recombination Counteracts Error Catastrophe. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Furio, V.; Moya, A.; Sanjuan, R. The cost of replication fidelity in an RNA virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10233–10237. [Google Scholar] [CrossRef]
- Elena, S.F.; Sanjuan, R. Adaptive value of high mutation rates of RNA viruses: Separating causes from consequences. J. Virol. 2005, 79, 11555–11558. [Google Scholar] [CrossRef]
- Furio, V.; Moya, A.; Sanjuan, R. The cost of replication fidelity in human immunodeficiency virus type 1. Proc. Biol. Sci. 2007, 274, 225–230. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide | Sequence (5′-3′) |
|---|---|
| 3D EcoRI-R | AAT TAA TTC ATC GAT GAA TTC GGG CC |
| 3D BglII-F | GGC AAA GAA GTG GAG ATC TTG GAT GC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Ellis, V.D., III; Woodman, A.; Zhao, Y.; Arnold, J.J.; Cameron, C.E. RNA-Dependent RNA Polymerase Speed and Fidelity are not the Only Determinants of the Mechanism or Efficiency of Recombination. Genes 2019, 10, 968. https://doi.org/10.3390/genes10120968
Kim H, Ellis VD III, Woodman A, Zhao Y, Arnold JJ, Cameron CE. RNA-Dependent RNA Polymerase Speed and Fidelity are not the Only Determinants of the Mechanism or Efficiency of Recombination. Genes. 2019; 10(12):968. https://doi.org/10.3390/genes10120968
Chicago/Turabian StyleKim, Hyejeong, Victor D. Ellis, III, Andrew Woodman, Yan Zhao, Jamie J. Arnold, and Craig E. Cameron. 2019. "RNA-Dependent RNA Polymerase Speed and Fidelity are not the Only Determinants of the Mechanism or Efficiency of Recombination" Genes 10, no. 12: 968. https://doi.org/10.3390/genes10120968
APA StyleKim, H., Ellis, V. D., III, Woodman, A., Zhao, Y., Arnold, J. J., & Cameron, C. E. (2019). RNA-Dependent RNA Polymerase Speed and Fidelity are not the Only Determinants of the Mechanism or Efficiency of Recombination. Genes, 10(12), 968. https://doi.org/10.3390/genes10120968