HisCoM-PAGE: Hierarchical Structural Component Models for Pathway Analysis of Gene Expression Data



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. SNUH—PDAC Microarray Data
2.1.2. The Cancer Genome Atlas - PDAC RNA-Seq Data
2.1.3. Simulation Data
2.2. Methods
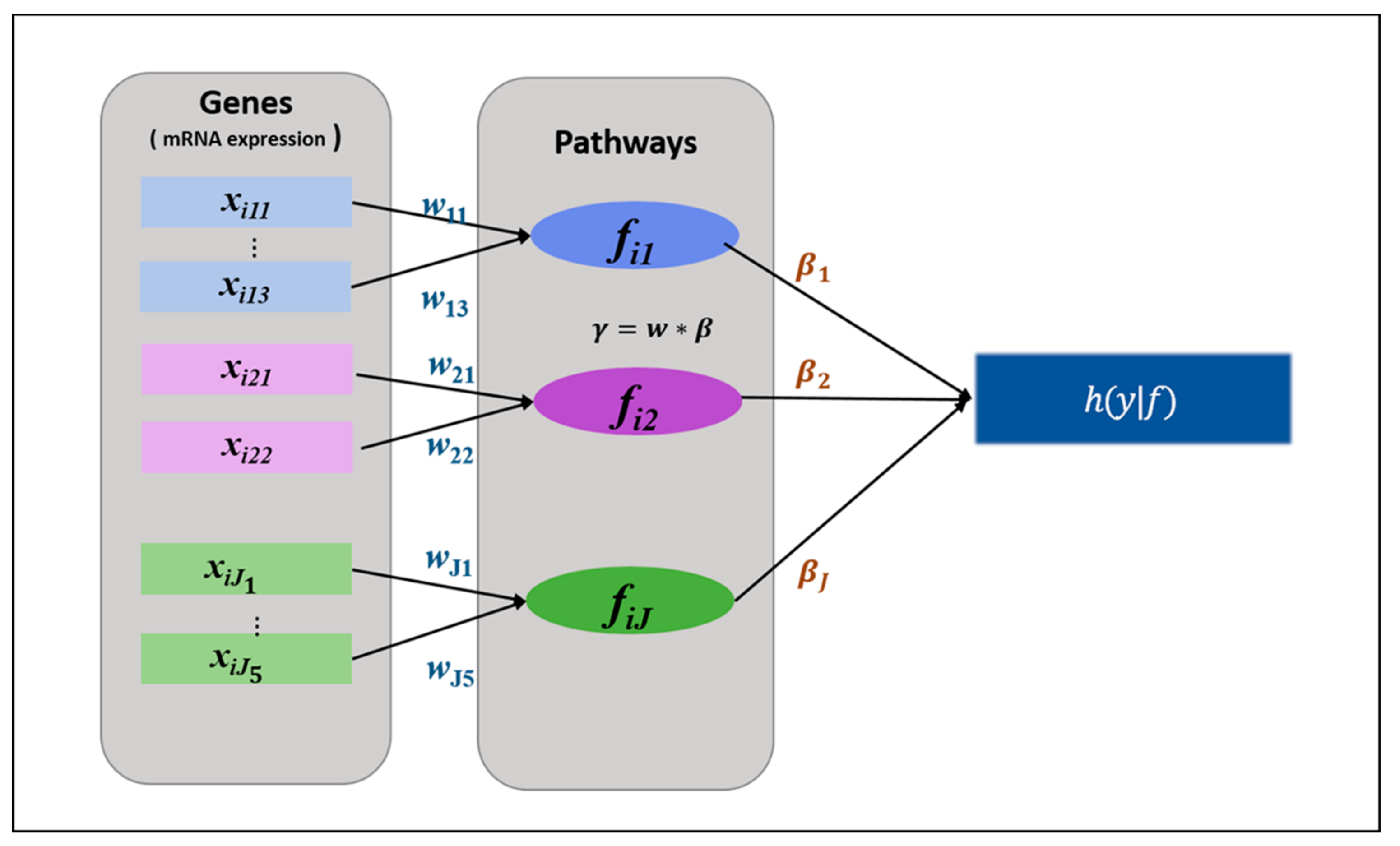
2.2.1. HisCoM-PAGE Method
- Initialize
- Compute η, u, A, and , based on the latest value of . Then maximize with the fixed Repeat these steps until converges.
- Compute η, u, A, and z, based on the latest value of . Then maximize the with the updated . Repeat these steps until converges.
- Iterate steps 2 and 3 until converges.
2.2.2. Comparison Methods
3. Results
3.1. Real Data Analysis Result
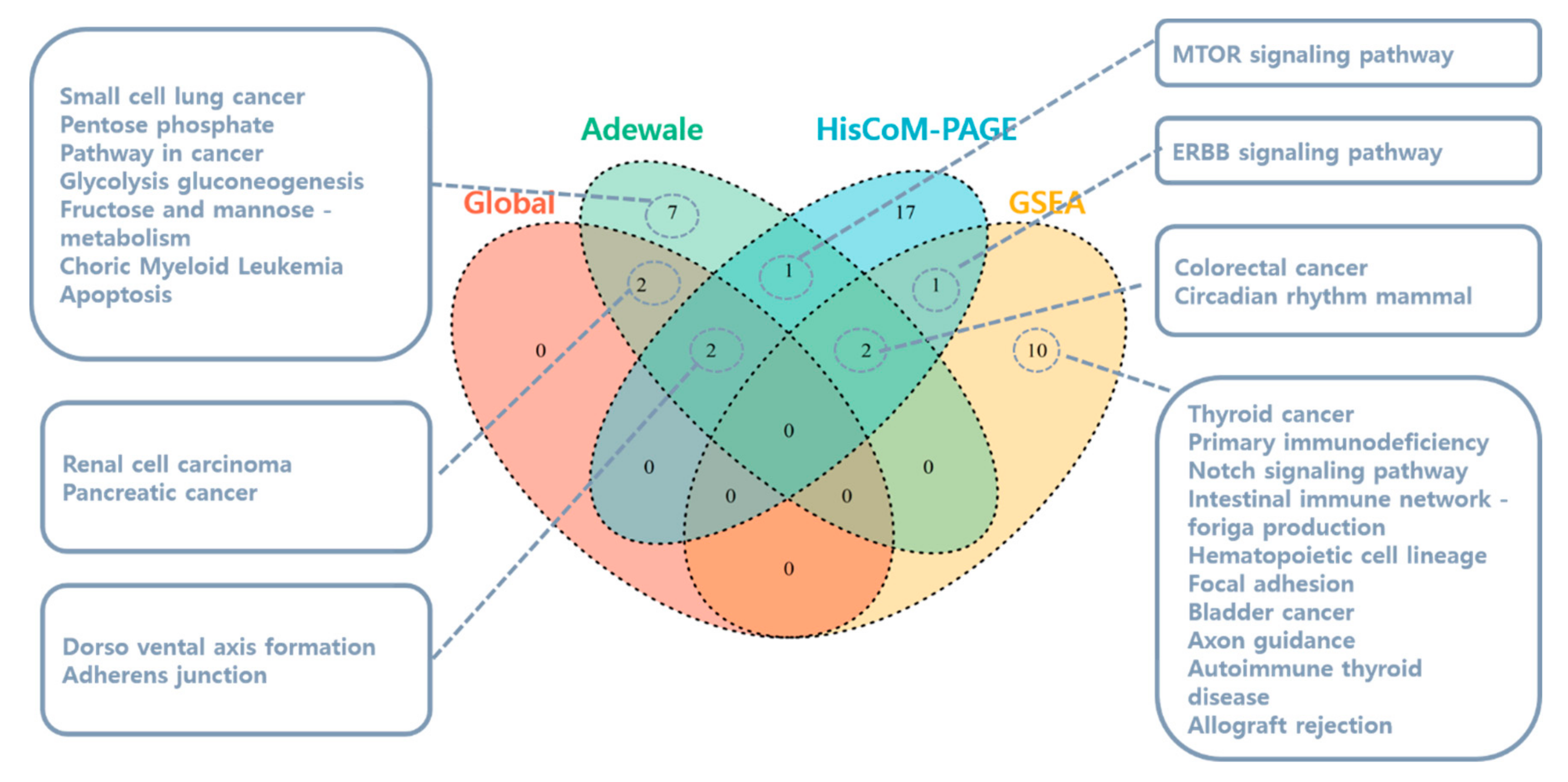
3.1.1. Pathway Analysis Using SNUH Microarray Data
3.1.2. Pathway Analysis Using TCGA RNA-Seq Data
3.1.3. PDAC-Related Genes Using SNUH Microarray Data
3.1.4. PDAC-related Genes Using TCGA RNA-Seq Data
3.2. Simulation Analysis Result
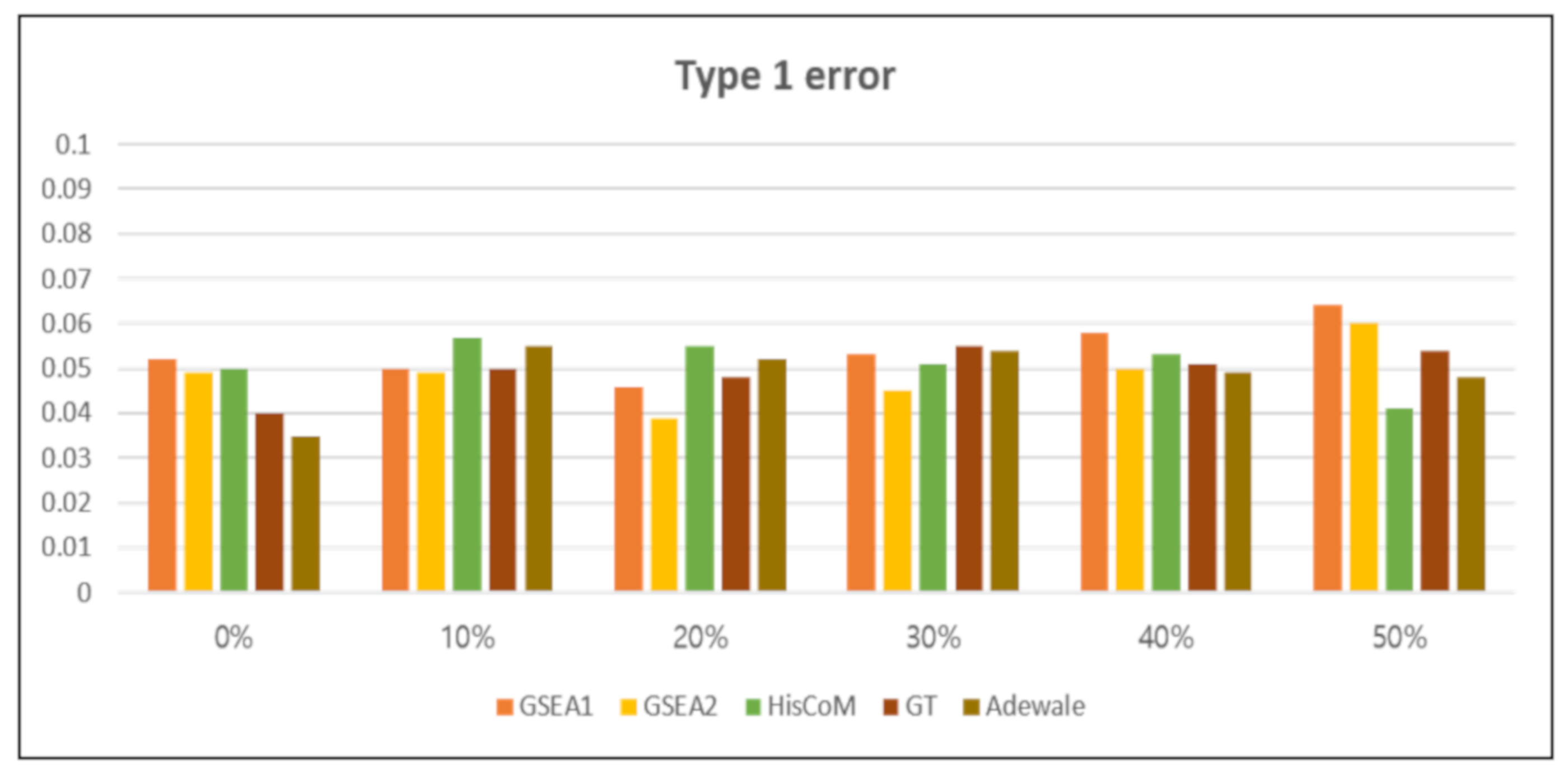
3.2.1. Type 1 Error
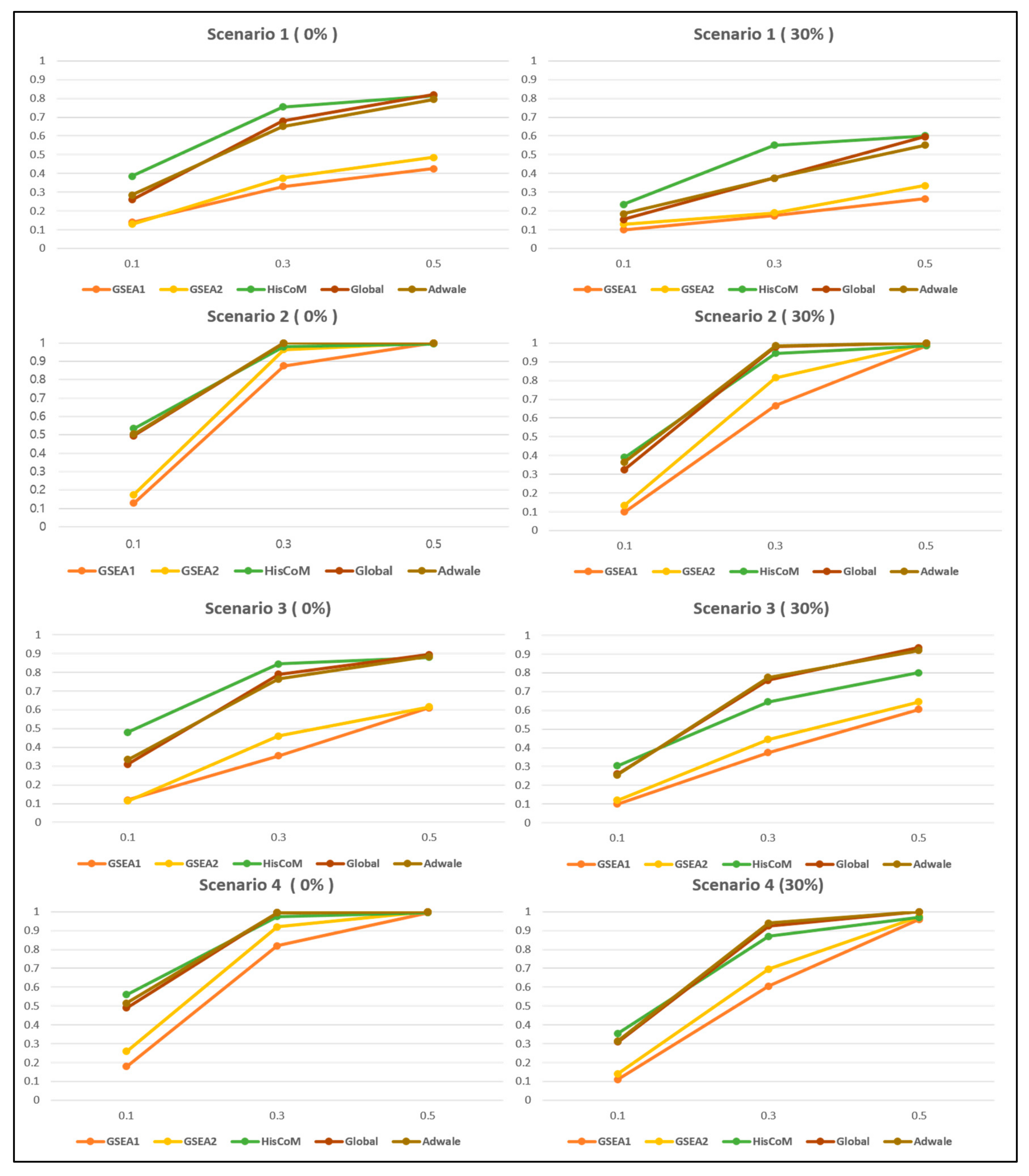
3.2.2. Comparison of Power
4. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lockhart, D.J.; Winzeler, E.A. Genomics, gene expression and DNA arrays. Nature 2000, 405, 827. [Google Scholar] [CrossRef] [PubMed]
- Casamassimi, A.; Federico, A.; Rienzo, M.; Esposito, S.; Ciccodicola, A. Transcriptome profiling in human diseases: New advances and perspectives. Int. J. Mol. Sci. 2017, 18, 1652. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Pusztai, L. Gene-expression signatures in breast cancer. N. Engl. J. Med. 2009, 360, 790–800. [Google Scholar] [CrossRef]
- Glazko, G.V.; Emmert-Streib, F. Unite and conquer: Univariate and multivariate approaches for finding differentially expressed gene sets. Bioinformatics 2009, 25, 2348–2354. [Google Scholar] [CrossRef] [PubMed]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten years of pathway analysis: Current approaches and outstanding challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef] [PubMed]
- Maciejewski, H. Gene set analysis methods: Statistical models and methodological differences. Brief. Bioinform. 2013, 15, 504–518. [Google Scholar] [CrossRef]
- Liu, Q.; Dinu, I.; Adewale, A.J.; Potter, J.D.; Yasui, Y. Comparative evaluation of gene-set analysis methods. BMC Bioinform. 2007, 8, 431. [Google Scholar] [CrossRef]
- Raman, P.; Maddipati, R.; Lim, K.H.; Tozeren, A. Pancreatic cancer survival analysis defines a signature that predicts outcome. PLoS ONE 2018, 13, e0201751. [Google Scholar] [CrossRef]
- Xu, J.; Shi, K.Q.; Chen, B.C.; Huang, Z.P.; Lu, F.Y.; Zhou, M.T. A nomogram based on preoperative inflammatory markers predicting the overall survival of pancreatic ductal adenocarcinoma. J. Gastroenterol. Hepatol. 2017, 32, 1394–1402. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yagi, S.; Kinoshita, H.; Sakamoto, Y.; Okada, K.; Uryuhara, K.; Morimoto, T.; Kaihara, S.; Hosotani, R. Long-term survival after resection of pancreatic cancer: A single-center retrospective analysis. World J. Gastroenterol. WJG 2015, 21, 262. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Simillion, C.; Liechti, R.; Lischer, H.E.; Ioannidis, V.; Bruggmann, R. Avoiding the pitfalls of gene set enrichment analysis with SetRank. BMC Bioinform. 2017, 18, 151. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Goeman, J.J.; Van De Geer, S.A.; De Kort, F.; Van Houwelingen, H.C. A global test for groups of genes: Testing association with a clinical outcome. Bioinformatics 2004, 20, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Goeman, J.J.; Oosting, J.; Cleton-Jansen, A.M.; Anninga, J.K.; Van Houwelingen, H.C. Testing association of a pathway with survival using gene expression data. Bioinformatics 2005, 21, 1950–1957. [Google Scholar] [CrossRef]
- Adewale, A.J.; Dinu, I.; Potter, J.D.; Liu, Q.; Yasui, Y. Pathway analysis of microarray data via regression. J. Comput. Biol. 2008, 15, 269–277. [Google Scholar] [CrossRef]
- Lee, S.; Choi, S.; Kim, Y.J.; Kim, B.J. Pathway-based approach using hierarchical components of collapsed rare variants. Bioinformatics 2016, 32, i586–i594. [Google Scholar] [CrossRef]
- Hwang, H.; Takane, Y. Generalized structured component analysis. Psychometrika 2004, 69, 81–99. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, S.; Choi, S.; Jang, J.Y.; Park, T. Hierarchical structural component modeling of microRNA-mRNA integration analysis. BMC Bioinform. 2018, 19, 75. [Google Scholar] [CrossRef]
- Choi, S.; Lee, S.; Kim, Y.; Hwang, H.; Park, T. HisCoM-GGI: Hierarchical structural component analysis of gene-gene interactions. J. Bioinform. Comput. Biol. 2018, 16, 1840026. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Liu, C.; Yuan, X.; Kang, S.; Miao, R.; Xiao, H.; Zhao, G.; Luo, H.; Bu, D.; Zhao, H.; et al. Large-scale prediction of long non-coding RNA functions in a coding–non-coding gene co-expression network. Nucleic Acids Res. 2011, 39, 3864–3878. [Google Scholar] [CrossRef] [PubMed]
- Calza, S.; Raffelsberger, W.; Ploner, A.; Sahel, J.; Leveillard, T.; Pawitan, Y. Filtering genes to improve sensitivity in oligonucleotide microarray data analysis. Nucleic Acids Res. 2007, 35, e102. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Cancer Genome Atlas Research Network. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Grimes, T.; Walker, A.R.; Datta, S.; Datta, S. Predicting survival times for neuroblastoma patients using RNA-seq expression profiles. Biol. Direct 2018, 13, 11. [Google Scholar] [CrossRef]
- Lee, S.; Kim, J.; Lee, S. A comparative study on gene-set analysis methods for assessing differential expression associated with the survival phenotype. BMC Bioinform. 2011, 12, 377. [Google Scholar] [CrossRef]
- Wan, F. Simulating survival data with predefined censoring rates for proportional hazards models. Stat. Med. 2017, 36, 838–854. [Google Scholar] [CrossRef]
- Hwang, H. Regularized generalized structured component analysis. Psychometrika 2009, 74, 517–530. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D. BioCarta. Biotech Softw. Internet Rep. Comput. Softw. J. Sci. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Shen, W.; Tao, G.Q.; Zhang, Y.; Cai, B.; Sun, J.; Tian, Z.Q. TGF-β in pancreatic cancer initiation and progression: Two sides of the same coin. Cell Biosci. 2017, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Truty, M.J.; Urrutia, R. Basics of TGF-β and pancreatic cancer. Pancreatology 2007, 7, 423–435. [Google Scholar] [CrossRef]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117. [Google Scholar] [CrossRef]
- Friess, H.; Yamanaka, Y.; Büchler, M.; Ebert, M.; Beger, H.G.; Gold, L.I.; Korc, M. Enhanced expression of transforming growth factor β isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993, 105, 1846–1856. [Google Scholar] [CrossRef]
- Wakefield, L.M.; Roberts, A.B. TGF-β signaling: Positive and negative effects on tumorigenesis. Curr. Opin. Genet. Dev. 2002, 12, 22–29. [Google Scholar] [CrossRef]
- Villanueva, A.; García, C.; Paules, A.B.; Vicente, M.; Megías, M.; Reyes, G.; de Villalonga, P.; Agell, N.; Lluís, F.; Bachs, O.; et al. Disruption of the antiproliferative TGF-β signaling pathways in human pancreatic cancer cells. Oncogene 1998, 17, 1969. [Google Scholar] [CrossRef]
- Javle, M.; Li, Y.; Tan, D.; Dong, X.; Chang, P.; Kar, S.; Li, D. Biomarkers of TGF-β signaling pathway and prognosis of pancreatic cancer. PLoS ONE 2014, 9, e85942. [Google Scholar] [CrossRef] [PubMed]
- Kundu, J.; Wahab, S.M.; Kundu, J.K.; Choi, Y.L.; Erkin, O.C.; Lee, H.S.; Park, S.G.; Shin, Y.K. Tob1 induces apoptosis and inhibits proliferation, migration and invasion of gastric cancer cells by activating Smad4 and inhibiting β-catenin signaling. Int. J. Oncol. 2012, 41, 839–848. [Google Scholar] [CrossRef]
- Wang, J.; Sen, S. MicroRNA functional network in pancreatic cancer: From biology to biomarkers of disease. J. Biosci. 2011, 36, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, J.; Cheng, J.; Lubahn, D.B. Genes targeted by the Hedgehog-signaling pathway can be regulated by Estrogen related receptor β. BMC Mol. Biol. 2015, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Jiang, W.; Wang, S.; Wang, L.; Xie, K. Role of Wnt/β-catenin signaling in drug resistance of pancreatic cancer. Curr. Pharm. Des. 2012, 18, 2464–2471. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, C.; Kong, Y.; Huang, H.; Wang, C.; Zhang, H. TGFβ signaling in pancreatic ductal adenocarcinoma. Tumor Biol. 2015, 36, 1613–1618. [Google Scholar] [CrossRef]
- Korc, M. Pathways for aberrant angiogenesis in pancreatic cancer. Mol. Cancer 2003, 2, 8. [Google Scholar] [CrossRef]
- Subramani, R.; Lopez-Valdez, R.; Arumugam, A.; Nandy, S.; Boopalan, T.; Lakshmanaswamy, R. Targeting insulin-like growth factor 1 receptor inhibits pancreatic cancer growth and metastasis. PLoS ONE 2014, 9, e97016. [Google Scholar] [CrossRef]
- Hamacher, R.; Schmid, R.M.; Saur, D.; Schneider, G. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol. Cancer 2008, 7, 64. [Google Scholar] [CrossRef]
- Bian, Y.; Yu, Y.; Wang, S.; Li, L. Up-regulation of fatty acid synthase induced by EGFR/ERK activation promotes tumor growth in pancreatic cancer. Biochem. Biophys. Res. Commun. 2015, 463, 612–617. [Google Scholar] [CrossRef]
- Boroughs, L.K.; Deberardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, E.V.; Xue, J.; Neelamegham, S.; Matta, K.L. The pattern of glycosyl-and sulfotransferase activities in cancer cell lines: A predictor of individual cancer-associated distinct carbohydrate structures for the structural identification of signature glycans. Carbohydr. Res. 2006, 341, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, S.; Guillaumond, F. LDL Receptor: An open route to feed pancreatic tumor cells. Mol. Cell. Oncol. 2016, 3, e1033586. [Google Scholar] [CrossRef] [PubMed]
- Gerrard, D.L.; Boyd, J.R.; Stein, G.S.; Jin, V.X.; Frietze, S. Disruption of Broad Epigenetic Domains in PDAC Cells by HAT Inhibitors. Epigenomes 2019, 3, 11. [Google Scholar] [CrossRef]
- Iriana, S.; Ahmed, S.; Gong, J.; Annamalai, A.A.; Tuli, R.; Hendifar, A.E. Targeting mTOR in pancreatic ductal adenocarcinoma. Front. Oncol. 2016, 6, 99. [Google Scholar] [CrossRef] [PubMed]
- Broussard, B.L.; Mikhaylina, A.; Heslin, M.J.; Arnoletti, J.P.; Frolov, A. Hedgehog (HH) and ErbB signaling as mediators of tumor-stroma interactions in pancreatic ductal adenocarcinoma (PDAC). J. Am. Coll. Surg. 2012, 215, S134–S135. [Google Scholar] [CrossRef]
- Pignatelli, M.; Ansari, T.W.; Gunter, P.; Liu, D.; Hirano, S.; Takeichi, M.; Klöppel, G.; Lemoine, N.R. Loss of membranous E-cadherin expression in pancreatic cancer: Correlation with lymph node metastasis, high grade, and advanced stage. J. Pathol. 1994, 174, 243–248. [Google Scholar] [CrossRef]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-cadherin–integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef]
- Ceyhan, G.O.; Giese, N.A.; Erkan, M.; Kerscher, A.G.; Wente, M.N.; Giese, T.; Büchler, M.W.; Friess, H. The neurotrophic factor artemin promotes pancreatic cancer invasion. Ann. Surg. 2006, 244, 274. [Google Scholar] [CrossRef]
- Yamazaki, K.; Masugi, Y.; Effendi, K.; Tsujikawa, H.; Hiraoka, N.; Kitago, M.; Shinoda, M.; Itano, O.; Tanabe, M.; Kitagawa, Y.; et al. Upregulated SMAD3 promotes epithelial–mesenchymal transition and predicts poor prognosis in pancreatic ductal adenocarcinoma. Lab. Investig. 2014, 94, 683. [Google Scholar] [CrossRef]
- Ungefroren, H.; Groth, S.; Sebens, S.; Lehnert, H.; Gieseler, F.; Fändrich, F. Differential roles of Smad2 and Smad3 in the regulation of TGF-β1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: Control by Rac1. Mol. Cancer 2011, 10, 67. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Wang, B.; Gu, S.; Li, X.; Sun, S. Expression of Beclin 1 and Bcl-2 in pancreatic neoplasms and its effect on pancreatic ductal adenocarcinoma prognosis. Oncol. Lett. 2017, 14, 7849–7861. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liang, Y.; Yin, Q.; Liu, S.; Wang, Q.; Tang, Y.; Cao, C. Clinical and prognostic significance of serum transforming growth factor-beta1 levels in patients with pancreatic ductal adenocarcinoma. Braz. J. Med. Biol. Res. 2016, 49, e5485. [Google Scholar] [CrossRef] [PubMed]
- Tomihara, H.; Yamada, D.; Eguchi, H.; Iwagami, Y.; Noda, T.; Asaoka, T.; Wada, H.; Kawamoto, K.; Gotoh, K.; Takeda, Y.; et al. MicroRNA-181b-5p, ETS1, and the c-Met pathway exacerbate the prognosis of pancreatic ductal adenocarcinoma after radiation therapy. Cancer Sci. 2017, 108, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Shibaji, T.; Nagao, M.; Ikeda, N.; Kanehiro, H.; Hisanaga, M.; Ko, S.; Fukumoto, A.; Nakajima, Y. Prognostic significance of HIF-1 alpha overexpression in human pancreatic cancer. Anticancer Res. 2003, 23, 4721–4727. [Google Scholar] [PubMed]
- Hoffmann, A.C.; Mori, R.; Vallbohmer, D.; Brabender, J.; Klein, E.; Drebber, U.; Baldus, S.E.; Cooc, J.; Azuma, M.; Metzger, R.; et al. High expression of HIF1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA, VEGF, and bFGF. Neoplasia 2008, 10, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.C.; Qiu, Z.J.; Liu, J.; Sun, J.; Jiang, T.; Huang, K.J.; Yao, M.; Huang, C. Expression of hypoxia-inducible factor-1 alpha and associated proteins in pancreatic ductal adenocarcinoma and their impact on prognosis. Int. J. Oncol. 2007, 30, 1359–1367. [Google Scholar] [CrossRef]
- Yao, J.; Liang, L.H.; Zhang, Y.; Ding, J.; Tian, Q.; Li, J.J.; He, X.H. GNAI1 suppresses tumor cell migration and invasion and is post-transcriptionally regulated by Mir-320a/c/d in hepatocellular carcinoma. Cancer Biol. Med. 2012, 9, 234. [Google Scholar]
- Wang, W.; Zhao, L.; Wei, X.; Wang, L.; Liu, S.; Yang, Y.; Wang, F.; Sun, G.; Zhang, J.; Ma, Y.; et al. MicroRNA-320a promotes 5-FU resistance in human pancreatic cancer cells. Sci. Rep. 2016, 6, 27641. [Google Scholar] [CrossRef]
- De Leeuw, C.A.; Neale, B.M.; Heskes, T.; Posthuma, D. The statistical properties of gene-set analysis. Nat. Rev. Genet. 2016, 17, 353. [Google Scholar] [CrossRef]
- Kao, P.Y.; Leung, K.H.; Chan, L.W.; Yip, S.P.; Yap, M.K. Pathway analysis of complex diseases for GWAS, extending to consider rare variants, multi-omics and interactions. Biochim. et Biophys. Acta (Bba)-Gen. Subj. 2017, 1861, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Janky, R.S.; Binda, M.M.; Allemeersch, J.; Govaere, O.; Swinnen, J.V.; Roskams, T.; Aerts, S.; Topal, B. Prognostic relevance of molecular subtypes and master regulators in pancreatic ductal adenocarcinoma. BMC Cancer 2016, 16, 632. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gong, B.; Bushel, P.R.; Thierry-Mieg, J.; Thierry-Mieg, D.; Xu, J.; Fang, H.; Hong, H.; Shen, J.; Su, Z.; et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat. Biotechnol. 2014, 32, 926. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef] [PubMed]
- Mantel, N. Evaluation of survival data and two new rank order statistics arising in its consideration, Cancer Chemotherapy Reports. Mantel162350Cancer Chemother. Rep. 1966, 50, 163–170. [Google Scholar]
- Kim, S.; Choi, S.; Yoon, J.H.; Kim, Y.; Lee, S.; Park, T. Drug response prediction model using a hierarchical structural component modeling method. BMC Bioinform. 2018, 19, 117. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Variable Description | Descriptive Statistics |
|---|---|---|
| Age | Age at diagnosis | 63.32(10.064) mean(se) |
| Sex | Male: 75, Female: 50 | |
| Positive Lymph Nodes (LN) | Number of cancers transmitted by Lymphocytes | (0,1,2) (1st Quantile, Median,3rd Quantile) |
| Size | Maximum Tumor Size (cm) | 3.574 (mean) |
| Differentiation | Clinico-pathologic characteristics and prognostic value of various histological types. | WD: 19, MD: 85, PD: 18, Other: 2 (NA: 1) |
| Jaundice | Yes: 89, No: 36 | |
| 7th staging T stage | AJCC 7th T staging criteria. The extent of the tumor. | 1th: 6, 2nd: 3, 3rd: 104, 4th: 12 |
| 7th staging N stage | AJCC 7th N staging criteria. The spread to nearby lymph nodes. | Yes: 71, No: 54 |
| Radiation therapy | Radiation therapy after surgery | Yes: 72, No: 53 |
| Chemotherapy | Chemotherapy after surgery | Yes: 94, No: 31 |
| Overall survival time | Median: 25 months |
| Pathway Database | Pathway Name | Microarray Data | RNA-seq Data | ||||
|---|---|---|---|---|---|---|---|
| |βpath| | p-Value | q-Value | |βpath| | p-Value | q-Value | ||
| BIOCARTA | Transforming Growth Factor-β (TGF-β)* | 0.017 | 0.00001 | 0.002 | 0.006 | 0.053 | 0.211 |
| Non-Typeable Haemophilus Influenzae (NTHI) | 0.014 | 0.00033 | 0.03 | 0.006 | 0.314 | 0.419 | |
| MITOCHONDRIA | 0.01 | 0.00054 | 0.03 | 0.007 | 0.197 | 0.394 | |
| Transducer Of ERBB2.1(TOB1)* | 0.016 | 0.00056 | 0.03 | 0.005 | 0.46 | 0.46 | |
| KEGG | BASAL CELL CARCINOMA | 0.015 | 0.0001 | 0.0074 | 0.0071 | 0.261 | 0.894 |
| COLORECTAL CANCER | 0.014 | 0.0001 | 0.0074 | 0.0085 | 0.843 | 0.959 | |
| CIRCADIAN RHYTHM MAMMAL | 0.01 | 0.0012 | 0.0306 | 0.0062 | 0.869 | 0.959 | |
| FATTY ACID METABOLISM* | 0.012 | 0.0009 | 0.0306 | 0.0088 | 0.868 | 0.959 | |
| GLYCOSPHINGOLIPID BIOSYNTHESIS GLOBO SERIES* | 0.01 | 0.0008 | 0.0306 | 0.0018 | 0.364 | 0.9055 | |
| INSULIN SIGNALING PATHWAY* | 0.016 | 0.0011 | 0.0306 | 0.013 | 0.951 | 0.959 | |
| STEROID HORMONE BIOSYNTHESIS* | 0.014 | 0.0015 | 0.0306 | 0.0037 | 0.541 | 0.959 | |
| Vascular Endothelial Growth Factor (VEGF) SIGNALING * | 0.014 | 0.0007 | 0.0306 | 0.0078 | 0.724 | 0.959 | |
| WNT SIGNALING* | 0.017 | 0.0014 | 0.0306 | 0.0104 | 0.415 | 0.905 | |
| ADHERENS JUNCTION* | 0.013 | 0.0018 | 0.0326 | 0.01 | 0.819 | 0.959 | |
| DILATED CARDIOMYOPATHY | 0.016 | 0.0023 | 0.0326 | 0.011 | 0.851 | 0.959 | |
| OTHER GLYCAN DEGRADATION | 0.009 | 0.0021 | 0.0326 | 0.0031 | 0.665 | 0.959 | |
| OXIDATIVE PHOSPHORYLATION* | 0.015 | 0.0022 | 0.0326 | 0.0099 | 0.178 | 0.894 | |
| AMYOTROPHIC LATERAL SCLEROSIS(ALS) | 0.01 | 0.0028 | 0.0369 | 0.0108 | 0.004 | 0.0852 | |
| DORSO VENTRAL AXIS FORMATION | 0.009 | 0.0031 | 0.0369 | 0.0036 | 0.344 | 0.905 | |
| SULFUR METABOLISM | 0.008 | 0.0032 | 0.0369 | 0.0046 | 0.212 | 0.894 | |
| CARDIAC MUSCLE CONTRACTION | 0.013 | 0.0038 | 0.0373 | 0.0086 | 0.732 | 0.959 | |
| ERBB SIGNALING* | 0.013 | 0.004 | 0.0373 | 0.0069 | 0.401 | 0.9055 | |
| GLYCEROPHOSPHOLIPID METABOLISM* | 0.015 | 0.0036 | 0.0373 | 0.0121 | 0.952 | 0.959 | |
| HYPERTROPHIC CARDIOMYOPATHY (HCM) | 0.015 | 0.004 | 0.0373 | 0.0127 | 0.959 | 0.959 | |
| GLIOMA | 0.012 | 0.0044 | 0.039 | 0.0081 | 0.711 | 0.959 | |
| Mammalian TOR (MTOR) SIGNALING* | 0.011 | 0.0055 | 0.0465 | 0.0098 | 0.014 | 0.112 | |
| HEDGEHOG SIGNALING * | 0.011 | 0.0058 | 0.047 | 0.0068 | 0.258 | 0.894 | |
| Pathway Database | Pathway Name | Gene | SNUH Microarray | TCGA RNA-seq | ||||
|---|---|---|---|---|---|---|---|---|
| |wgene × βpath| | p-Value | q-Value | |wgene × βpath| | p-Value | q-Value | |||
| BIOCARTA | Non-Typeable Haemophilus Influenzae (NTHI) | SMAD3 | 0.032 | 0.00001 | 0.004 | 0.004 | 0.1246 | 0.298 |
| Transducer Of ERBB2.1(TOB1) | SMAD3 | 0.032 | 0.00001 | 0.004 | 0.004 | 0.1134 | 0.298 | |
| Transforming Growth Factor-β (TGF-β) | SMAD3 | 0.032 | 0.00001 | 0.004 | 0.004 | 0.1114 | 0.298 | |
| CHEMICAL | BCL2L1 | 0.024 | 0.00003 | 0.004 | 0.006 | 0.0762 | 0.259 | |
| IL-2 receptor beta chain (IL2RB) | BCL2L1 | 0.024 | 0.00003 | 0.004 | 0.006 | 0.0866 | 0.266 | |
| RAS | BCL2L1 | 0.024 | 0.00003 | 0.004 | 0.006 | 0.0783 | 0.259 | |
| Bcl-2 antagonist of cell death (BAD) | BCL2L1 | 0.024 | 0.00003 | 0.004 | 0.006 | 0.0777 | 0.259 | |
| MITOCHONDRIA | BCL2L1 | 0.024 | 0.00003 | 0.004 | 0.006 | 0.0753 | 0.259 | |
| CCCTC-binding factor (CTCF) | TGF-β1 | 0.019 | 0.00005 | 0.004 | 0.008 | 0.9707 | 0.982 | |
| Inflammatory Response(INFLAM) | TGF-β1 | 0.019 | 0.00005 | 0.004 | 0.008 | 0.9715 | 0.982 | |
| Erythrocyte Differentiation (ERYTH) | TGF-β1 | 0.019 | 0.00005 | 0.004 | 0.008 | 0.9716 | 0.982 | |
| MAP Kinase(MAPK) | TGF-β1 | 0.019 | 0.00005 | 0.004 | 0.008 | 0.9726 | 0.982 | |
| Anaplastic lymphoma kinase(ALK) | TGF-β1 | 0.018 | 0.00006 | 0.004 | 0.008 | 0.9695 | 0.982 | |
| G1 | TGF-β1 | 0.018 | 0.00006 | 0.004 | 0.008 | 0.9706 | 0.982 | |
| P38MAPK | TGF-β1 | 0.019 | 0.00006 | 0.004 | 0.008 | 0.9718 | 0.982 | |
| TOB1 | TGF-β1 | 0.018 | 0.00006 | 0.004 | 0.008 | 0.971 | 0.982 | |
| NKT | TGF-β1 | 0.018 | 0.00006 | 0.004 | 0.008 | 0.971 | 0.982 | |
| Interleukin-1 receptor (IL1R) | TGF-β1 | 0.018 | 0.00006 | 0.004 | 0.008 | 0.971 | 0.982 | |
| TGF-β | TGF-β1 | 0.018 | 0.00006 | 0.004 | 0.008 | 0.971 | 0.982 | |
| KERATINOCYTE | ETS1 | 0.015 | 0.00008 | 0.005 | 0.001 | 0.3516 | 0.643 | |
| E-26-specific (ETS) | ETS1 | 0.015 | 0.0001 | 0.006 | 0.001 | 0.3588 | 0.643 | |
| P53HYPOXIA | HIF1A | 0.016 | 0.00047 | 0.028 | 0.0002 | 0.4766 | 0.762 | |
| Hypoxia-Inducible Factor(HIF) | HIF1A | 0.016 | 0.00047 | 0.028 | 0.0001 | 0.4767 | 0.762 | |
| Erythropoietin mediated neuroprotection through NF-kB (EPONFKB) | HIF1A | 0.016 | 0.0005 | 0.028 | 0.0001 | 0.4786 | 0.762 | |
| Vascular Endothelial Growth Factor (VEGF) | HIF1A | 0.015 | 0.0006 | 0.033 | 0.00005 | 0.9824 | 0.982 | |
| DEATH | TNFRSF10B | 0.018 | 0.00064 | 0.033 | 0.002 | 0.647 | 0.897 | |
| Formyl methionyl leucyl phenilalanine (FMLP) | GNA15 | 0.015 | 0.00074 | 0.037 | 0.006 | 0.0485 | 0.24 | |
| IL1R | IL1RAP | 0.01 | 0.00095 | 0.041 | 0.002 | 0.2839 | 0.581 | |
| SET | GZMA | 0.015 | 0.001 | 0.041 | - | - | - | |
| Phosphoinositides (PTDINS) | PFKP | 0.011 | 0.0011 | 0.041 | 0.00008 | 0.5064 | 0.778 | |
| Extrinsic Prothrombin Activation (EXTRINSIC) | TFPI | 0.013 | 0.00115 | 0.041 | 0.002 | 0.6457 | 0.897 | |
| Acute Myocardial Infarction (AMI) | TFPI | 0.013 | 0.00116 | 0.041 | 0.002 | 0.6457 | 0.897 | |
| protease-activated receptors-1 (PAR1) | GNAI1 | 0.017 | 0.00118 | 0.041 | 0.007 | 0.0502 | 0.24 | |
| Endothelial differentiation gene- 1 (EDG1) | GNAI1 | 0.017 | 0.00119 | 0.041 | 0.007 | 0.0464 | 0.24 | |
| G protein-coupled receptors (GPCR) | GNAI1 | 0.017 | 0.00119 | 0.041 | 0.007 | 0.0499 | 0.24 | |
| SPPA | GNAI1 | 0.017 | 0.00122 | 0.041 | 0.007 | 0.0481 | 0.24 | |
| Bioactive Peptide Induced Signaling (BIOPEPTIDES) | GNAI1 | 0.017 | 0.00122 | 0.041 | 0.007 | 0.0476 | 0.24 | |
| CXC chemokine receptor type-4 (CXCR4) | GNAI1 | 0.017 | 0.00122 | 0.041 | 0.007 | 0.0447 | 0.24 | |
| Mannose 6-phosphate receptors (MPR) | GNAI1 | 0.017 | 0.00122 | 0.041 | 0.008 | 0.0432 | 0.24 | |
| Glycogen synthase kinase-3 (GSK3) | GNAI1 | 0.017 | 0.00123 | 0.041 | 0.008 | 0.0432 | 0.24 | |
| Peroxisome proliferator-activated receptor alpha (PPARA) | ACOX1 | 0.015 | 0.00122 | 0.041 | 0.003 | 0.3221 | 0.63 | |
| VEGF | VEGFA | 0.01 | 0.00146 | 0.047 | 0.003 | 0.1576 | 0.339 | |
| Nitric Oxide-1(NO1) | VEGFA | 0.01 | 0.00147 | 0.047 | 0.003 | 0.1575 | 0.339 | |
| KEGG | CELL CYCLE | SMAD3 | 0.023 | 0.0001 | 0.047 | 0.003 | 0.099 | 0.099 |
| WNT SIGNALING | SMAD3 | 0.023 | 0.0001 | 0.047 | 0.003 | 0.096 | 0.099 | |
| TGF-β | SMAD3 | 0.023 | 0.0001 | 0.047 | 0.003 | 0.0957 | 0.099 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mok, L.; Kim, Y.; Lee, S.; Choi, S.; Lee, S.; Jang, J.-Y.; Park, T. HisCoM-PAGE: Hierarchical Structural Component Models for Pathway Analysis of Gene Expression Data. Genes 2019, 10, 931. https://doi.org/10.3390/genes10110931
Mok L, Kim Y, Lee S, Choi S, Lee S, Jang J-Y, Park T. HisCoM-PAGE: Hierarchical Structural Component Models for Pathway Analysis of Gene Expression Data. Genes. 2019; 10(11):931. https://doi.org/10.3390/genes10110931
Chicago/Turabian StyleMok, Lydia, Yongkang Kim, Sungyoung Lee, Sungkyoung Choi, Seungyeoun Lee, Jin-Young Jang, and Taesung Park. 2019. "HisCoM-PAGE: Hierarchical Structural Component Models for Pathway Analysis of Gene Expression Data" Genes 10, no. 11: 931. https://doi.org/10.3390/genes10110931
APA StyleMok, L., Kim, Y., Lee, S., Choi, S., Lee, S., Jang, J.-Y., & Park, T. (2019). HisCoM-PAGE: Hierarchical Structural Component Models for Pathway Analysis of Gene Expression Data. Genes, 10(11), 931. https://doi.org/10.3390/genes10110931