PipeMEM: A Framework to Speed Up BWA-MEM in Spark with Low Overhead


Abstract
1. Introduction
- developing a multi-node framework that supports BWA-MEM and supports the future development of a single-node alignment tool.
- the result of the alignment should be adaptable to the existing prevalent genome analysis tools in a big data environment, i.e., GATK.
- RDD creation step is in charge of data distribution.
- Mapper calls the BWA program with the help of JNI.
- Reducer merges the results produced by BWA in mapper together.
2. Methods
2.1. Framework Structure of PipeMEM
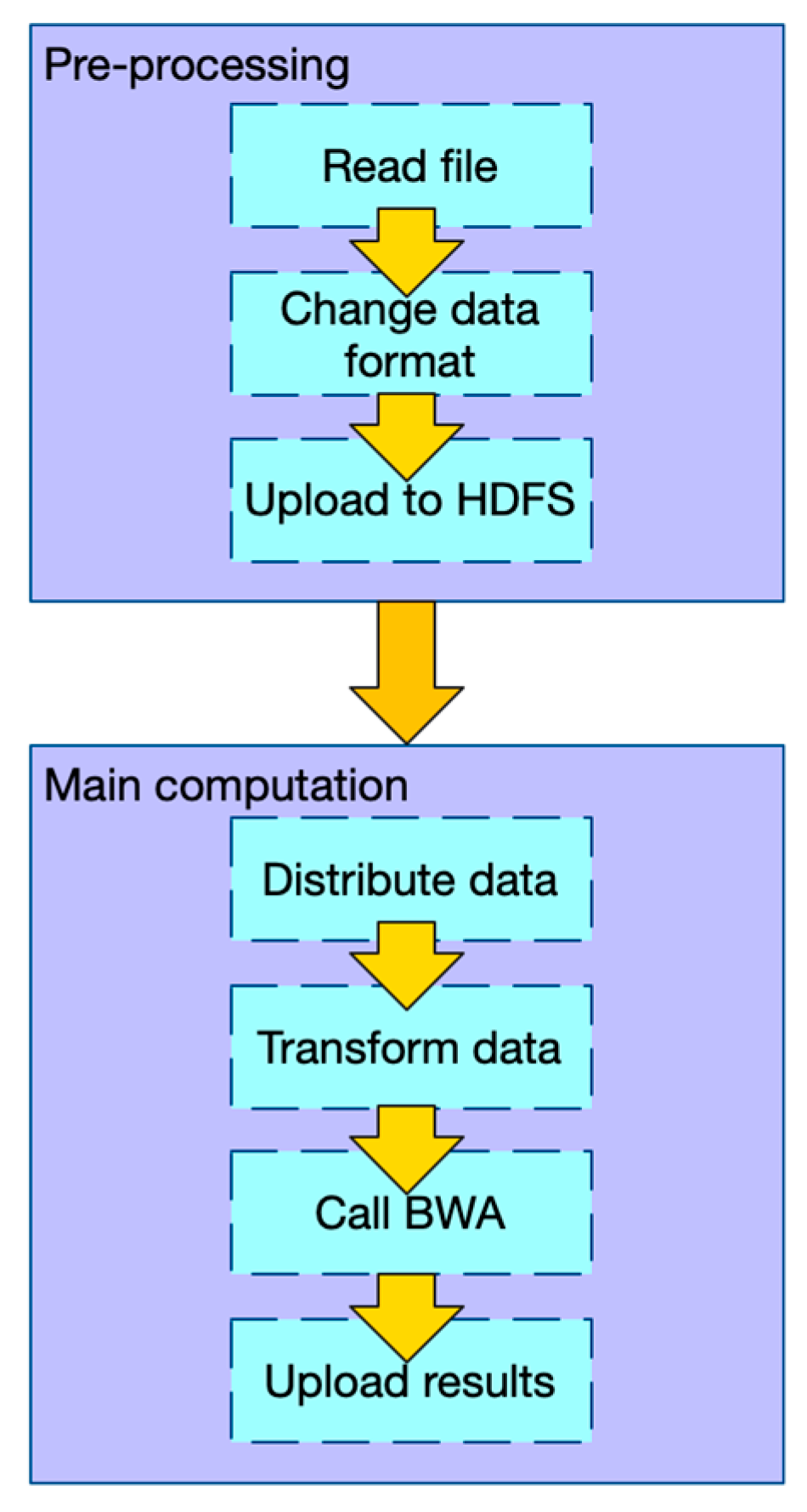
2.1.1. Workflow of PipeMEM
- In the pre-processing stage: (1) Change the data format of input files so that there is no need to iterate and merge input data in the main computation step. (2) Upload data to HDFS.
- In the main computation stage: (1) Distribute data from HDFS to different nodes. (2) Transform the data into the original format, so that the BWA-MEM program can process it. (3) Call BWA-MEM. (4) Upload the results to HDFS.
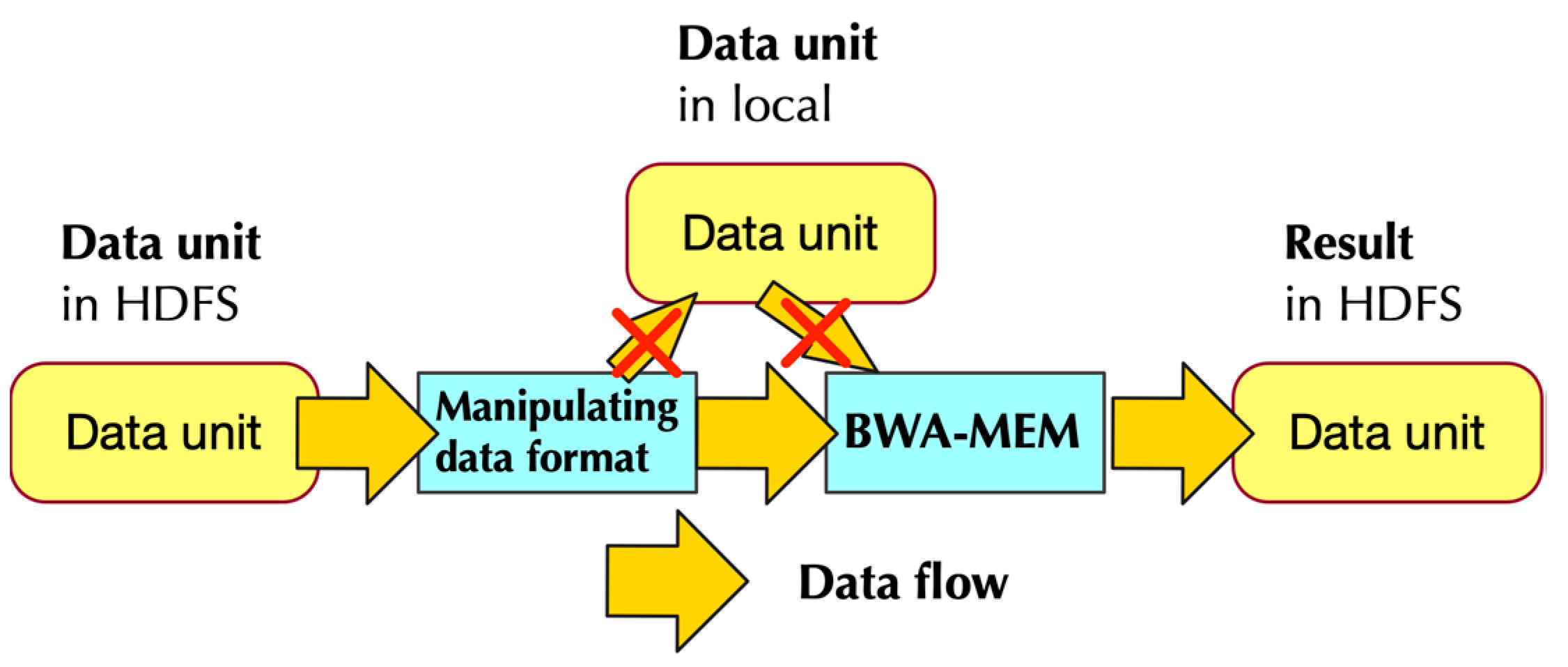
2.1.2. Data Flow of PipeMEM
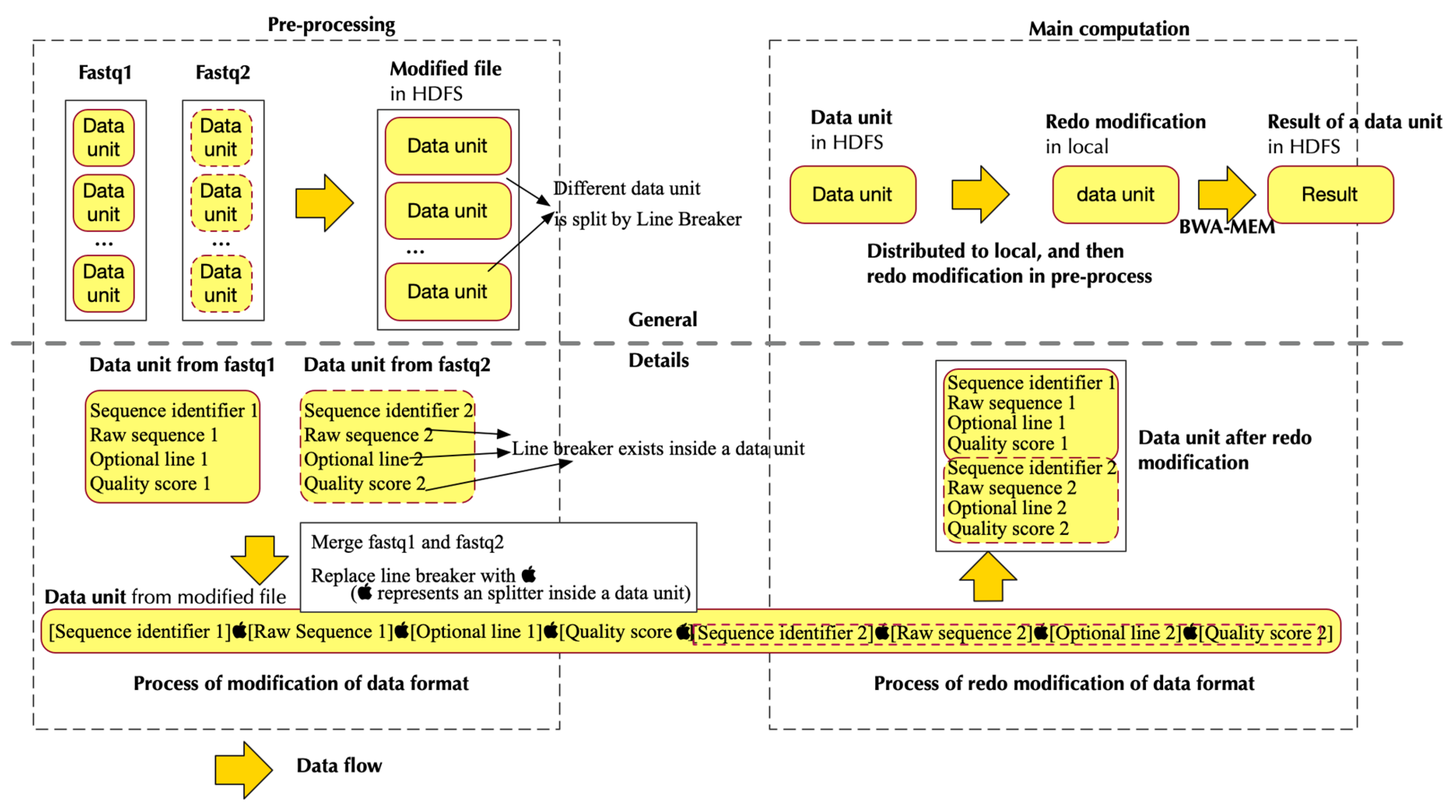
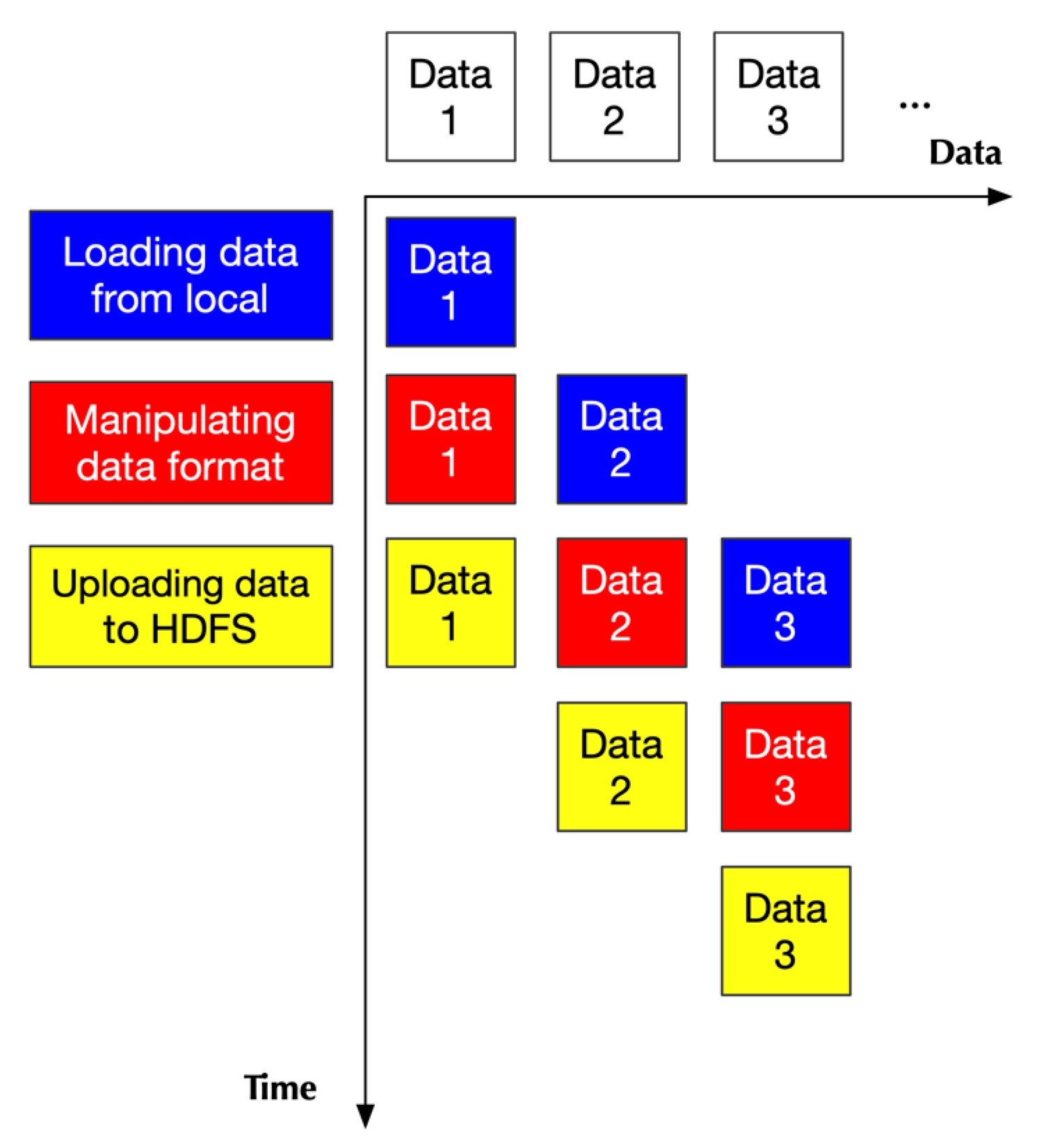
2.2. Pipeline in Pre-Processing
2.2.1. Design Principle
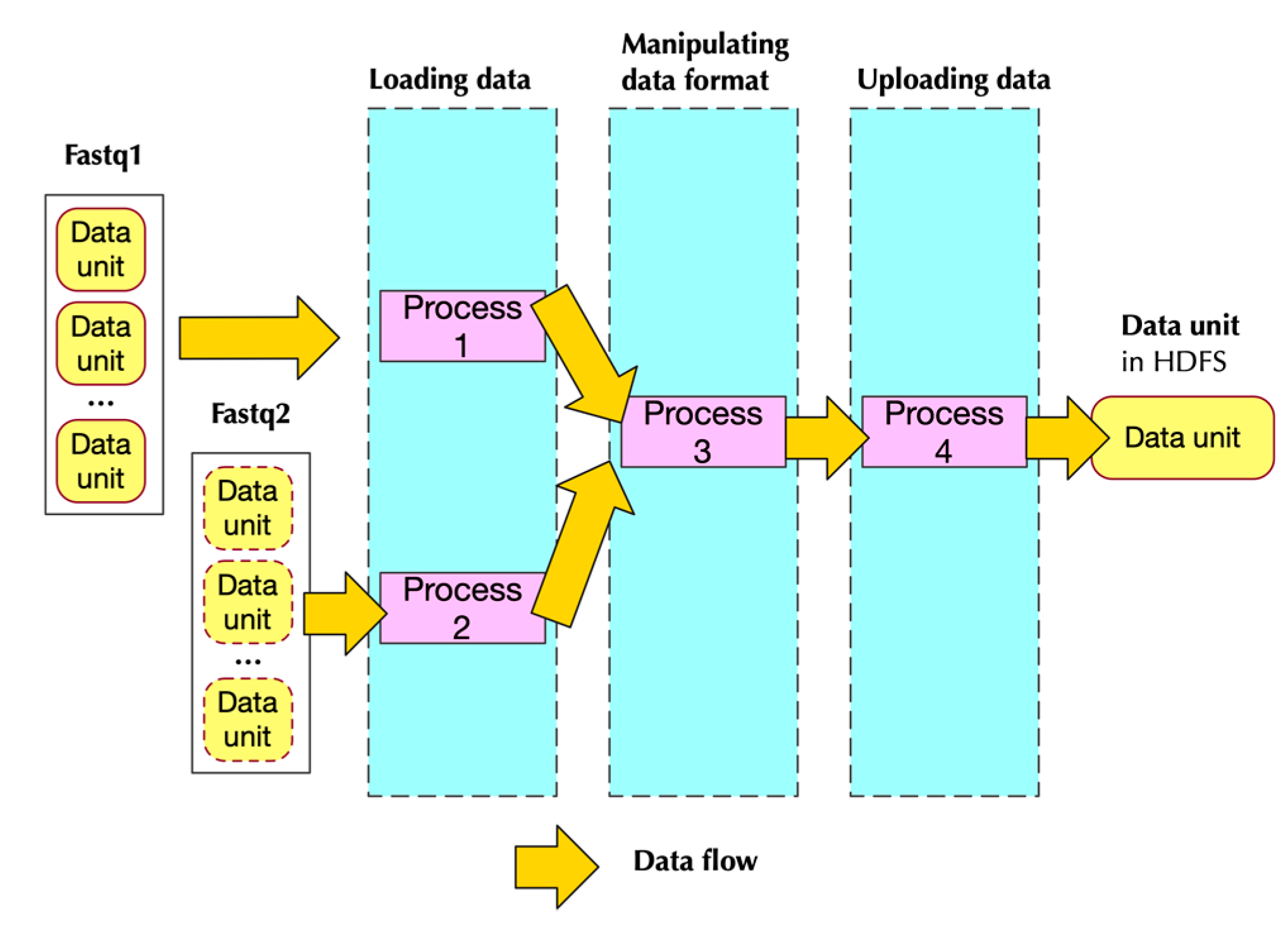
2.2.2. Implementation
- Two of them handling loading data
- One handling data format transformation
- One handling uploading data to HDFS
2.3. In-Memory-Computation in Main Computation
2.3.1. Design Principle
2.3.2. Implementation
- Separate data from HDFS and distribute them to different nodes to create RDDs
- Pipe the data in RDD to a program that modifies the format of input data. This step generates data format that could be utilized by BWA-MEM
- Call single-node BWA-MEM
- Pipe the result of BWA-MEM to generate new RDDs, and these RDDs would then store the data in HDFS.
2.4. Experimental Setup
2.4.1. Metrics
Throughput
Overhead
- Overhead in launching more workers
- Overhead in synchronization
- Overhead in network communication
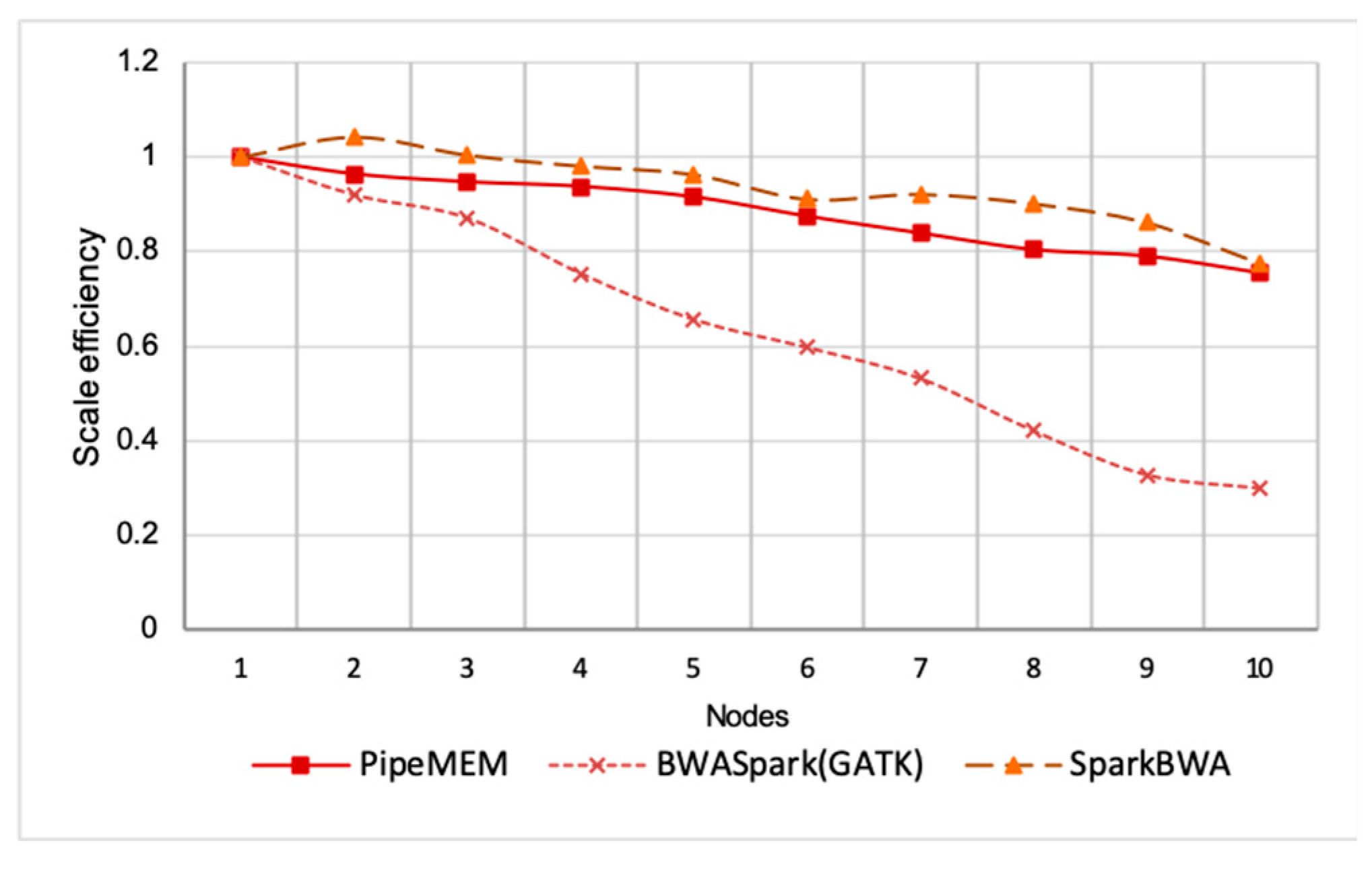
Scale Efficiency
2.4.2. Dataset and Experimental Environment
3. Results and Discussion
3.1. Pre-Processing
3.2. Main Computation
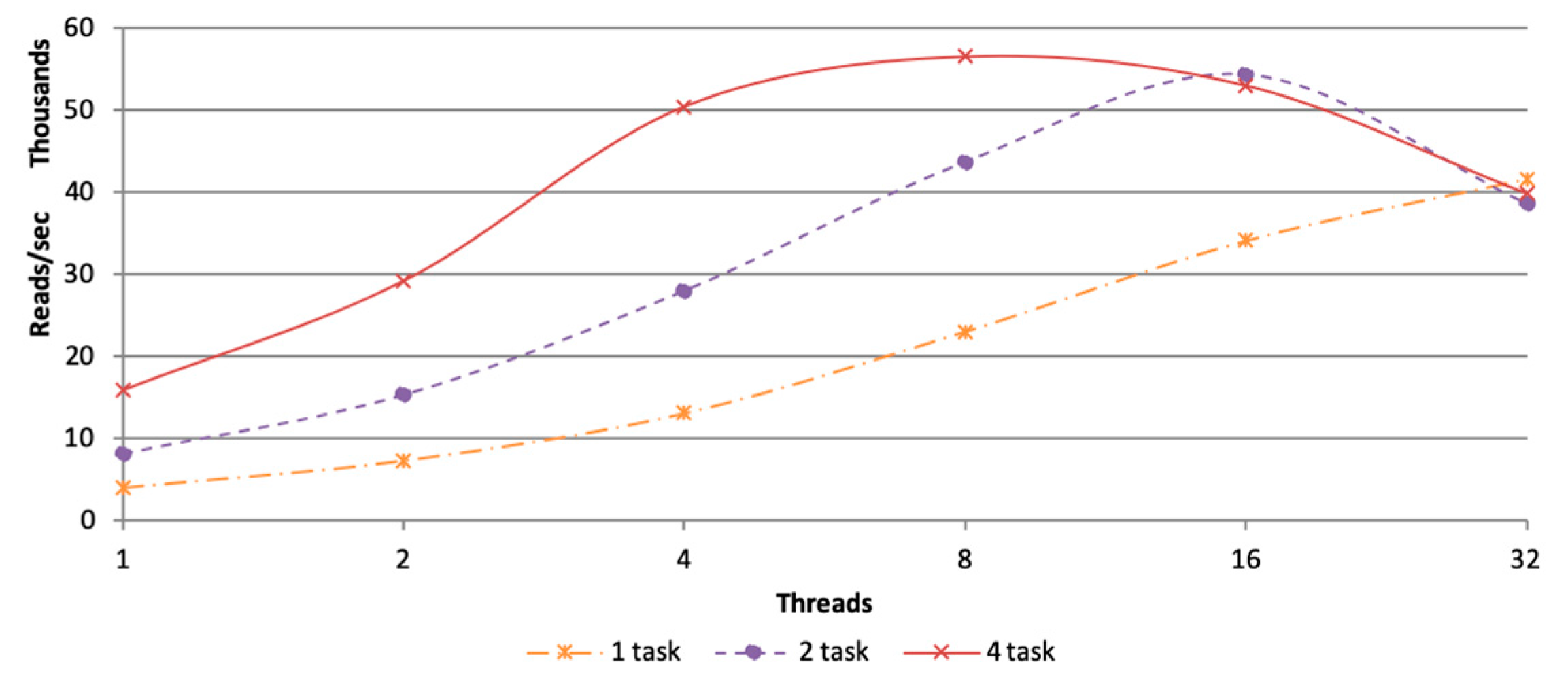
3.2.1. Parameter Setting of Tasks and Threads
3.2.2. In Memory Computation
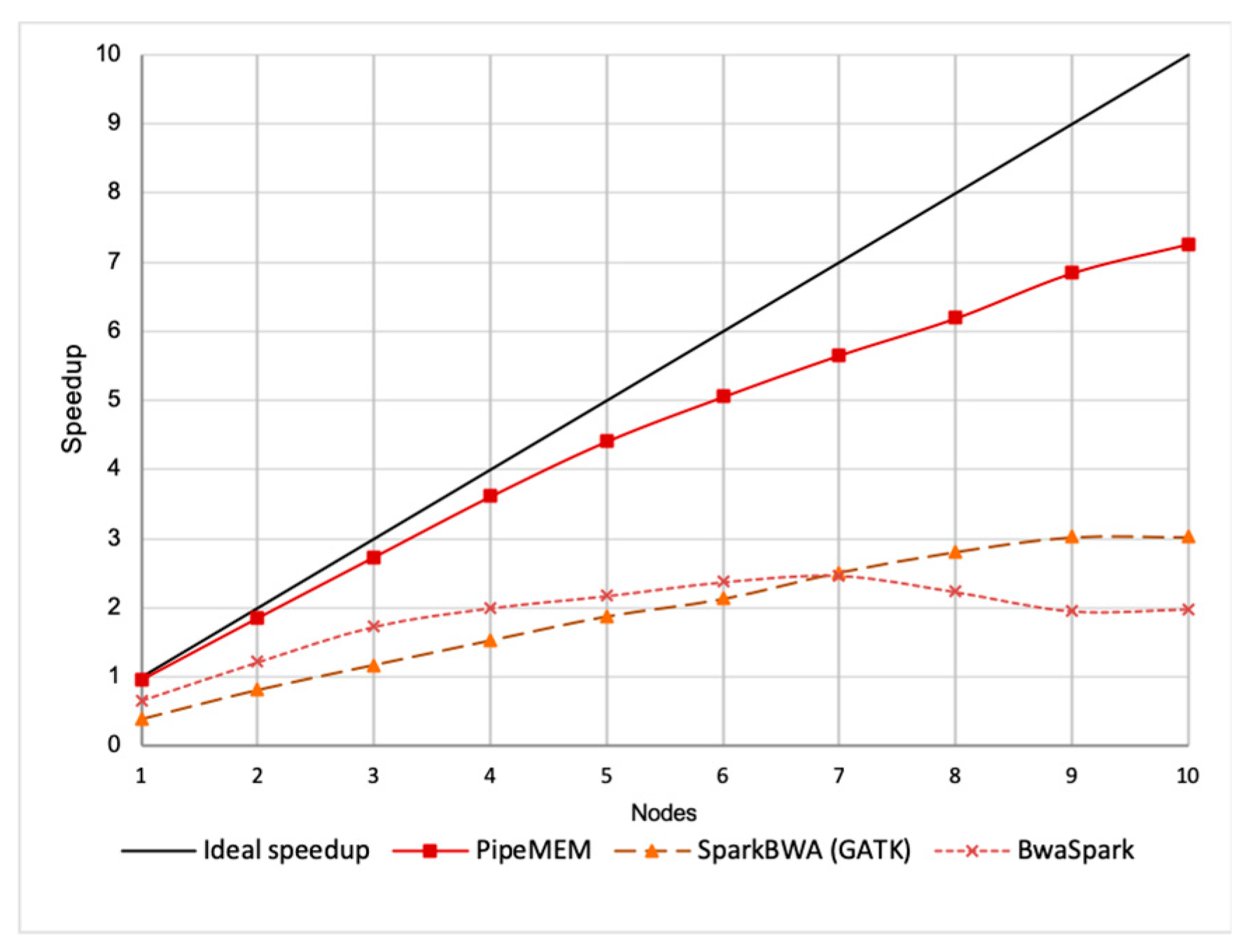
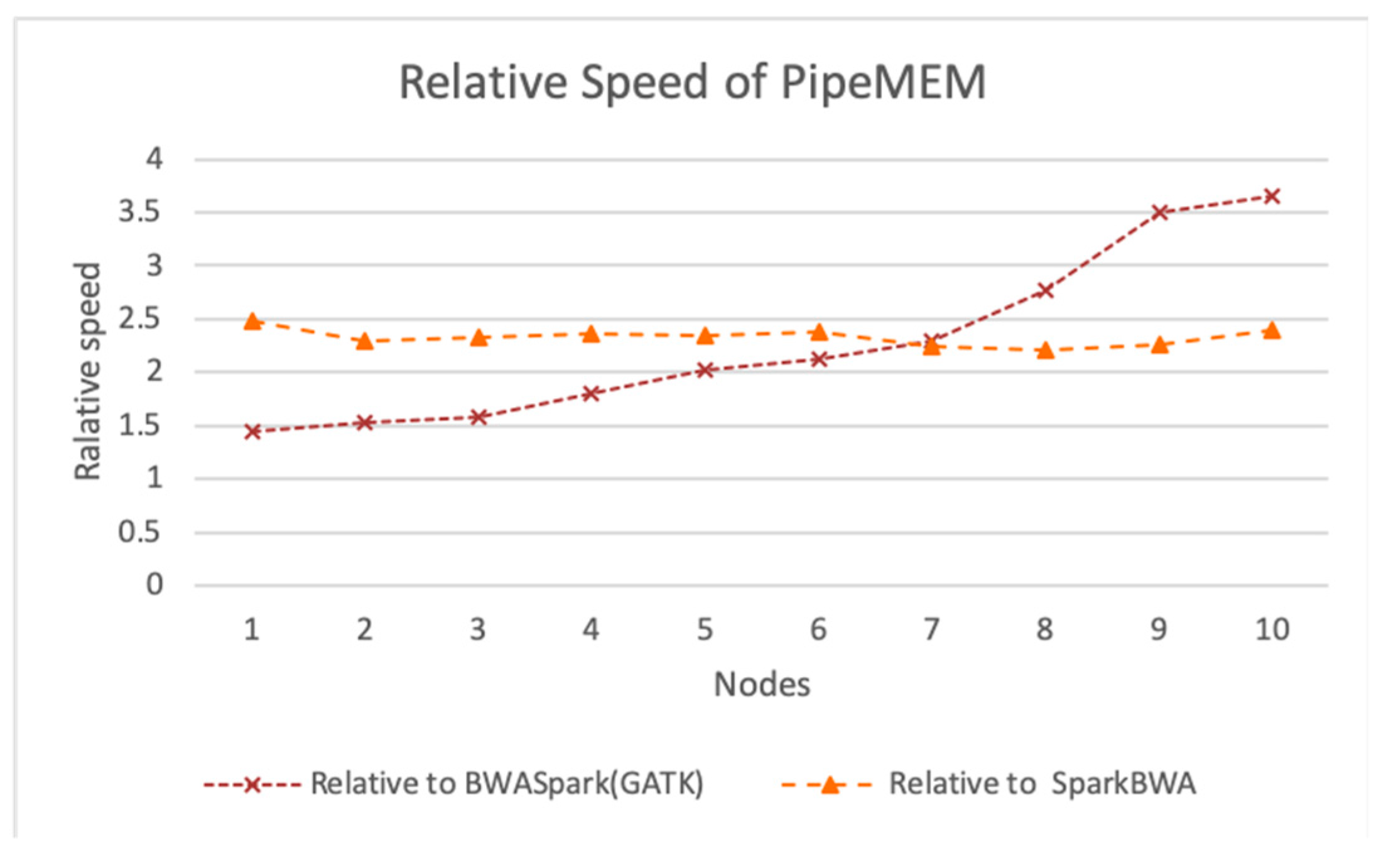
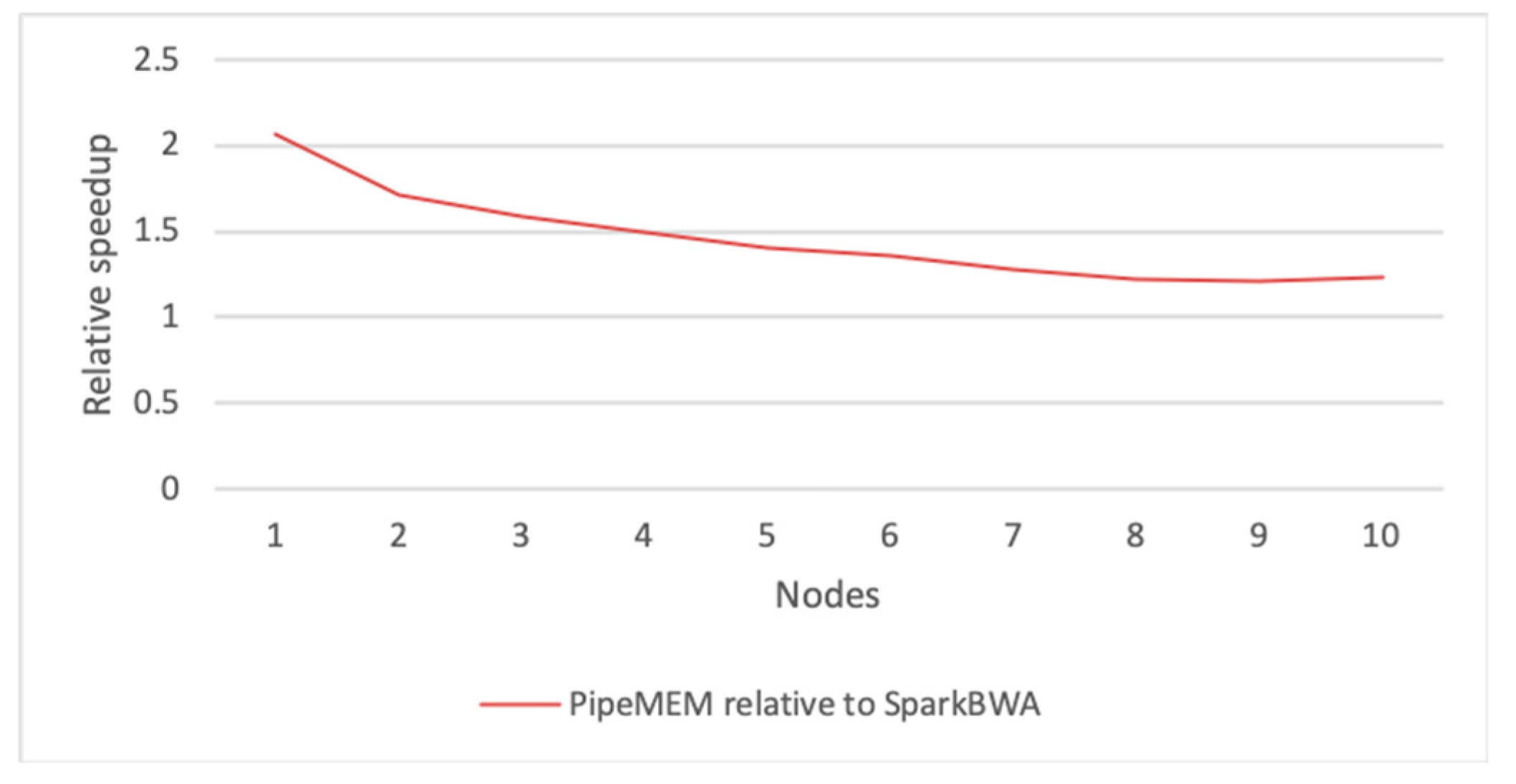
3.3. Comparison of PipeMEM, BWASpark(GATK), and SparkBWA
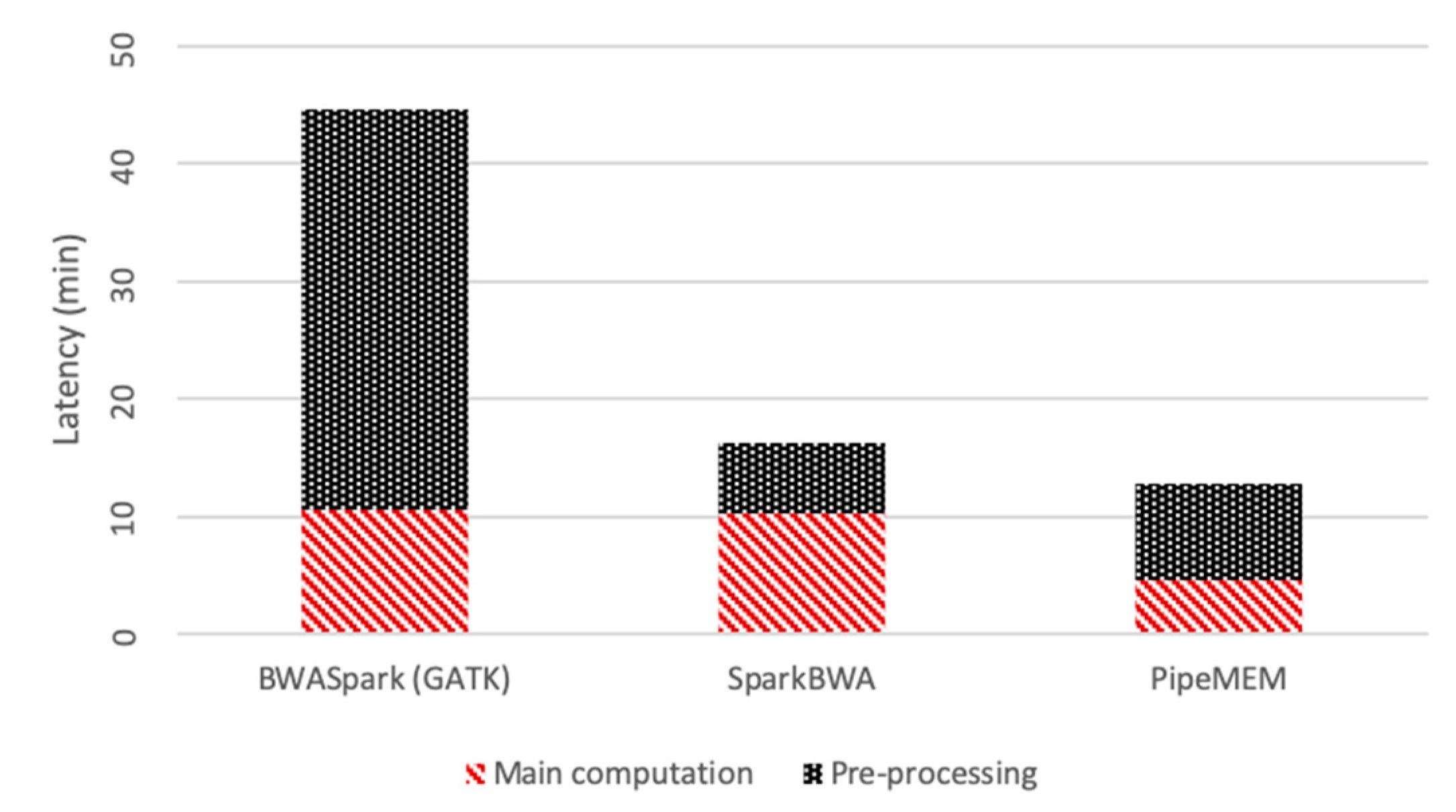
3.3.1. Pre-Processing
3.3.2. Main Computation
3.3.3. Integrate Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Liu, Y.; Schmidt, B.; Maskell, D.L. CUSHAW: A CUDA compatible short read aligner to large genomes based on the Burrows–Wheeler transform. Bioinformatics 2012, 28, 1830–1837. [Google Scholar] [CrossRef] [PubMed]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [PubMed]
- Feuerriegel, S.; Schleusener, V.; Beckert, P.; Kohl, T.A.; Miotto, P.; Cirillo, D.M.; Cabibbe, A.M.; Niemann, S.; Fellenberg, K. PhyResSE: Web tool delineating Mycobacterium tuberculosis antibiotic resistance and lineage from whole-genome sequencing data. J. Clin. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Layer, R.M.; Faust, G.G.; Lindberg, M.R.; Rose, D.B.; Garrison, E.P.; Marth, G.T.; Quinlan, A.R.; Hall, I.M. SpeedSeq: Ultra-fast personal genome analysis and interpretation. Nat. Methods 2015, 12, 966. [Google Scholar] [CrossRef]
- Torri, F.; Dinov, I.D.; Zamanyan, A.; Hobel, S.; Genco, A.; Petrosyan, P.; Clark, A.P.; Liu, Z.; Eggert, P.; Pierce, J.; et al. Next generation sequence analysis and computational genomics using graphical pipeline workflows. Genes 2012, 3, 545–575. [Google Scholar] [CrossRef]
- Genome Analysis Toolkit. Available online: https://software.broadinstitute.org/gatk/ (accessed on 15 August 2019).
- Ping, L. Speeding up large-scale next generation sequencing data analysis with pBWA. J. Appl. Bioinform. Comput. Biol. 2012, 1. [Google Scholar] [CrossRef]
- Darling, A.E.; Carey, L.; Feng, W.C. The Design, Implementation and Evaluation of mpiBLAST; Los Alamos National Laboratory: Los Alamos, NM, USA, 2003. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Georganas, E.; Buluç, A.; Chapman, J.; Oliker, L.; Rokhsar, D.; Yelick, K. Meraligner: A fully parallel sequence aligner. In Proceedings of the 2015 IEEE International Parallel and Distributed Processing Symposium (IPDPS), Hyderabad, India, 25–29 May 2015; pp. 561–570. [Google Scholar]
- Duan, X.; Xu, K.; Chan, Y.; Hundt, C.; Schmidt, B.; Balaji, P.; Liu, W. S-Aligner: Ultrascalable Read Mapping on Sunway Taihu Light. In Proceedings of the 2017 IEEE International Conference on Cluster Computing (CLUSTER), Honolulu, HI, USA, 5–8 September 2017; pp. 36–46. [Google Scholar]
- Zhao, M.; Lee, W.-P.; Garrison, E.P.; Marth, G.T. SSW library: An SIMD Smith-Waterman C/C++ library for use in genomic applications. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Waterman, M. Identification of common molecular subsequence. Mol. Biol. 1981, 147, 195–197. [Google Scholar]
- Weese, D.; Holtgrewe, M.; Reinert, K. RazerS 3: Faster, fully sensitive read mapping. Bioinformatics 2012, 28, 2592–2599. [Google Scholar] [CrossRef] [PubMed]
- González-Domínguez, J.; Hundt, C.; Schmidt, B. parSRA: A framework for the parallel execution of short read aligners on compute clusters. J. Comput. Sci. 2018, 25, 134–139. [Google Scholar] [CrossRef]
- Leo, S.; Santoni, F.; Zanetti, G. Biodoop: Bioinformatics on hadoop. In Proceedings of the 2009 International Conference Parallel Processing Workshops, Vienna, Austria, 22–25 September 2009; pp. 415–422. [Google Scholar]
- Nordberg, H.; Bhatia, K.; Wang, K.; Wang, Z. BioPig: A Hadoop-based analytic toolkit for large-scale sequence data. Bioinformatics 2013, 29, 3014–3019. [Google Scholar] [CrossRef]
- Jourdren, L.; Bernard, M.; Dillies, M.-A.; Le Crom, S. Eoulsan: A cloud computing-based framework facilitating high throughput sequencing analyses. Bioinformatics 2012, 28, 1542–1543. [Google Scholar] [CrossRef]
- Wiewiórka, M.S.; Messina, A.; Pacholewska, A.; Maffioletti, S.; Gawrysiak, P.; Okoniewski, M.J. SparkSeq: Fast, scalable and cloud-ready tool for the interactive genomic data analysis with nucleotide precision. Bioinformatics 2014, 30, 2652–2653. [Google Scholar] [CrossRef]
- Simonyan, V.; Mazumder, R. High-Performance Integrated Virtual Environment (HIVE) tools and applications for big data analysis. Genes 2014, 5, 957–981. [Google Scholar] [CrossRef]
- Abuín, J.M.; Pichel, J.C.; Pena, T.F.; Amigo, J. BigBWA: Approaching the Burrows–Wheeler aligner to Big Data technologies. Bioinformatics 2015, 31, 4003–4005. [Google Scholar] [CrossRef]
- Pireddu, L.; Leo, S.; Zanetti, G. SEAL: A distributed short read mapping and duplicate removal tool. Bioinformatics 2011, 27, 2159–2160. [Google Scholar] [CrossRef]
- Abuín, J.M.; Pichel, J.C.; Pena, T.F.; Amigo, J. SparkBWA: Speeding up the alignment of high-throughput DNA sequencing data. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- BWASpark. Available online: https://gatkforums.broadinstitute.org/gatk/discussions/tagged/bwaspark (accessed on 15 August 2019).
- McCool, M.; Robison, A.; Reinders, J. Structured Parallel Programming: Patterns for Efficient Computation; Elsevier: Waltham, MA, USA, 2012. [Google Scholar]
- Hennessy, J.L.; Patterson, D.A. Computer Architecture: A quantitative Approach; Elsevier: Waltham, MA, USA, 2011. [Google Scholar]
- McSherry, F.; Isard, M.; Murray, D.G. Scalability! but at what COST? In Proceedings of the HotOS, Kartause Ittingen, Switzerland, 18–20 May 2015; Volume 15, p. 14. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PipeMEM | BWASpark (GATK) | SparkBWA | |
|---|---|---|---|
| Pre-processing | (1) Change the data format (2) Upload data to HDFS | (1) Change the data format (fastq to sam) (2) Create a self-defined index file (3) Upload data to HDFS | (1) Upload data to HDFS |
| Main Computation | (1) Distribute data (2) Transform data back to the original format (3) BWA-MEM (4) Upload results to HDFS | (1) District sequence from the original file (2) Distribute data (sequence) (3) Modified-BWA-MEM (4) Merge results with original data (5) Store results to HDFS | (1) Iterate and then merge fastq files (2) Distribute data (3) BWA-MEM (4) Upload results to HDFS |
| Tag of Dataset | Number of Paired Read | Read Length (bp) | Size (GB) | Comment |
|---|---|---|---|---|
| 40 pD1 | 350,000 | 51 | 0.98 | Cut from NA12750 |
| D2 | 60,000,000 | 100 | 31 | Cut from NA12878 |
| Item | Task | Thread | Comment |
|---|---|---|---|
| PipeMEM | 4 | 8 | |
| BWASpark (GATK) | 16 | - | Every task corresponds to a thread |
| SparkBWA | 2 | 16 | Use Spark-Join instead of SortHDFS |
| D2 | PipeMEM | PipeMEM (pipeline) | Speedup |
|---|---|---|---|
| Pre-processing (min) | 23.31 | 8.16 | 2.86× |
| D2 | PipeMEM | BWASpark (GATK) | SparkBWA |
|---|---|---|---|
| Pre-processing (min) | 8.16 | 34.06 | 5.9 |
| Nodes (min) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | Average |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Local disk access | 27.60 | 14.00 | 9.70 | 7.30 | 6.04 | 5.26 | 4.82 | 4.32 | 3.95 | 3.70 | - |
| In-memory (optimize) | 27.00 | 14.00 | 9.50 | 7.20 | 5.90 | 5.15 | 4.60 | 4.20 | 3.80 | 3.58 | - |
| Differential | 0.60 | 0.00 | 0.20 | 0.10 | 0.14 | 0.11 | 0.22 | 0.12 | 0.15 | 0.12 | 0.18 |
| D2 | PipeMEM | BWASpark (GATK) | SparkBWA |
|---|---|---|---|
| Overhead | 3.70% | 33.65% | 61.05% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Liu, C.; Dong, S. PipeMEM: A Framework to Speed Up BWA-MEM in Spark with Low Overhead. Genes 2019, 10, 886. https://doi.org/10.3390/genes10110886
Zhang L, Liu C, Dong S. PipeMEM: A Framework to Speed Up BWA-MEM in Spark with Low Overhead. Genes. 2019; 10(11):886. https://doi.org/10.3390/genes10110886
Chicago/Turabian StyleZhang, Lingqi, Cheng Liu, and Shoubin Dong. 2019. "PipeMEM: A Framework to Speed Up BWA-MEM in Spark with Low Overhead" Genes 10, no. 11: 886. https://doi.org/10.3390/genes10110886
APA StyleZhang, L., Liu, C., & Dong, S. (2019). PipeMEM: A Framework to Speed Up BWA-MEM in Spark with Low Overhead. Genes, 10(11), 886. https://doi.org/10.3390/genes10110886