Genome-Wide Small RNA Sequencing Identifies MicroRNAs Deregulated in Non-Small Cell Lung Carcinoma Harboring Gain-of-Function Mutant p53


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Western Blotting
2.3. RNA Isolation, Small RNA Library Preparation, and Deep Sequencing
2.4. Quantitative Real Time PCR
2.5. Prediction of Novel MiRNAs
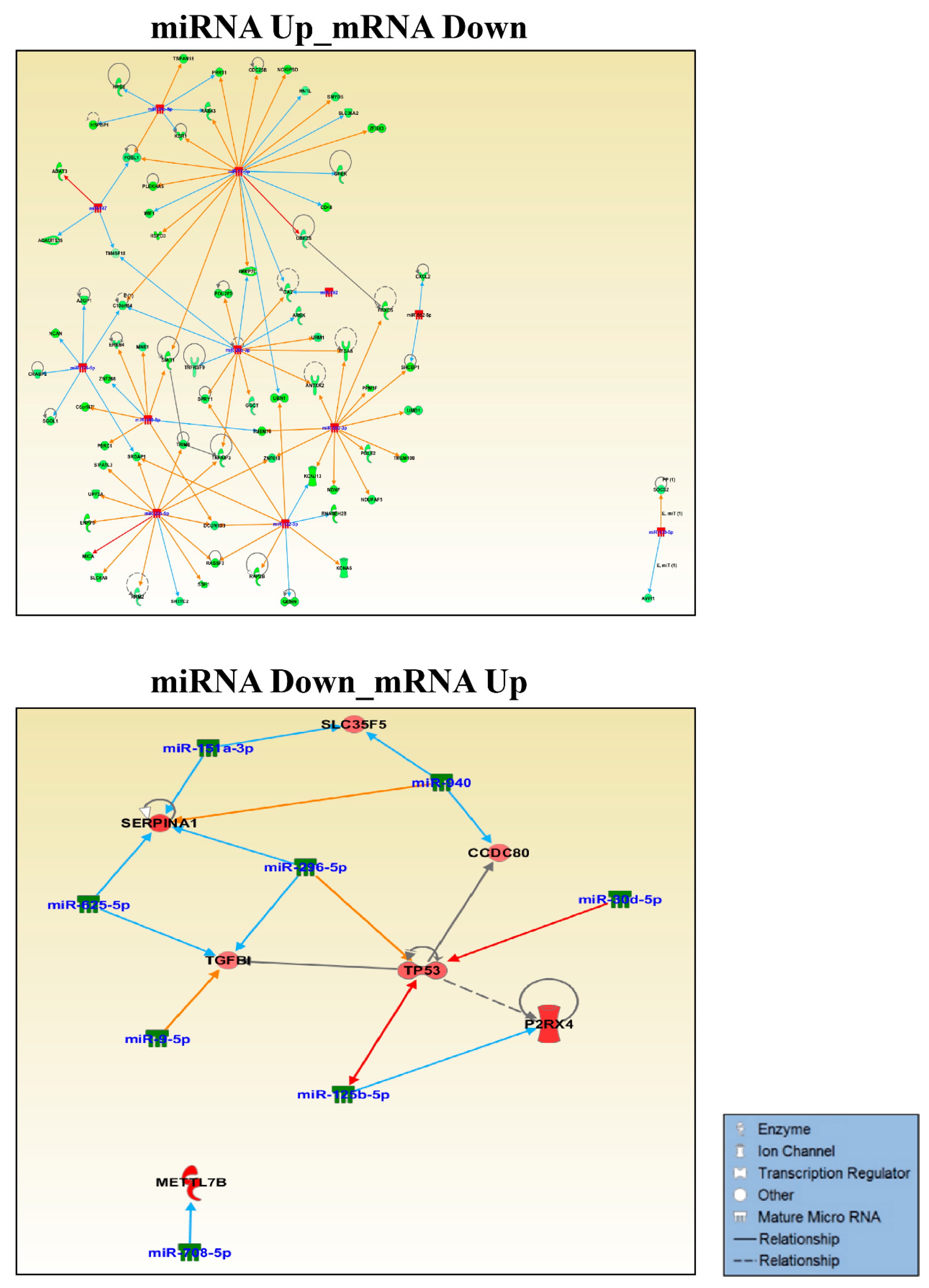
2.6. miRNA Target Prediction and Pathway Analysis in IPA
2.7. Analysis of Publicly Available MiRNA Datasets
2.8. Cloning of Novel MiRNA
2.9. Cell Proliferation Assay
2.10. Intracellular Ki-67 Staining
2.11. Colony Formation Assay
2.12. Monolayer Wound Healing Assay
2.13. Availability of Data and Materials
3. Results
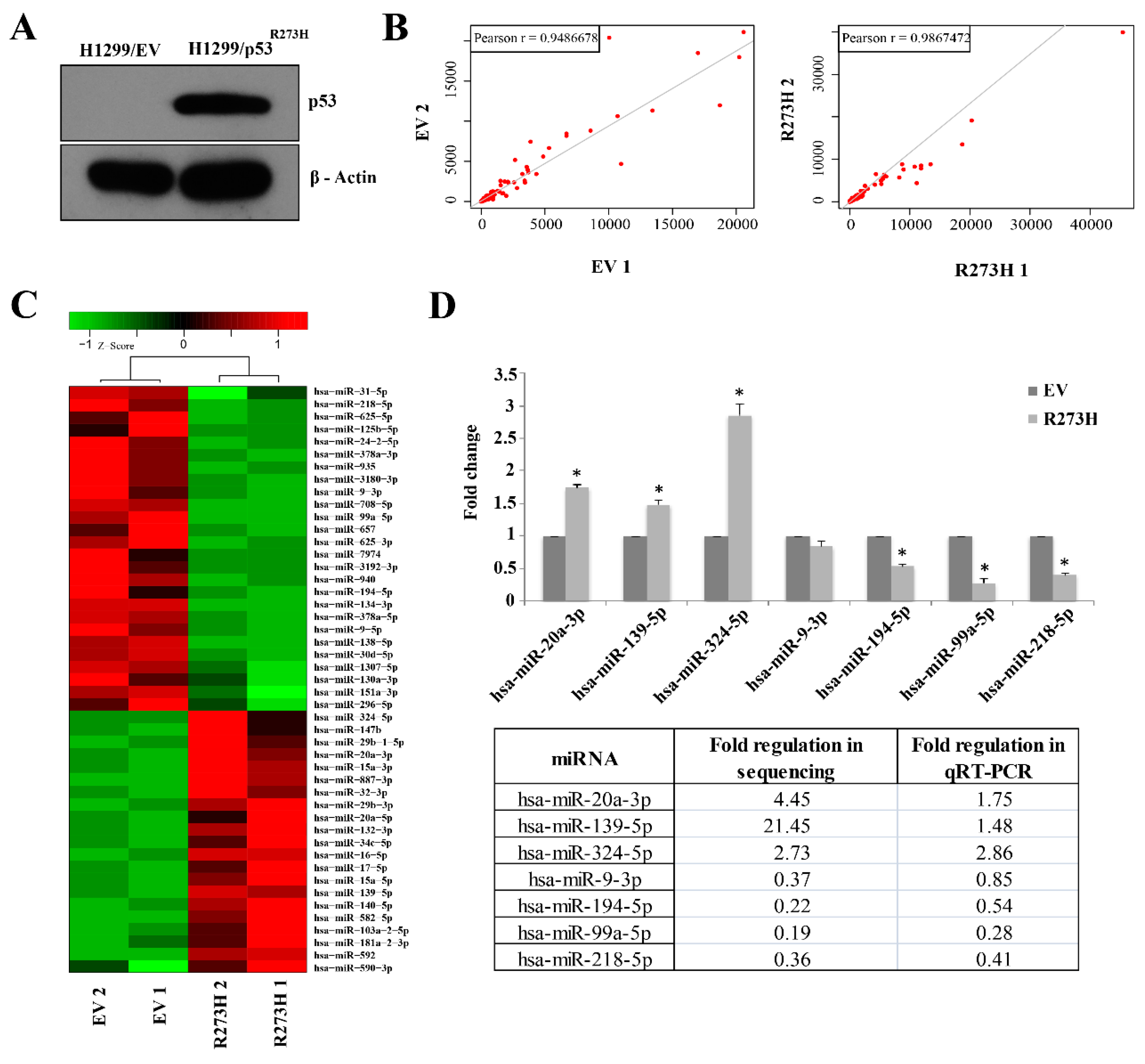
3.1. Small RNA Sequencing Identifies MiRNAs Deregulated in GOF Mutant p53 Cancer Cells
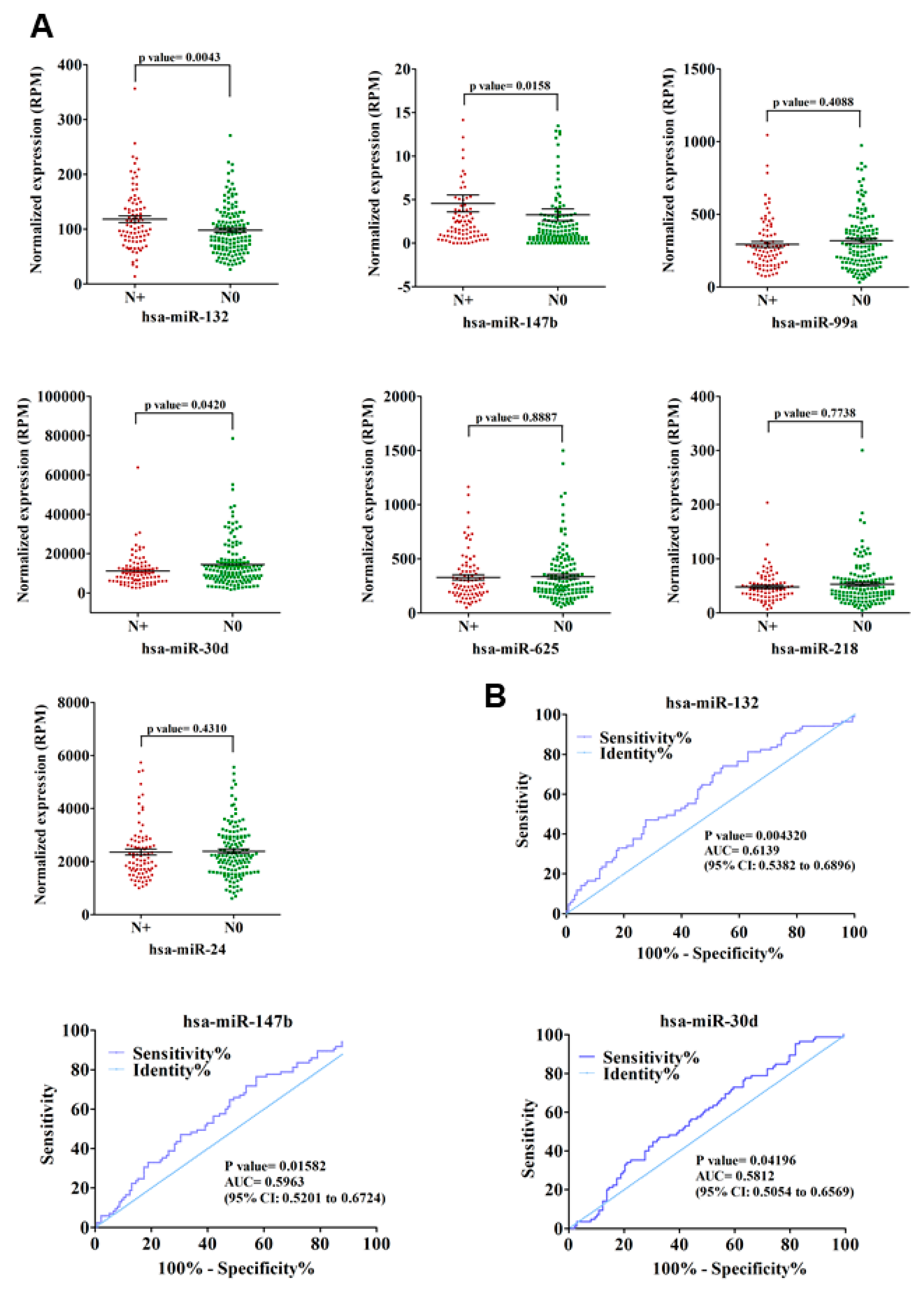
3.2. Validation of GOF Mutant p53-Regulated MiRNAs in Lung Adenocarcinoma (LUAD) Patients
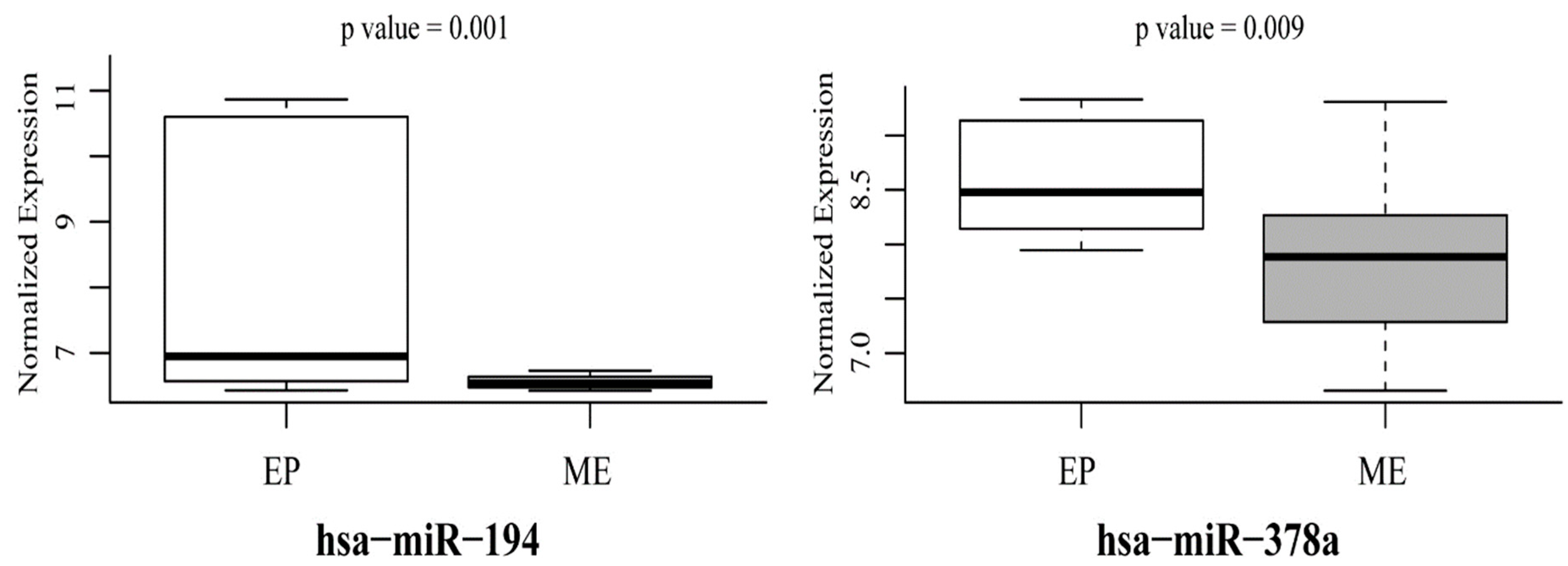
3.3. Mutant p53-Regulated MiR-194 and MiR-378a Predict EMT Phenotype in Cancer Cells
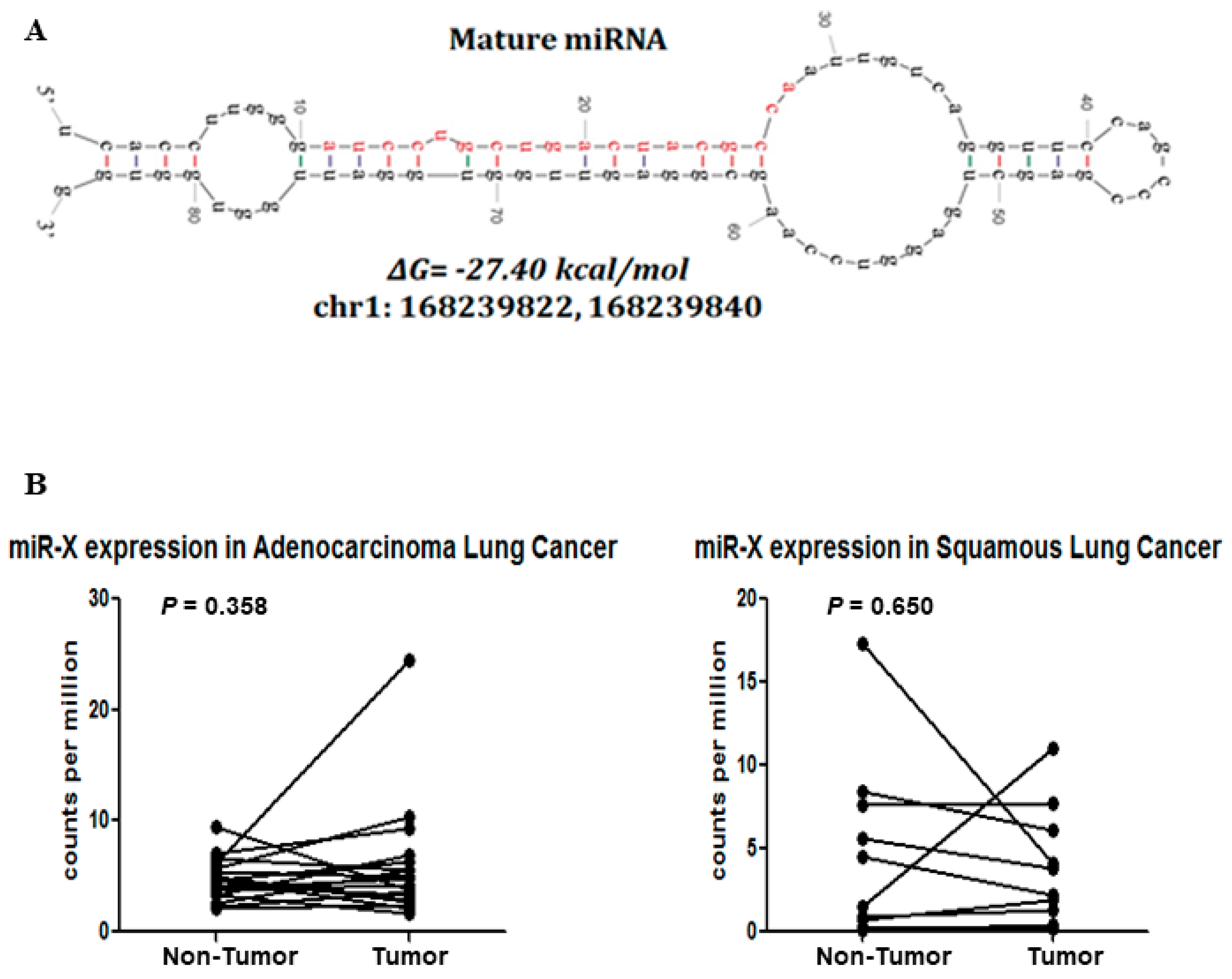
3.4. Discovery of a Novel MiRNA
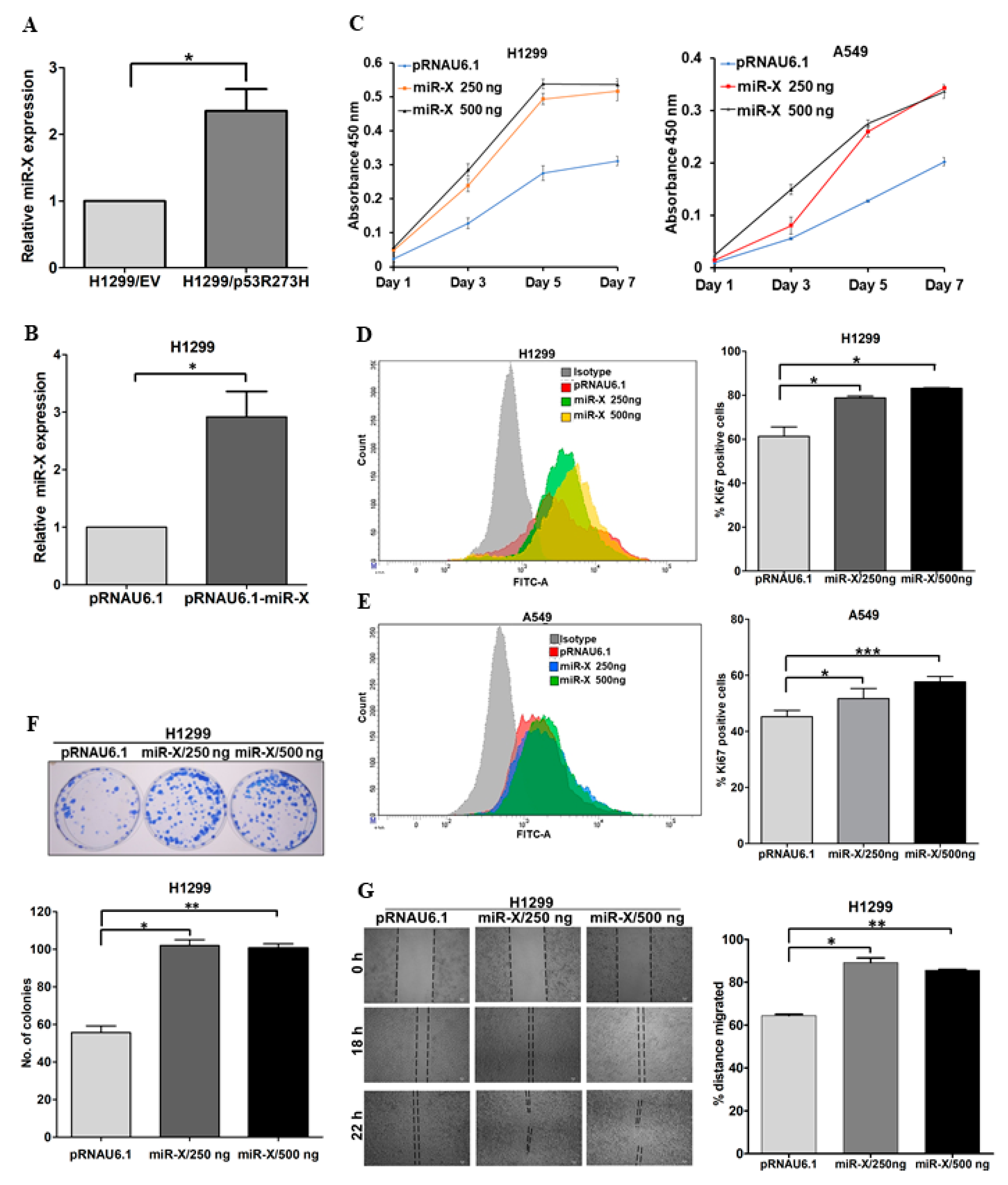
3.5. Novel miR-X Promotes Oncogenic Properties in Lung Adenocarcinoma Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef]
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Datta, A.; Ghatak, D.; Das, S.; Banerjee, T.; Paul, A.; Butti, R.; Gorain, M.; Ghuwalewala, S.; Roychowdhury, A.; Alam, S.K.; et al. p53 gain-of-function mutations increase Cdc7-dependent replication initiation. EMBO Rep. 2017, 18, 2030–2050. [Google Scholar] [CrossRef]
- Singh, S.; Vaughan, C.A.; Frum, R.A.; Grossman, S.R.; Deb, S.; Palit Deb, S. Mutant p53 establishes targetable tumor dependency by promoting unscheduled replication. J. Clin. Investig. 2017, 127, 1839–1855. [Google Scholar] [CrossRef]
- Bajaj, S.; Alam, S.K.; Roy, K.S.; Datta, A.; Nath, S.; Roychoudhury, S. E2 Ubiquitin-conjugating Enzyme, UBE2C Gene, Is Reciprocally Regulated by Wild-type and Gain-of-Function Mutant p53. J. Biol. Chem. 2016, 291, 14231–14247. [Google Scholar] [CrossRef]
- Strano, S.; Dell’Orso, S.; Di Agostino, S.; Fontemaggi, G.; Sacchi, A.; Blandino, G. Mutant p53: An oncogenic transcription factor. Oncogene 2007, 26, 2212–2219. [Google Scholar] [CrossRef]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef]
- Strano, S.; Fontemaggi, G.; Costanzo, A.; Rizzo, M.G.; Monti, O.; Baccarini, A.; Del Sal, G.; Levrero, M.; Sacchi, A.; Oren, M.; et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J. Biol. Chem. 2002, 277, 18817–18826. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Parkin, D.M.; Bray, F.I.; Devesa, S.S. Cancer burden in the year 2000. The global picture. Eur. J. Cancer 2001, 37, S4–S66. [Google Scholar] [CrossRef]
- Ahrendt, S.A.; Hu, Y.; Buta, M.; McDermott, M.P.; Benoit, N.; Yang, S.C.; Wu, L.; Sidransky, D. p53 mutations and survival in stage I non-small-cell lung cancer: Results of a prospective study. J. Natl. Cancer Inst. 2003, 95, 961–970. [Google Scholar] [CrossRef]
- Mogi, A.; Kuwano, H. TP53 mutations in nonsmall cell lung cancer. J. Biomed. Biotechnol. 2011, 2011, 583929. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Hamajima, N.; Ogawa, M.; Takahashi, T. Prognostic significance of p53 alterations in patients with non-small cell lung cancer: A meta-analysis. Clin. Cancer Res. 2000, 6, 4055–4063. [Google Scholar]
- Scian, M.J.; Stagliano, K.E.; Anderson, M.A.; Hassan, S.; Bowman, M.; Miles, M.F.; Deb, S.P.; Deb, S. Tumor-derived p53 mutants induce NF-kappaB2 gene expression. Mol. Cell. Biol. 2005, 25, 10097–10110. [Google Scholar] [CrossRef]
- Yeudall, W.A.; Vaughan, C.A.; Miyazaki, H.; Ramamoorthy, M.; Choi, M.Y.; Chapman, C.G.; Wang, H.; Black, E.; Bulysheva, A.A.; Deb, S.P.; et al. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis 2012, 33, 442–451. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Garzon, R.; Calin, G.A.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, C.; Wu, R.; Hu, W. Tumor suppressor p53 meets microRNAs. J. Mol. Cell Biol. 2011, 3, 44–50. [Google Scholar] [CrossRef]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef]
- Tarasov, V.; Jung, P.; Verdoodt, B.; Lodygin, D.; Epanchintsev, A.; Menssen, A.; Meister, G.; Hermeking, H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: Mir-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007, 6, 1586–1593. [Google Scholar] [CrossRef]
- Donzelli, S.; Fontemaggi, G.; Fazi, F.; Di Agostino, S.; Padula, F.; Biagioni, F.; Muti, P.; Strano, S.; Blandino, G. MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing mutant p53 gain of function. Cell Death Differ. 2012, 19, 1038–1048. [Google Scholar] [CrossRef]
- Masciarelli, S.; Fontemaggi, G.; Di Agostino, S.; Donzelli, S.; Carcarino, E.; Strano, S.; Blandino, G. Gain-of-function mutant p53 downregulates miR-223 contributing to chemoresistance of cultured tumor cells. Oncogene 2014, 33, 1601–1608. [Google Scholar] [CrossRef]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 2013, 32, 3286–3295. [Google Scholar] [CrossRef]
- Neilsen, P.M.; Noll, J.E.; Mattiske, S.; Bracken, C.P.; Gregory, P.A.; Schulz, R.B.; Lim, S.P.; Kumar, R.; Suetani, R.J.; Goodall, G.J.; et al. Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene 2013, 32, 2992–3000. [Google Scholar] [CrossRef]
- Subramanian, M.; Francis, P.; Bilke, S.; Li, X.L.; Hara, T.; Lu, X.; Jones, M.F.; Walker, R.L.; Zhu, Y.; Pineda, M.; et al. A mutant p53/let-7i-axis-regulated gene network drives cell migration, invasion and metastasis. Oncogene 2015, 34, 1094–1104. [Google Scholar] [CrossRef]
- Wang, W.; Cheng, B.; Miao, L.; Mei, Y.; Wu, M. Mutant p53-R273H gains new function in sustained activation of EGFR signaling via suppressing miR-27a expression. Cell Death Dis. 2013, 4, e574. [Google Scholar] [CrossRef]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef]
- Van Oijen, M.G.; Slootweg, P.J. Gain-of-function mutations in the tumor suppressor gene p53. Clin. Cancer Res. 2000, 6, 2138–2145. [Google Scholar]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Neilsen, P.M.; Noll, J.E.; Suetani, R.J.; Schulz, R.B.; Al-Ejeh, F.; Evdokiou, A.; Lane, D.P.; Callen, D.F. Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget 2011, 2, 1203–1217. [Google Scholar] [CrossRef]
- Campling, B.G.; el-Deiry, W.S. Clinical implications of p53 mutations in lung cancer. Methods Mol. Med. 2003, 75, 53–77. [Google Scholar] [CrossRef]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef]
- Kogan-Sakin, I.; Tabach, Y.; Buganim, Y.; Molchadsky, A.; Solomon, H.; Madar, S.; Kamer, I.; Stambolsky, P.; Shelly, A.; Goldfinger, N.; et al. Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ. 2011, 18, 271–281. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef]
- Roger, L.; Jullien, L.; Gire, V.; Roux, P. Gain of oncogenic function of p53 mutants regulates E-cadherin expression uncoupled from cell invasion in colon cancer cells. J. Cell Sci. 2010, 123, 1295–1305. [Google Scholar] [CrossRef]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; Lee, H.H.; et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef]
- Sokilde, R.; Kaczkowski, B.; Podolska, A.; Cirera, S.; Gorodkin, J.; Moller, S.; Litman, T. Global microRNA analysis of the NCI-60 cancer cell panel. Mol. Cancer Ther. 2011, 10, 375–384. [Google Scholar] [CrossRef]
- Dong, P.; Kaneuchi, M.; Watari, H.; Hamada, J.; Sudo, S.; Ju, J.; Sakuragi, N. MicroRNA-194 inhibits epithelial to mesenchymal transition of endometrial cancer cells by targeting oncogene BMI-1. Mol. Cancer 2011, 10, 99. [Google Scholar] [CrossRef]
- Zhang, G.J.; Zhou, H.; Xiao, H.X.; Li, Y.; Zhou, T. MiR-378 is an independent prognostic factor and inhibits cell growth and invasion in colorectal cancer. BMC Cancer 2014, 14, 109. [Google Scholar] [CrossRef]
- Morin, R.D.; O’Connor, M.D.; Griffith, M.; Kuchenbauer, F.; Delaney, A.; Prabhu, A.L.; Zhao, Y.; McDonald, H.; Zeng, T.; Hirst, M.; et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008, 18, 610–621. [Google Scholar] [CrossRef]
- Lee, Y.S.; Shibata, Y.; Malhotra, A.; Dutta, A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 2009, 23, 2639–2649. [Google Scholar] [CrossRef]
- Maute, R.L.; Schneider, C.; Sumazin, P.; Holmes, A.; Califano, A.; Basso, K.; Dalla-Favera, R. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Strano, S.; Blandino, G. Transcriptional regulation by mutant p53 and oncogenesis. Sub-Cell. Biochem. 2014, 85, 91–103. [Google Scholar] [CrossRef]
- Datta, A.; Dey, S.; Das, P.; Alam, S.K.; Roychoudhury, S. Transcriptome profiling identifies genes and pathways deregulated upon floxuridine treatment in colorectal cancer cells harboring GOF mutant p53. Genom. Data 2016, 8, 47–51. [Google Scholar] [CrossRef]
- Pichiorri, F.; Suh, S.S.; Rocci, A.; De Luca, L.; Taccioli, C.; Santhanam, R.; Zhou, W.; Benson, D.M., Jr.; Hofmainster, C.; Alder, H.; et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 2010, 18, 367–381. [Google Scholar] [CrossRef]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef]
- Shin, S.; Cha, H.J.; Lee, E.M.; Jung, J.H.; Lee, S.J.; Park, I.C.; Jin, Y.W.; An, S. MicroRNAs are significantly influenced by p53 and radiation in HCT116 human colon carcinoma cells. Int. J. Oncol. 2009, 34, 1645–1652. [Google Scholar]
- Padua, D.; Massague, J. Roles of TGFbeta in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Yu, J.; Bulk, E.; Ji, P.; Hascher, A.; Tang, M.; Metzger, R.; Marra, A.; Serve, H.; Berdel, W.E.; Wiewroth, R.; et al. The EPHB6 receptor tyrosine kinase is a metastasis suppressor that is frequently silenced by promoter DNA hypermethylation in non-small cell lung cancer. Clin. Cancer Res. 2010, 16, 2275–2283. [Google Scholar] [CrossRef]
- Boyd, D.B. Insulin and cancer. Integr. Cancer Ther. 2003, 2, 315–329. [Google Scholar] [CrossRef]
- Londin, E.; Loher, P.; Telonis, A.G.; Quann, K.; Clark, P.; Jing, Y.; Hatzimichael, E.; Kirino, Y.; Honda, S.; Lally, M.; et al. Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc. Natl. Acad. Sci. USA 2015, 112, E1106–E1115. [Google Scholar] [CrossRef]
- Liang, C.; Li, Y.; Luo, J. A Novel Method to Detect Functional microRNA Regulatory Modules by Bicliques Merging. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 13, 549–556. [Google Scholar] [CrossRef]
- Qin, S.; Jin, P.; Zhou, X.; Chen, L.; Ma, F. The Role of Transposable Elements in the Origin and Evolution of MicroRNAs in Human. PLoS ONE 2015, 10, e0131365. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | H1299/EV Replicate 1 | H1299/EV Replicate 2 | H1299/R273H Replicate 1 | H1299/R273H Replicate 2 | |
|---|---|---|---|---|---|
| Total Raw Reads | 1,442,378 | 751,538 | 1,292,998 | 874,753 | |
| Reads after pre-processing | 753,866 | 340,244 | 566,961 | 500,523 | |
| Mapping to References (miRBase, rRNA, tRNA & Adaptor) | Total Reads | 582,481 | 261,368 | 462,283 | 420,018 |
| Percentage | 77.27 | 76.82 | 81.54 | 83.92 | |
| Reads mapped to miRNA | Total Reads | 540,145 | 251,022 | 453,934 | 408,055 |
| Percentage | 71.65 | 73.78 | 80.06 | 81.53 | |
| Reads mapped to tRNA | Total Reads | 40,071 | 9763 | 7932 | 11,191 |
| Percentage | 5.32 | 2.87 | 1.40 | 2.24 | |
| Reads mapped to rRNA | Total Reads | 2235 | 575 | 406 | 767 |
| Percentage | 0.30 | 0.17 | 0.07 | 0.15 | |
| Reads mapped to adaptor | Total Reads | 30 | 8 | 11 | 5 |
| Percentage | 0.0040 | 0.0024 | 0.0019 | 0.0010 | |
| Total expressed miRNA (1 or more reads) | 692 | 512 | 507 | 518 | |
| miRNA (3 or more reads) | 363 | 245 | 299 | 306 | |
| H1299/EV vs H1299/mutant p53R273H | ||
|---|---|---|
| Molecular and Cellular Functions | ||
| Name | p-Value | # Molecules |
| Cell Cycle | 7.36E-06-4.52E-02 | 8 |
| Cellular Development | 7.50E-05-4.82E-02 | 13 |
| Cellular Growth and Proliferation | 7.50E-05-4.82E-02 | 12 |
| Cellular Movement | 2.72E-04-3.14E-02 | 10 |
| DNA Replication, Recombination and Repair | 3.67E-04-4.82E-02 | 5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Datta, A.; Das, P.; Dey, S.; Ghuwalewala, S.; Ghatak, D.; Alam, S.K.; Chatterjee, R.; Roychoudhury, S. Genome-Wide Small RNA Sequencing Identifies MicroRNAs Deregulated in Non-Small Cell Lung Carcinoma Harboring Gain-of-Function Mutant p53. Genes 2019, 10, 852. https://doi.org/10.3390/genes10110852
Datta A, Das P, Dey S, Ghuwalewala S, Ghatak D, Alam SK, Chatterjee R, Roychoudhury S. Genome-Wide Small RNA Sequencing Identifies MicroRNAs Deregulated in Non-Small Cell Lung Carcinoma Harboring Gain-of-Function Mutant p53. Genes. 2019; 10(11):852. https://doi.org/10.3390/genes10110852
Chicago/Turabian StyleDatta, Arindam, Pijush Das, Sanjib Dey, Sangeeta Ghuwalewala, Dishari Ghatak, Sk. Kayum Alam, Raghunath Chatterjee, and Susanta Roychoudhury. 2019. "Genome-Wide Small RNA Sequencing Identifies MicroRNAs Deregulated in Non-Small Cell Lung Carcinoma Harboring Gain-of-Function Mutant p53" Genes 10, no. 11: 852. https://doi.org/10.3390/genes10110852
APA StyleDatta, A., Das, P., Dey, S., Ghuwalewala, S., Ghatak, D., Alam, S. K., Chatterjee, R., & Roychoudhury, S. (2019). Genome-Wide Small RNA Sequencing Identifies MicroRNAs Deregulated in Non-Small Cell Lung Carcinoma Harboring Gain-of-Function Mutant p53. Genes, 10(11), 852. https://doi.org/10.3390/genes10110852