Genomic Insights into the Carbon and Energy Metabolism of a Thermophilic Deep-Sea Bacterium Deferribacter autotrophicus Revealed New Metabolic Traits in the Phylum Deferribacteres

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Sequencing and Assembly
2.2. Genome Annotation
2.3. Phylogenetic Analysis
2.4. Growth Experiments on Carbon Monoxide Utilization
2.5. Data Availability
3. Results and Discussion
3.1. General Genome Properties, Genome Mobility, and Genomic Islands
3.2. Central Metabolism
3.3. Autotrophic CO2 Fixation
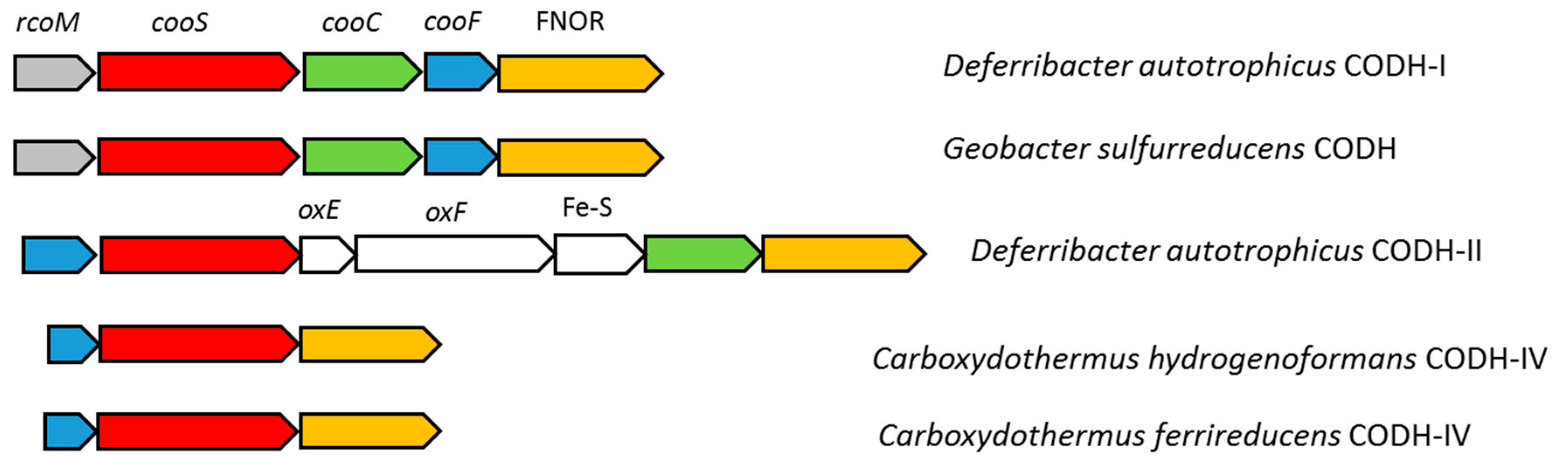
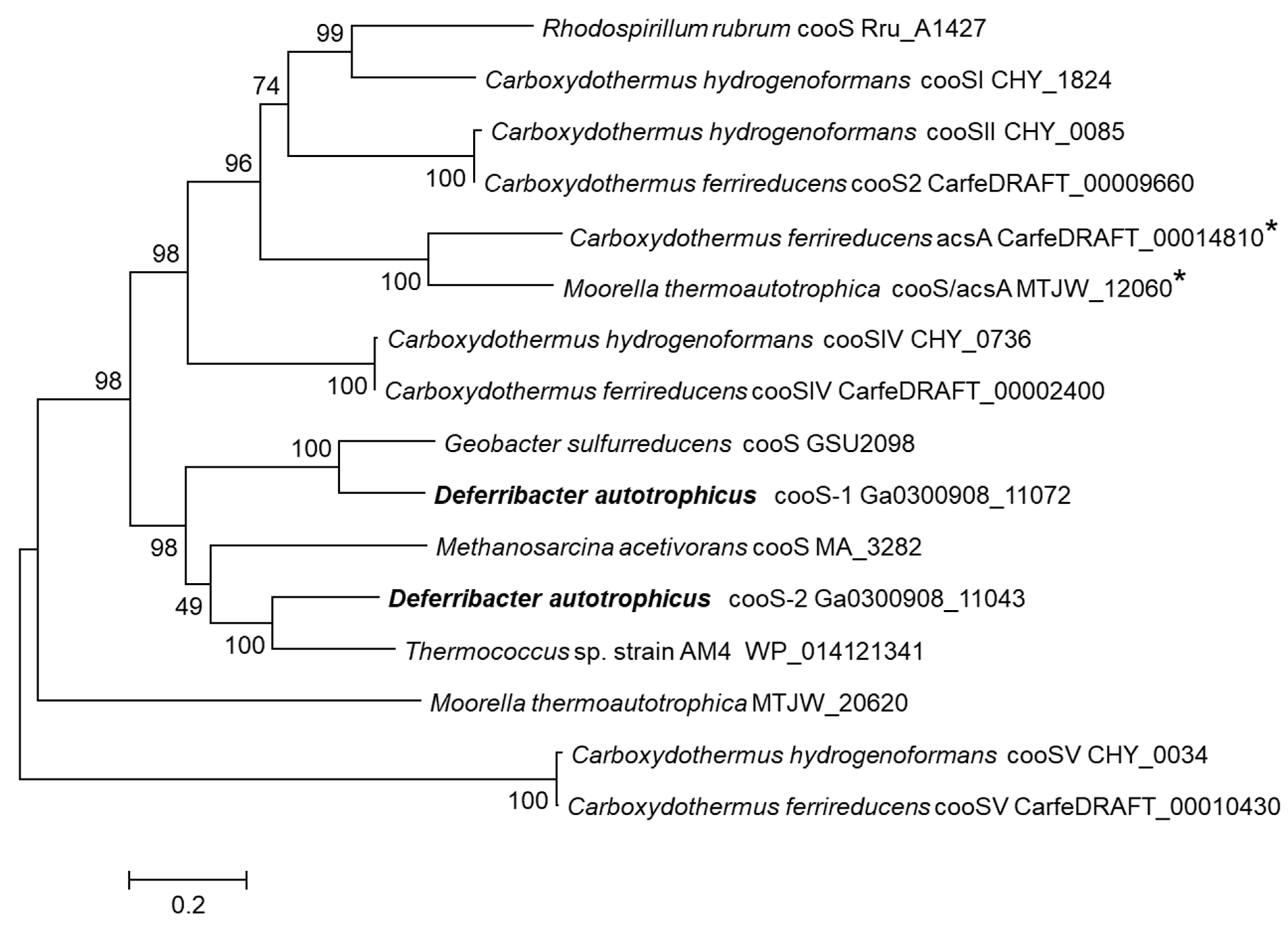
3.4. Carbon Monoxide Utilization
- 4CO + NO3− + 2H2O = 4CO2 + NH3 + OH−
- [CO/NH3] = 4.00; ΔG°’ = −593.6 kJ per reaction; ΔG°’ = −148.4 kJ·mol−1 CO.
3.5. Nitrate Reduction
3.6. Iron Reduction
3.7. Sulfur Reduction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Garrity, G.M.; Holt, J.G. Deferribacteres phy. nov. In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2015; ISBN 9781118960608. [Google Scholar] [CrossRef]
- Greene, A.C.; Patel, B.K.C.; Sheehy, A.J. Deferribacter thermophilus gen. nov., sp. nov., a novel thermophilic manganese and iron-reducing bacterium isolated from a petroleum reservoir. Int. J. Syst. Bacteriol. 1997, 47, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Kobayashi, H.; Nealson, K.H.; Horikoshi, K. Deferribacter desulfuricans sp. nov., a novel sulfur-, nitrate- and arsenate-reducing thermophile isolated from a deep-sea hydrothermal vent. Int. J. Syst. Evol. Microbiol. 2003, 53, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Miroshnichenko, M.L.; Slobodkin, A.I.; Kostrikina, N.A.; L’Haridon, S.; Nercessian, O.; Spring, S.; Stackebrandt, E.; Bonch-Osmolovskaya, E.A.; Jeanthon, C. Deferribacter abyssi sp. nov., an anaerobic thermophile from deep-sea hydrothermal vents of the Mid-Atlantic Ridge. Int. J. Syst. Evol. Microbiol. 2003, 53, 1637–1641. [Google Scholar] [CrossRef] [PubMed]
- Slobodkina, G.B.; Kolganova, T.V.; Chernyh, N.A.; Querellou, J.; Bonch-Osmolovskaya, E.A.; Slobodkin, A.I. Deferribacter autotrophicus sp. nov., an iron(III)-reducing bacterium from a deep-sea hydrothermal vent. Int. J. Syst. Evol. Microbiol. 2009, 5, 1508–1512. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Slobodkin, A.; Campbell, B.; Cary, S.C.; Bonch-Osmolovskaya, E.; Jeanthon, C. Evidence for the presence of thermophilic Fe(III)-reducing microorganisms in deep-sea hydrothermal vents at 13 degrees N (East Pacific Rise). FEMS Microbiol. Ecol. 2001, 36, 235–243. [Google Scholar]
- Takai, K.; Nunoura, T.; Ishibashi, J.I.; Lupton, J.; Suzuki, R.; Hamasaki, H.; Ueno, Y.; Kawagucci, S.; Gamo, T.; Suzuki, Y.; et al. Variability in the microbial communities and hydrothermal fluid chemistry at the newly discovered Mariner hydrothermal field, southern Lau Basin. J. Geophys. Res.-Biogeosci. 2008, 113, 2031. [Google Scholar] [CrossRef]
- Takai, K.; Nunoura, T.; Horikoshi, K.; Shibuya, T.; Nakamura, K.; Suzuki, Y.; Stott, M.; Massoth, G.J.; Christenson, B.W.; deRonde, C.E.; et al. Variability in microbial communities in black smoker chimneys at the NW caldera vent field, Brothers volcano, Kermadec arc. Geomicrobiol. J. 2009, 26, 552–569. [Google Scholar] [CrossRef]
- Takaki, Y.; Shimamura, S.; Nakagawa, S.; Fukuhara, Y.; Horikawa, H.; Ankai, A.; Harada, T.; Hosoyama, A.; Oguchi, A.; Fukui, S.; et al. Bacterial lifestyle in a deep-sea hydrothermal vent chimney revealed by the genome sequence of the thermophilic bacterium Deferribacter desulfuricans SSM1. DNA Res. 2010, 17, 123–137. [Google Scholar] [CrossRef]
- Leggett, R.M.; Clavijo, B.J.; Clissold, L.; Clark, M.D.; Caccamo, M. NextClip: An analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics 2014, 30, 566–568. [Google Scholar] [CrossRef]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.; Korobeynikov, A.; Lapidus, A.; Prjibelsky, A.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling genomes and mini-metagenomes from highly chimeric reads. In Proceedings of the Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics), RECOMB 2013, Beijing, China, 7–10 April 2013; Volume 7821, pp. 158–170. [Google Scholar]
- Chen, I.-M.A.; Chu, K.; Palaniappan, K.; Pillay, M.; Ratner, A.; Huang, J.; Huntemann, M.; Varghese, N.; White, J.R.; Seshadri, R.; et al. IMG/M v.5.0: An integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 2019, 47, 666–677. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, 52–58. [Google Scholar] [CrossRef]
- Varani, A.M.; Siguier, P.; Gourbeyre, E.; Charneau, V.; Chandler, M. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 2011, 12, 30. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, 246–251. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, 30–35. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Rissman, A.I.; Mau, B.; Biehl, B.S.; Darling, A.E.; Glasner, J.D.; Perna, N.T. Reordering contigs of draft genomes using the Mauve Aligner. Bioinformatics 2009, 25, 2071–2073. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Medlar, A.J.; Törönen, P.; Holm, L. AAI-profiler: Fast proteome-wide exploratory analysis reveals taxonomic identity, misclassification and contamination. Nucleic Acids Res. 2018, 46, 479–485. [Google Scholar] [CrossRef]
- Søndergaard, D.; Pedersen, C.N.S.; Greening, C. HydDB: A web tool for hydrogenase classification and analysis. Sci. Rep. 2016, 6, 34212. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Kublanov, I.V.; Gavrilov, S.N.; Rojo, D.; Roman, P.; Golyshin, P.; Slepak, V.Z.; Smedile, F.; Ferrer, M.; Messina, E.; et al. Elemental sulfur and acetate can support life of a novel strictly anaerobic haloarchaeon. ISME J. 2016, 10, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Slobodkin, A.I.; Sokolova, T.G.; Lysenko, A.M.; Wiegel, J. Reclassification of Thermoterrabacterium ferrireducens as Carboxydothermus ferrireducens comb. nov., and emended description of the genus Carboxydothermus. Int. J. Syst. Evol. Microbiol. 2006, 56, 2349–2351. [Google Scholar] [CrossRef]
- Slobodkina, G.B.; Mardanov, A.V.; Ravin, N.V.; Frolova, A.A.; Chernyh, N.A.; Bonch-Osmolovskaya, E.A.; Slobodkin, A.I. Respiratory ammonification of nitrate coupled to anaerobic oxidation of elemental sulfur in deep-sea autotrophic thermophilic bacteria. Front. Microbiol. 2017, 8, 87. [Google Scholar] [CrossRef]
- Antipov, D.; Hartwick, N.; Shen, M.; Raiko, M.; Lapidus, A.; Pevzner, P.A. plasmidSPAdes: Assembling plasmids from whole genome sequencing data. Bioinformatics 2016, 32, 493. [Google Scholar] [CrossRef]
- Lechner, M.; Findeiss, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-) orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Aksenova, H.Y.; Rainey, F.A.; Janssen, P.H.; Zavarzin, G.A.; Morgan, H.W. Spirochaeta thermophila sp. nov., an obligately anaerobic, polysaccharolytic, extremely thermophilic bacterium. Int. J. Syst. Bacteriol. 1992, 42, 175–177. [Google Scholar] [CrossRef]
- Pohlschroeder, M.; Leschine, S.B.; Canale-Parola, E. Spirochaeta caldaria sp. nov., a thermophilic bacterium that enhances cellulose degradation by Clostridium thermocellum. Arch. Microbiol. 1994, 161, 17–24. [Google Scholar] [CrossRef]
- Imachi, H.; Sakai, S.; Hirayama, H.; Nakagawa, S.; Nunoura, T.; Takai, K.; Horikoshi, K. Exilispira thermophila gen. nov., sp. nov., an anaerobic, thermophilic spirochaete isolated from a deep-sea hydrothermal vent chimney. Int. J. Syst. Bacteriol. 2008, 58, 2258–2265. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A. The acetate switch. Microbiol. Mol. Biol. Rev. 2005, 69, 12–50. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, P.-L.; Lovley, D.R. Role of the NiFe hydrogenase Hya in oxidative stress defense in Geobacter sulfurreducens. J Bacteriol. 2012, 194, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.R.; King, G.M. Isolation and characterization of Acidobacterium ailaaui sp. nov., a novel member of Acidobacteria subdivision 1, from a geothermally heated Hawaiian microbial mat. Int. J. Syst. Evol. Microbiol. 2016, 66, 5328–5335. [Google Scholar]
- Pinske, C.; Jaroschinsky, M.; Linek, S.; Kelly, C.L.; Sargent, F.; Sawers, R.G. Physiology and bioenergetics of [NiFe]-hydrogenase 2-catalyzed H2-consuming and H2-producing reactions in Escherichia coli. J. Bacteriol. 2015, 197, 296–306. [Google Scholar] [CrossRef]
- Beaton, S.E.; Evans, R.M.; Finney, A.J.; Lamont, C.M.; Armstrong, F.A.; Sargent, F.; Carr, S.B. The structure of hydrogenase-2 from Escherichia coli: Implications for H2-driven proton pumping. Biochem. J. 2018, 16, 1353–1370. [Google Scholar] [CrossRef]
- Fukuda, E.; Wakagi, T. Substrate recognition by 2-oxoacid: Ferredoxin oxidoreductase from Sulfolobus sp. strain 7. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 2002, 1597, 74–80. [Google Scholar] [CrossRef]
- Nishizawa, Y.; Yabuki, T.; Fukuda, E.; Wakagi, T. Gene expression and characterization of two 2-oxoacid: Ferredoxin oxidoreductases from Aeropyrum pernix K1. FEBS Lett. 2005, 579, 2319–2322. [Google Scholar] [CrossRef]
- Nunoura, T.; Chikaraishi, Y.; Izaki, R.; Suwa, T.; Sato, T.; Harada, T.; Mori, K.; Kato, Y.; Miyazaki, M.; Shimamura, S.; et al. A primordial and reversible TCA cycle in a facultatively chemolithoautotrophic thermophile. Science 2018, 359, 559–563. [Google Scholar] [CrossRef]
- Mall, A.; Sobotta, J.; Huber, C.; Tschirner, C.; Kowarschik, S.; Bačnik, K.; Mergelsberg, M.; Boll, M.; Hügler, M.; Eisenreich, W.; et al. Reversibility of citrate synthase allows autotrophic growth of a thermophilic bacterium. Science 2018, 359, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.H.; Liesack, W.; Schink, B. Geovibrio thiophilus sp. nov., a novel sulfur-reducing bacterium belonging to the phylum Deferribacteres. Int. J. Syst. Evol. Microbiol. 2002, 52, 1341–1347. [Google Scholar] [PubMed]
- Fröstl, J.M.; Seifritz, C.; Drake, H.L. Effect of nitrate on the autotrophic metabolism of the acetogens Clostridium thermoautotrophicum and Clostridium thermoaceticum. J. Bacteriol. 1996, 178, 4597–4603. [Google Scholar] [CrossRef] [PubMed]
- Geelhoed, J.S.; Henstra, A.M.; Stams, A.J.M. Carboxydotrophic growth of Geobacter sulfurreducens. Appl. Microbiol. Biotechnol. 2016, 100, 997–1007. [Google Scholar] [CrossRef]
- Inoue, M.; Nakamoto, I.; Omae, K.; Oguro, T.; Ogata, H.; Yoshida, T.; Sako, Y. Structural and phylogenetic diversity of anaerobic carbon-monoxide dehydrogenases. Front. Microbiol. 2019, 9, 3353. [Google Scholar] [CrossRef] [PubMed]
- Henstra, A.M.; Stams, A.J. Novel physiological features of Carboxydothermus hydrogenoformans and Thermoterrabacterium ferrireducens. Appl. Environ. Microbiol. 2004, 70, 7236–7240. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, T.G.; Jeanthon, C.; Kostrikina, N.A.; Chernyh, N.A.; Lebedinsky, A.V.; Stackebrandt, E.; Bonch-Osmolovskaya, E.A. The first evidence of anaerobic CO oxidation coupled with H2 production by a hyperthermophile archaeon isolated from a deep-sea hydrothermal vent. Extremophiles 2004, 8, 317–323. [Google Scholar] [CrossRef]
- Techtmann, S.; Colman, A.S.; Lebedinsky, A.V.; Sokolova, T.G.; Robb, F.T. Evidence for horizontal gene transfer of anaerobic carbon monoxide dehydrogenases. Front. Microbiol. 2012, 3, 132. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 7, 1870–1874. [Google Scholar] [CrossRef]
- Wu, M.; Ren, Q.; Durkin, A.S.; Daugherty, S.C.; Brinkac, L.M.; Dodson, R.J.; Madupu, R.; Sullivan, S.A.; Kolonay, J.F.; Nelson, W.C.; et al. Life in hot carbon monoxide: The complete genome sequence of Carboxydothermus hydrogenoformans Z-2901. PLoS Genet. 2005, 1, e65. [Google Scholar] [CrossRef]
- Kpebe, A.; Benvenuti, M.; Guendon, C.; Rebai, A.; Fernandez, V.; Le Laz, S.; Etienne, E.; Guigliarelli, B.; García-Molina, G.; de Lacey, A.L.; et al. A new mechanistic model for an O2-protected electron-bifurcating hydrogenase, Hnd from Desulfovibrio fructosovorans. BBA Bioenerg. 2018, 1859, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.; Klotz, M.G. Diversity and evolution of bioenergetics systems involved in microbial nitrogen compound transformations. Biochim. Biophys. Acta 2013, 1827, 114–135. [Google Scholar] [CrossRef] [PubMed]
- Mardanov, A.V.; Beletsky, A.V.; Kadnikov, V.V.; Slobodkin, A.I.; Ravin, N.V. Genome analysis of Thermosulfurimonas dismutans, the first thermophilic sulfur-disproportionating bacterium of the phylum Thermodesulfobacteria. Front. Microbiol. 2016, 7, 950. [Google Scholar] [CrossRef] [PubMed]
- Kublanov, I.V.; Sigalova, O.M.; Gavrilov, S.N.; Lebedinsky, A.V.; Rinke, C.; Kovaleva, O.; Chernyh, N.A.; Ivanova, N.; Daum, C.; Reddy, T.B.K.; et al. Genomic analysis of Caldithrix abyssi, the thermophilic anaerobic bacterium of the novel bacterial phylum Calditrichaeota. Front. Microbiol. 2017, 8, 195. [Google Scholar] [CrossRef] [PubMed]
- Hanson, T.E.; Campbell, B.J.; Kalis, K.M.; Campbell, M.A.; Klotz, M.G. Nitrate ammonification by Nautilia profundicola AmH: Experimental evidence consistent with a free hydroxylamine intermediate. Front. Microbiol. 2013, 4, 180. [Google Scholar] [CrossRef]
- Simon, J.; Kern, M.; Hermann, B.; Einsle, O.; Butt, J.N. Physiological function and catalytic versatility of bacterial multihaem cytochromes c involved in nitrogen and sulfur cycling. Biochem. Soc. Trans. 2011, 39, 1864–1870. [Google Scholar] [CrossRef]
- Hein, S.; Witt, S.; Simon, J. Clade II nitrous oxide respiration of Wolinella succinogenes depends on the NosG, -C1, -C2, -H electron transport module, NosB and a Rieske/cytochrome bc complex. Environ. Microbiol. 2017, 12, 4913–4925. [Google Scholar] [CrossRef]
- Garg, N.; Taylor, A.J.; Kelly, D.J. Bacterial periplasmic nitrate and trimethylamine-N-oxide respiration coupled to menaquinol-cytochrome c reductase (Qcr): Implications for electrogenic reduction of alternative electron acceptors. Sci. Rep. 2018, 8, 15478. [Google Scholar] [CrossRef]
- Shi, L.; Dong, H.; Reguera, G.; Beyenal, H.; Lu, A.; Liu, J.; Yu, H.Q.; Fredrickson, J.K. Extracellular electron transfer mechanisms between microorganisms and minerals. Nat. Rev. Microbiol. 2016, 14, 651–662. [Google Scholar] [CrossRef]
- White, G.F.; Edwards, M.J.; Gomez-Perez, L.; Richardson, D.J.; Butt, J.N.; Clarke, T.A. Mechanisms of bacterial extracellular electron exchange. Adv. Microb. Physiol. 2016, 68, 87–138. [Google Scholar] [CrossRef]
- Holmes, D.E.; Dang, Y.; Walker, D.J.F.; Lovley, D.R. The electrically conductive pili of Geobacter species are a recently evolved feature for extracellular electron transfer. Microb. Genom. 2016, 2, e000072. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Squier, T.C.; Zachara, J.M.; Fredrickson, J.K. Respiration of metal (hydr)oxides by Shewanella and Geobacter: A key role for multihaem c-type cytochromes. Mol. Microbiol. 2007, 65, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Fredrickson, J.K.; Zachara, J.M. Genomic analyses of bacterial porin-cytochrome gene clusters. Front. Microbiol. 2014, 5, 657. [Google Scholar] [CrossRef] [PubMed]
- Levar, C.E.; Chan, C.H.; Mehta-Kolte, M.G.; Bond, D.R. An inner membrane cytochrome required only for reduction of high redox potential extracellular electron acceptors. MBio 2014, 5, e02034-14. [Google Scholar] [CrossRef]
- Zacharoff, L.; Chan, C.H.; Bond, D.R. Reduction of low potential electron acceptors requires the CbcL inner membrane cytochrome of Geobacter sulfurreducens. Bioelectrochemistry 2016, 107, 7–13. [Google Scholar] [CrossRef]
- Wang, F.; Gu, Y.; O’Brien, J.P.; Yi, S.M.; Yalcin, S.E.; Srikanth, V.; Shen, C.; Vu, D.; Ing, N.L.; Hochbaum, A.I.; et al. Structure of microbial nanowires reveals stacked hemes that transport electrons over micrometers. Cell 2019, 177, 361–369. [Google Scholar] [CrossRef]
- Light, S.H.; Su, L.; Rivera-Lugo, R.; Cornejo, J.A.; Louie, A.; Iavarone, A.T.; Ajo-Franklin, C.M.; Portnoy, D.A. A flavin-based extracellular electron transfer mechanism in diverse Gram-positive bacteria. Nature 2018, 562, 140–144. [Google Scholar] [CrossRef]
- Hedderich, R.; Klimmek, O.; Kröger, A.; Dirmeier, R.; Keller, M.; Stetter, K.O. Anaerobic respiration with elemental sulfur and with disulfides. FEMS Microbiol. Rev. 1999, 22, 353–381. [Google Scholar] [CrossRef]
- Schauder, R.; Müller, E. Polysulfide as a possible substrate for sulfur-reducing bacteria. Arch. Microbiol. 1993, 160, 377–382. [Google Scholar] [CrossRef]
- Duval, S.; Ducluzeau, A.L.; Nitschke, W.; Schoepp-Cothenet, B. Enzyme phylogenies as markers for the oxidation state of the environment: The case of respiratory arsenate reductase and related enzymes. BMC Evol. Biol. 2008, 8, 206. [Google Scholar] [CrossRef]
- Jormakka, M.; Yokoyama, K.; Yano, T.; Tamakoshi, M.; Akimoto, S.; Shimamura, T.; Curmi, P.; Iwata, S. Molecular mechanism of energy conservation in polysulfide respiration. Nat. Struct. Mol. Biol. 2008, 7, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Schut, G.J.; Bridger, S.L.; Adams, M.W.W. Insights into the metabolism of elemental sulfur by the hyperthermophilic archaeon Pyrococcus furiosus: Characterization of a coenzyme A-dependent NAD(P)H sulfur oxidoreductase. J. Bacteriol. 2007, 189, 4431–4441. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Schicho, R.N.; Kelly, R.M.; Adams, M.W. Hydrogenase of the hyperthermophile Pyrococcus furiosus is an elemental sulfur reductase or sulfhydrogenase: Evidence for a sulfur-reducing hydrogenase ancestor. Proc. Natl. Acad. Sci. USA 1993, 90, 5341–5344. [Google Scholar] [CrossRef] [PubMed]
- Bridger, S.L.; Clarkson, S.M.; Stirrett, K.; DeBarry, M.B.; Lipscomb, G.L.; Schut, G.J.; Westpheling, J.; Scott, R.A.; Adams, M.W. Deletion strains reveal metabolic roles for key elemental sulfur-responsive proteins in Pyrococcus furiosus. J. Bacteriol. 2011, 193, 6498–6504. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.W.; Holden, J.F.; Menon, A.L.; Schut, G.J.; Grunden, A.M.; Hou, C.; Hutchins, A.M.; Jenney, F.E.; Kim, C.; Ma, K.; et al. Key role for sulfur in peptide metabolism and in regulation of three hydrogenases in the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 2001, 183, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Goris, T.; Schubert, T.; Gadkari, J.; Wubet, T.; Tarkka, M.; Buscot, F.; Adrian, L.; Diekert, G. Insights into organohalide respiration and the versatile catabolism of Sulfurospirillum multivorans gained from comparative genomics and physiological studies. Environ. Microbiol. 2014, 16, 3562–3580. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D. autotrophicus SL50T | D. desulfuricans SSM1 [9] | ||
|---|---|---|---|
| Chromosome Contigs * | Chromosome | Plasmid | |
| Size | 2,543,746 | 2,234,389 | 308,544 |
| GC-content | 32.6 | 31.1 | 24.5 |
| Protein coding genes | |||
| total | 2451 | 2396 | 287 |
| pseudogenes | 23 | 12 | 0 |
| RNA genes | 53 | ||
| rRNA operons | 2 | 2 | - |
| tRNA | 42 | 44 | - |
| small ncRNA | 4 | 2 | - |
| IS elements | 66 * | 7 | 34 |
| IS element families | IS110, IS21, IS256, ISL3, IS3, IS5, IS1634, IS607 | IS256, IS5, ISL3 | IS200/IS605, IS256, IS5 |
| CRISPR-Cas systems | |||
| CRISPR repeat stretches | 3 | 1 | 1 |
| CRISPR spacers | 26 | ||
| Complete Cas gene clusters | 0 | 1 | 1 |
| Cas-related genes | 5 | ||
| Genomic islands | |||
| Number of GIs | 12 | 5 | - |
| Total length of GIs | 164,610 | 72,606 | - |
| Microorganism 1 | Nitrate Reduction | Product | napMADGH | narGHJI | nrfA | hao | hcp | otr | S0 Reduction | psrABC |
|---|---|---|---|---|---|---|---|---|---|---|
| Calditerrivibrio nitroreducens | + | NH4+ | + | - | + | - | + | - | - | + |
| Deferribacter autotrophicus | + | NH4+ | + | - | - | + | + | + | + | +++ |
| Deferribacter desulfuricans | + | NO2− | + | + | - | + | + | - | + | +++ |
| Denitrovibrio acetiphilus | + | NH4+ | + | + | - | + | + | - | - | ++ |
| Flexistipes sinusarabici | nt | nt | + | - | - | + | + | - | - | ++ |
| Geovibrio thiophilus | + | NO2−, NH4+ | + | + | - | - | + | - | + | ++ |
| Mucispirillum schaedleri | - | - | + | - | + | - | - | - | nt | - |
| Seleniivibrio woodruffii | - | - | + | + | + | + | + | - | - | +++ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slobodkin, A.; Slobodkina, G.; Allioux, M.; Alain, K.; Jebbar, M.; Shadrin, V.; Kublanov, I.; Toshchakov, S.; Bonch-Osmolovskaya, E. Genomic Insights into the Carbon and Energy Metabolism of a Thermophilic Deep-Sea Bacterium Deferribacter autotrophicus Revealed New Metabolic Traits in the Phylum Deferribacteres. Genes 2019, 10, 849. https://doi.org/10.3390/genes10110849
Slobodkin A, Slobodkina G, Allioux M, Alain K, Jebbar M, Shadrin V, Kublanov I, Toshchakov S, Bonch-Osmolovskaya E. Genomic Insights into the Carbon and Energy Metabolism of a Thermophilic Deep-Sea Bacterium Deferribacter autotrophicus Revealed New Metabolic Traits in the Phylum Deferribacteres. Genes. 2019; 10(11):849. https://doi.org/10.3390/genes10110849
Chicago/Turabian StyleSlobodkin, Alexander, Galina Slobodkina, Maxime Allioux, Karine Alain, Mohamed Jebbar, Valerian Shadrin, Ilya Kublanov, Stepan Toshchakov, and Elizaveta Bonch-Osmolovskaya. 2019. "Genomic Insights into the Carbon and Energy Metabolism of a Thermophilic Deep-Sea Bacterium Deferribacter autotrophicus Revealed New Metabolic Traits in the Phylum Deferribacteres" Genes 10, no. 11: 849. https://doi.org/10.3390/genes10110849
APA StyleSlobodkin, A., Slobodkina, G., Allioux, M., Alain, K., Jebbar, M., Shadrin, V., Kublanov, I., Toshchakov, S., & Bonch-Osmolovskaya, E. (2019). Genomic Insights into the Carbon and Energy Metabolism of a Thermophilic Deep-Sea Bacterium Deferribacter autotrophicus Revealed New Metabolic Traits in the Phylum Deferribacteres. Genes, 10(11), 849. https://doi.org/10.3390/genes10110849