The Role of Co-Deleted Genes in Neurofibromatosis Type 1 Microdeletions: An Evolutive Approach

 ,
,
Abstract
1. Introduction
2. Methodology
2.1. Background
2.2. Genes and Organisms Selected for Analysis
2.3. Sequence Alignment and Phylogenetic Analyses
2.4. Selection Analysis
3. Results
3.1. Phylogenetic Relationships of the Co-Deleted Genes
3.2. Selection Analysis of the Co-Deleted Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rasmussen, S.A.; Friedman, J.M. NF1 gene and neurofibromatosis 1. Am. J. Epidemiol. 2000, 151, 33–40. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health Consensus Development Conference Statement. Neurofibromatosis. Arch. Neurol. Chic. 1998, 45, 575–578. [Google Scholar]
- The Human Gene Mutation Database. Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 16 March 2017).
- Messiaen, L.M.; Wimmer, K. NF1 mutational spectrum. Neurofibromatoses. Monogr. Hum. Genet. 2008, 16, 63–77. [Google Scholar]
- Venturin, M.; Guarnieri, P.; Natacci, F.; Stabile, M.; Tenconi, R.; Clementi, M.; Hernandez, C.; Thompson, P.; Upadhyaya, M.; Larizza, L.; et al. Mental retardation and cardiovascular malformations in NF1 microdeleted patients point to candidate genes in 17q11.2. J. Med. Genet. 2004, 41, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Mensink, K.A.; Ketterling, R.P.; Flynn, H.C.; Knudson, R.A.; Lindor, N.M.; Heese, B.A.; Spinner, R.J.; Babovic-Vuksanovic, D. Connective tissue dysplasia in five new patients with NF1 microdeletions: further expansion of phenotype and review of the literature. J. Med. Genet. 2006, 43, e8. [Google Scholar] [CrossRef] [PubMed]
- De Raedt, T.; Brems, H.; Wolkenstein, P.; Vidaud, D.; Pilotti, S.; Perrone, F.; Mautner, V.; Frahm, S.; Sciot, R.; Legius, E. Elevated risk for MPNST in NF1 microdeletion patients. Am. J. Hum. Genet. 2003, 72, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Descheemaeker, M.J.; Roelandts, K.; De Raedt, T.; Brems, H.; Fryns, J.P.; Legius, E. Intelligence in individuals with a neurofibromatosis type 1 microdeletion. Am. J. Med. Genet. 2004, 131, 325–326. [Google Scholar] [CrossRef]
- Zhang, J.; Tong, H.; Fu, X.; Zhang, Y.; Liu, J.; Cheng, R.; Liang, J.; Peng, J.; Sun, Z.; Liu, H.; et al. Molecular characterization of NF1 and neurofibromatosis type 1 genotype- phenotype correlations in a Chinese population. Sci. Rep. 2005, 5, 11291. [Google Scholar] [CrossRef]
- Bartelt-Kirbach, B.; Wuepping, M.; Dodrimont-Lattke, M.; Kaufmann, D. Expression analysis of genes lying in the NF1 microdeletion interval points to four candidate modifiers for neurofibroma formation. Neurogenetics 2009, 10, 79–85. [Google Scholar] [CrossRef]
- Pasmant, E.; Masliah-Planchon, J.; Lévy, P.; Laurendeau, I.; Ortonne, N.; Parfait, B.; Valeyrie-Allanore, L.; Leroy, K.; Wolkenstein, P.; Vidaud, M.; et al. Identification of genes potentially involved in the increased risk of malignancy in NF1-microdeleted patients. Mol. Med. 2011, 17, 79–87. [Google Scholar] [CrossRef]
- Yang, F.; Xu, Y.P.; Li, J.; Duan, S.S.; Fu, Y.J.; Zhang, Y.; Zhao, Y.; Qiao, W.T.; Chen, Q.M.; Geng, Y.Q.; et al. Cloning and characterization of a novel intracellular protein p48.2 that negatively regulates cell cycle progression. Int. J. Biochem. Cell Biol. 2009 41, 2240–2250. [CrossRef]
- Sabbagh, A.; Pasmant, E.; Laurendeau, I.; Parfait, B.; Barbarot, S.; Guillot, B.; Combemale, P.; Ferkal, S.; Vidaud, M.; Aubourg, P. Members of the NF France Network. Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum. Mol. Genet. 2009, 18, 2768–2778. [Google Scholar] [CrossRef] [PubMed]
- Mautner, V.F.; Kluwe, L.; Friedrich, R.E.; Roehl, A.C.; Bammert, S.; Högel, J.; Spöri, H.; Cooper, D.N.; Kehrer-Sawatzki, H. Clinical characterization of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 Mb type-1 NF1 deletions. J. Med. Genet. 2010, 47, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.C.; Bustamante, A.G.; Clark, S.; Glanowski, T.B.; Sackton, M.J.; Hubisz, A.; Fledel-Alon, A.; Tanenbaum, D.M.; Civello, D.; White, T.J.; et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005, 3, e170. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.D.; Fledel-Alon, A.; Williamson, S.; Nielsen, R.; Hubisz, M.T.; Glanowski, S.; Tanenbaum, D.M.; White, T.J.; Sninsky, J.J.; Hernandez, R.D.; et al. Natural selection on protein-coding genes in the human genome. Nature 2005, 437, 1153–1157. [Google Scholar] [CrossRef]
- Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Rosset, C.; Vairo, F.; Bandeira, I.C.; Fonini, M.; Netto, C.B.O.; Ashton-Prolla, P. Clinical and molecular characterization of neurofibromatosis in southern Brazil. Expert. Rev. Mol. Diagn. 2018, 18, 577–586. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Posada, D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 2005, 21, 2104–2105. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree. 2013. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 12 June 2019).
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Anisimova, M.; Yang, Z. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol. Biol. Evol. 2007, 24, 1219–1228. [Google Scholar] [CrossRef]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef]
- Knudson, A.G. Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Ruggieri, M.; Polizzi, A.; Spalice, A.; Salpietro, V.; Caltabiano, R.; D’Orazi, V.; Pavone, P.; Pirrone, C.; Magro, G.; Platania, N.; et al. The natural history of spinal neurofibromatosis: a critical review of clinical and genetic features. Clin. Genet. 2015, 87, 401–410. [Google Scholar] [CrossRef]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef]
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.; Liu, Y.; Cochran, M.; Abbott, M.A.; Atkin, J.; et al. High incidence of Noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: genotype-phenotype correlation. Hum. Mutat. 2015, 36, 1052–1063. [Google Scholar] [CrossRef]
- Jenne, D.E.; Tinschert, S.; Reimann, H.; Lasinger, W.; Thiel, G.; Hameister, H.; Kehrer-Sawatzki, H. Molecular characterization and gene content of breakpoint boundaries in patients with neurofibromatosis type 1 with 17q11.2 microdeletions. Am. J. Hum. Genet. 2001, 69, 516–527. [Google Scholar] [CrossRef] [PubMed][Green Version]
- López-Correa, C.; Dorschner, M.; Brems, H.; Lázaro, C.; Clementi, M.; Upadhyaya, M.; Dooijes, D.; Moog, U.; Kehrer-Sawatzki, H.; Rutkowski, J.L.; et al. Recombination hotspot in NF1 microdeletion patients. Hum. Mol. Genet. 2001, 10, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Messiaen, L.; Vogt, J.; Bengesser, K.; Fu, C.; Mikhail, F.; Serra, E.; Garcia-Linares, C.; Cooper, D.N.; Lazaro, C.; Kehrer-Sawatzki, H. Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1). Hum. Mutat. 2011, 32, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Hillmer, M.; Wagner, D.; Summerer, A.; Daiber, M.; Mautner, V.F.; Messiaen, L.; Cooper, D.N.; Kehrer-Sawatzki, H. Fine mapping of meiotic NAHR-associated crossovers causing large NF1 deletions. Hum. Mol. Genet. 2016, 25, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Vogt, J.; Mussotter, T.; Bengesser, K.; Claes, K.; Högel, J.; Chuzhanova, N.; Fu, C.; van den Ende, J.; Mautner, V.F.; Cooper, D.N.; et al. Identification of recurrent type-2 NF1 microdeletions reveals a mitotic nonallelic homologous recombination hotspot underlying a human genomic disorder. Hum. Mutat. 2012, 33, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 microdeletions in neurofibromatosis type 1: from genotype to phenotype. Hum Mutat 2010, 31, e1506–e1518. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.; Cilliers, D.; Coleman, K.; Tatton-Brown, K.; Barker, K.; Bernhard, B.; Burn, J.; Huson, S.; Josifova, D.; Lacombe, D.; et al. Mutations in RNF135, a gene within the NF1 microdeletion region, cause phenotypic abnormalities including overgrowth. Nat. Genet. 2007, 39, 963–965. [Google Scholar] [CrossRef]
- Nalepa, G.; Rolf, M.; Harper, Y.W. Drug discovery in the ubiquitin-proteasome system. Nat. Rev. Drug. Discov. 2006, 5, 596–613. [Google Scholar] [CrossRef]
- Squazzo, S.L.; O’Geen, H.; Komashko, V.M.; Krig, S.R.; Jin, V.X.; Jang, S.W.; Margueron, R.; Reinberg, D.; Green, R.; Farnham, P.J. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res. 2006, 16, 890–900. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, Y.; Jones, S.; Sausen, M.; McMahon, K.; Sharma, R.; Wang, Q.; Belzberg, A.J.; Chaichana, K.; Gallia, G.L.; et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1170–1172. [Google Scholar] [CrossRef]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Yoshida, A.; Ichikawa, H.; Mori, T.; Nakamura, M.; Kawai, A.; Hiraoka, N. Immunohistochemistry for trimethylated H3K27 in the diagnosis of malignant peripheral nerve sheath tumours. Histopathology 2017, 70, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.K.; Phillips, G.R.; Roth, A.D.; Pedraza, L.; Shan, W.; Belkaid, W.; Mi, S.; Fex-Svenningsen, A.; Florens, L.; Yates, J.R., 3rd; et al. Glial membranes at the node of Ranvier prevent neurite outgrowth. Science 2005, 310, 1813–1817. [Google Scholar] [CrossRef] [PubMed]
- Venturin, M.; Carra, S.; Gaudenzi, G.; Brunelli, S.; Gallo, G.R.; Moncini, S.; Cotelli, F.; Riva, P. ADAP2 in heart development: a candidate gene for the occurrence of cardiovascular malformations in NF1 microdeletion syndrome. J. Med. Genet. 2014, 51, 436–443. [Google Scholar] [CrossRef]
- Piddubnyak, V.; Rigou, P.; Michel, L.; Rain, J.C.; Geneste, O.; Wolkenstein, P.; Vidaud, D.; Hickman, J.A.; Mauviel, A.; Poyet, J.L. Positive regulation of apoptosis by HCA66, a new Apaf-1 interacting protein, and its putative role in the physiopathology of NF1 microdeletion syndrome patients. Cell Death Differ. 2007, 14, 1222–1233. [Google Scholar] [CrossRef]
- Lee, K.Y.; Fu, H.; Aladjem, M.I.; Myung, K. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J. Cell Biol. 2013, 200, 31–44. [Google Scholar] [CrossRef]
- Bell, D.W.; Sikdar, N.; Lee, K.Y.; Price, J.C.; Chatterjee, R.; Park, H.D.; Fox, J.; Ishiai, M.; Rudd, M.L.; Pollock, L.M.; et al. Predisposition to cancer caused by genetic and functional defects of mammalian Atad5. PLoS Genet 2011, 7, e1002245. [Google Scholar] [CrossRef]



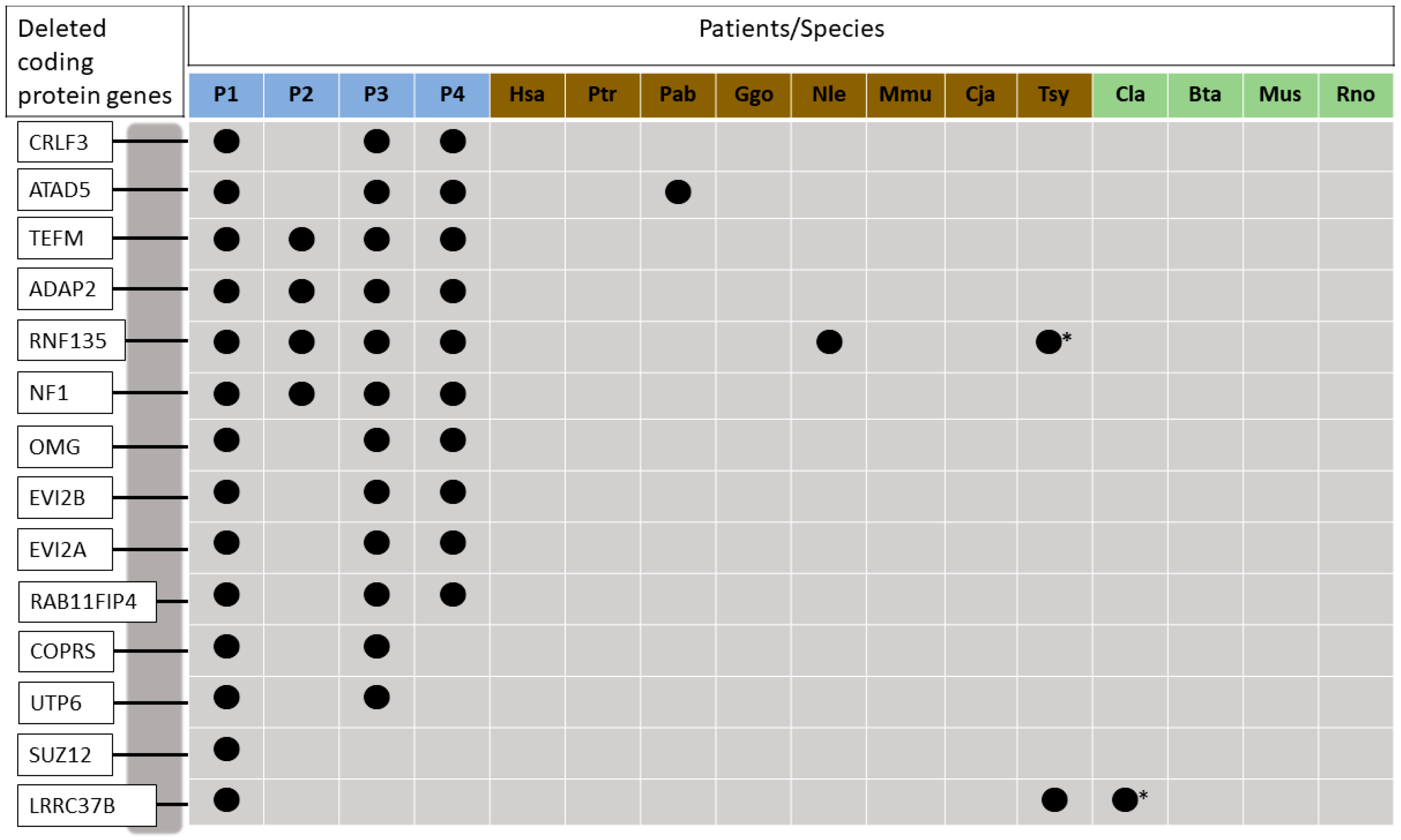{kind=link}
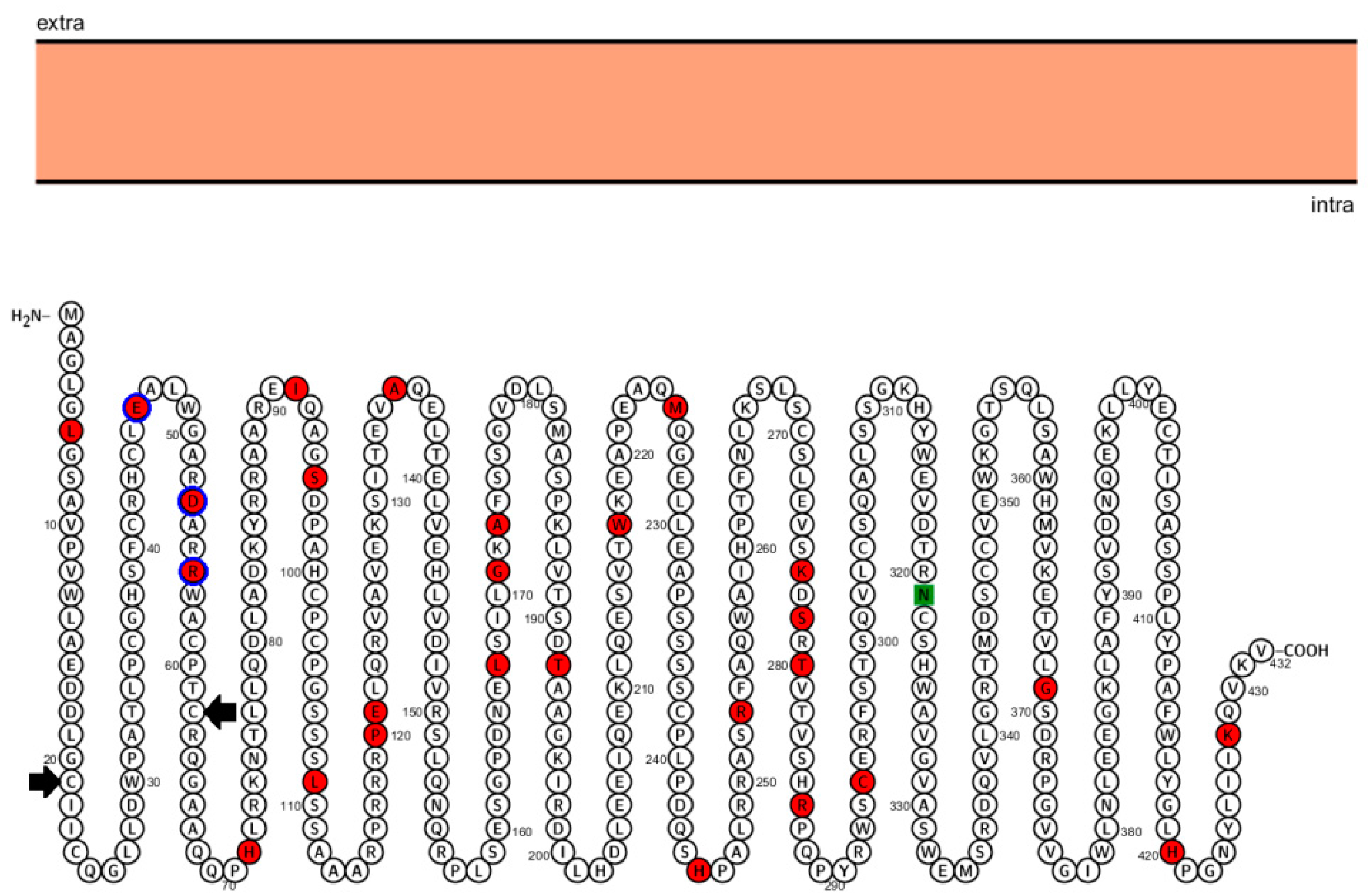{kind=link}
| Gene Family | Model/Likelihood | Comparison | Parameters/Significance |
|---|---|---|---|
| CRLF3 | M0/-3789,882728 | M3 vs. M0 | 2ΔL = 31.37 (df = 4)/p << 0.001 * |
| M1/-3774,195335 | M2 vs. M1 | 2ΔL = −6.51(df = 2)/p < 0.99 | |
| M2/-3777,450354 | M8 vs. M7 | 2ΔL = 0.79(df = 2)/p < 0.5 | |
| M3/-3774,195335 | |||
| M7/-3774,904179 | |||
| M8/-3774,50421 | |||
| ATAD5 | M0/-22006,87329 | M3 vs. M0 | 2ΔL = 301.11 (df = 4)/p < 0.001 * |
| M1/-21865,9053 | M2 vs. M1 | 2ΔL = 0 (df = 2)/p < 0.99 | |
| M2/-21865,9053 | M8 vs. M7 | 2ΔL = 4.26 (df = 2)/p < 0.5 | |
| M3/-21856,31708 | |||
| M7/-21859,57933 | |||
| M8/-21857,4487 | |||
| ADAP2 | M0/-4320,93576 | M3 vs. M0 | 2ΔL = 83.94 (df = 4)/p < 0.001 * |
| M1/-4283,476747 | M2 vs. M1 | 2ΔL = 0 (df = 2)/p < 0.99 | |
| M2/-4283,476747 | M8 vs. M7 | 2ΔL = 0.176 (df = 2)/p < 0.99 | |
| M3/-4278,962315 | |||
| M7/-4279,346509 | |||
| M8/-4279,258106 | |||
| RNF135 | M0/-5802,616454 | M3 vs. M0 | 2ΔL = 116.5 (df = 4)/p < 0.001 * |
| M1/-5745,76494 | M2 vs. M1 | 2ΔL = 2.80 (df = 2)/p < 0.1 | |
| M2/-5744,363479 | M8 vs. M7 | 2ΔL = 11.65 (df = 2)/p < 0.005 * | |
| M3/-5744,351541 | |||
| M7/-5750,260024 | |||
| M8/-5744,430178 | |||
| NF1 | M0/-20275,12645 | M3 vs. M0 | 2ΔL = 73.08 (df = 4)/p < 0.001 * |
| M1/-20240,46408 | M2 vs. M1 | 2ΔL = −4 × 10−6 (df = 2)/p < 0.99 | |
| M2/-20240,46408 | M8 vs. M7 | 2ΔL = 1.57 (df = 2)/p < 0.99 | |
| M3/-20238,58367 | M3 vs. M0 | ||
| M7/-20239,50865 | |||
| M8/-20238,71939 | |||
| UTP6 | M0/-6507,150936 | M3 vs. M0 | 2ΔL = 154.60 (df = 4)/p < 0.001 * |
| M1/-6433,008949 | M2 vs. M1 | 2ΔL = 0 (df = 2)/p < 0.99 | |
| M2/-6433,008949 | M8 vs. M7 | 2ΔL = 8.52 (df = 2)/p < 0.025 * | |
| M3/-6429,850049 | |||
| M7/-6434,194703 | |||
| M8/-6429,931052 | |||
| SUZ12 | M0/-4810,097946 | M3 vs. M0 | 2ΔL = 14.64 (df = 4)/p < 0.005 * |
| M1/-4803,610839 | M2 vs. M1 | 2ΔL = 0 (df = 2)/p < 0.99 | |
| M2/-4803,610839 | M8 vs. M7 | 2ΔL = −0.000288 (df = 2)/p < 0.99 | |
| M3/-4802,77363 | |||
| M7/-4802,776934 | |||
| M8/-4802,777078 | |||
| OMG | M0/-3421,827547 | M3 vs. M0 | 2ΔL = 20.99 (df = 4)/p < 0.001 * |
| M1/-3412,432711 | M2 vs. M1 | 2ΔL = 0 (df = 2)/p < 0.99 | |
| M2/-3393,13422 | M8 vs. M7 | 2ΔL = 0.018 (df = 2)/p < 0.99 | |
| M3/-3412,246309 | |||
| M7/-3412,265355 | |||
| M8/-3412,256186 | |||
| LRRC37B | M0/-16071,4708 | M3 vs. M0 | 2ΔL = 120.7 (df = 4)/p < 0.001 * |
| M1/-16018,91532 | M2 vs. M1 | 2ΔL = 9.87 (df = 2)/p < 0.005 * | |
| M2/-16013,97878 | M8 vs. M7 | 2ΔL = 13.30 (df = 2)/p < 0.001 * | |
| M3/-16011,07676 | |||
| M7/-16018,02362 | |||
| M8/-16011,36949 | |||
| EVI2A | M0/-2792,694611 | M3 vs. M0 | 2ΔL = 44.45 (df = 4)/p < 0.001 * |
| M1/-2771,253237 | M2 vs. M1 | 2ΔL = 1.32 (df = 2)/p < 0.99 | |
| M2/-2770,590286 | M8 vs. M7 | 2ΔL = 4.28 (df = 2)/p < 0.1 | |
| M3/-2770,464944 | |||
| M7/-2772,654596 | |||
| M8/-2770,510367 | |||
| EVI2B | M0/-5998,097719 | M3 vs. M0 | 2ΔL = 71.86 (df = 4)/p < 0.001 * |
| M1/-5964,792227 | M2 vs. M1 | 2ΔL = 1.85 (df = 2)/p < 0.1 | |
| M2/-5963,862998 | M8 vs. M7 | 2ΔL = 6.25 (df = 2)/p < 0.025 * | |
| M3/-5962,167576 | |||
| M7/-5965,886294 | |||
| M8/-5962,759212 | |||
| RAB11FIP4 | M0/-8519,576811 | M3 vs. M0 | 2ΔL = 214.76 (df = 4)/p < 0.001 * |
| M1/-8500,408316 | M2 vs. M1 | 2ΔL = 0 (df = 2)/p < 0.99 | |
| M2/-8500,408316 | M8 vs. M7 | 2ΔL = 3.27 (df = 2)/p < 0.1 | |
| M3/-8412,192643 | |||
| M7/-8412,081211 | |||
| M8/-8410,442254 | |||
| TEFM | M0/-4523,914053 | M3 vs. M0 | 2ΔL = 95.54 (df = 4)/p < 0.001 * |
| M1/-4479,041925 | M2 vs. M1 | 2ΔL = 2.70 (df = 2)/p < 0.1 | |
| M2/-4477,690271 | M8 vs. M7 | 2ΔL = 3.91 (df = 2)/p < 0.1 | |
| M3/-4476,141837 | |||
| M7/-4478,690601 | |||
| M8/-4476,734719 | |||
| CORPS | M0/-2021,525059 | M3 vs. M0 | 2ΔL = 39.62 (df = 4)/p < 0.001 * |
| M1/-2001,718453 | M2 vs. M1 | 2ΔL = 0 (df = 2/p < 0.99 | |
| M2/-2001,718453 | M8 vs. M7 | 2ΔL = 0.26 (df = 2)/p < 0.5 | |
| M3/-2001,71364 | |||
| M7/-2001,856893 | |||
| M8/-2001,724449 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brussa Reis, L.; Turchetto-Zolet, A.C.; Fonini, M.; Ashton-Prolla, P.; Rosset, C. The Role of Co-Deleted Genes in Neurofibromatosis Type 1 Microdeletions: An Evolutive Approach. Genes 2019, 10, 839. https://doi.org/10.3390/genes10110839
Brussa Reis L, Turchetto-Zolet AC, Fonini M, Ashton-Prolla P, Rosset C. The Role of Co-Deleted Genes in Neurofibromatosis Type 1 Microdeletions: An Evolutive Approach. Genes. 2019; 10(11):839. https://doi.org/10.3390/genes10110839
Chicago/Turabian StyleBrussa Reis, Larissa, Andreia Carina Turchetto-Zolet, Maievi Fonini, Patricia Ashton-Prolla, and Clévia Rosset. 2019. "The Role of Co-Deleted Genes in Neurofibromatosis Type 1 Microdeletions: An Evolutive Approach" Genes 10, no. 11: 839. https://doi.org/10.3390/genes10110839
APA StyleBrussa Reis, L., Turchetto-Zolet, A. C., Fonini, M., Ashton-Prolla, P., & Rosset, C. (2019). The Role of Co-Deleted Genes in Neurofibromatosis Type 1 Microdeletions: An Evolutive Approach. Genes, 10(11), 839. https://doi.org/10.3390/genes10110839