A Method for the Structure-Based, Genome-Wide Analysis of Bacterial Intergenic Sequences Identifies Shared Compositional and Functional Features


 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Databases
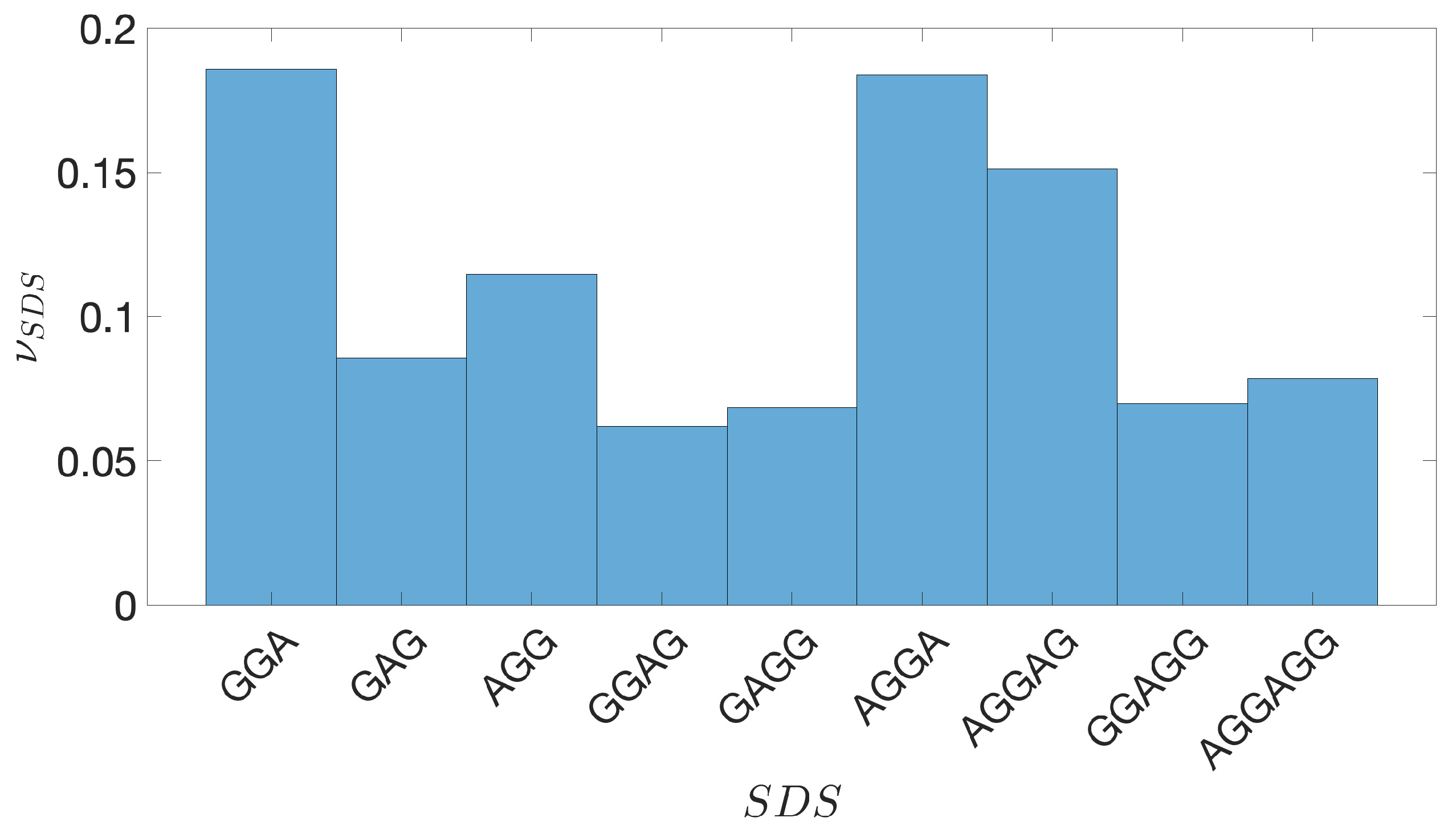
2.2. Shine-Dalgarno Sequences
2.3. Spectral Clustering
- aligning each sequence with all the others (pairwise alignment) thus obtaining a matrix whose entries are similarity scores;
- analyzing the eigenvalues of the Laplacian matrix, computed by the similarity matrix, for determining the appropriate number of clusters;
- making use of the eigenvectors of the Laplacian matrix to work out the k-means algorithm, which allows us to associate each IGS to the selected clusters.
2.3.1. Sequences Alignment
2.3.2. The Normalized Laplacian Matrix
2.3.3. Clustering Algorithm
2.3.4. Silhouette
2.4. Base Composition Analysis
2.5. About STRING
2.5.1. Coexpression
2.5.2. Cooccurrence
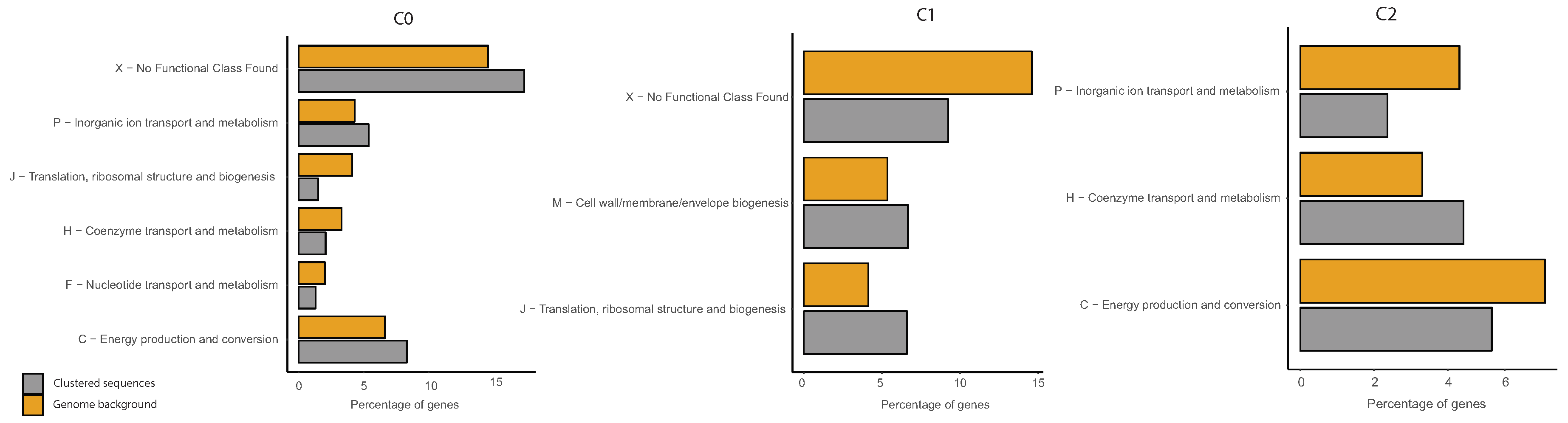
2.6. COG Categories Enrichment
3. Results and Discussion
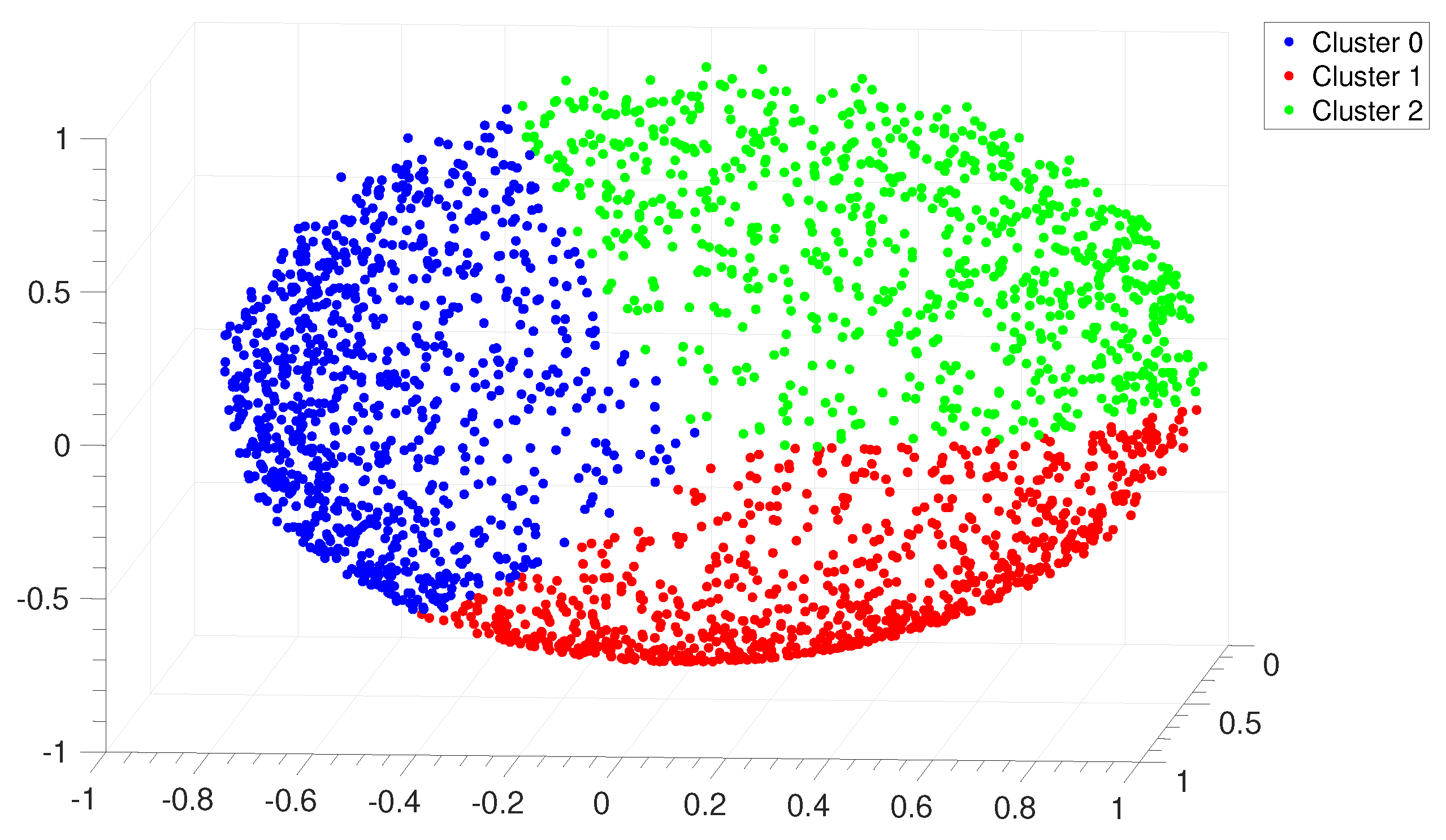
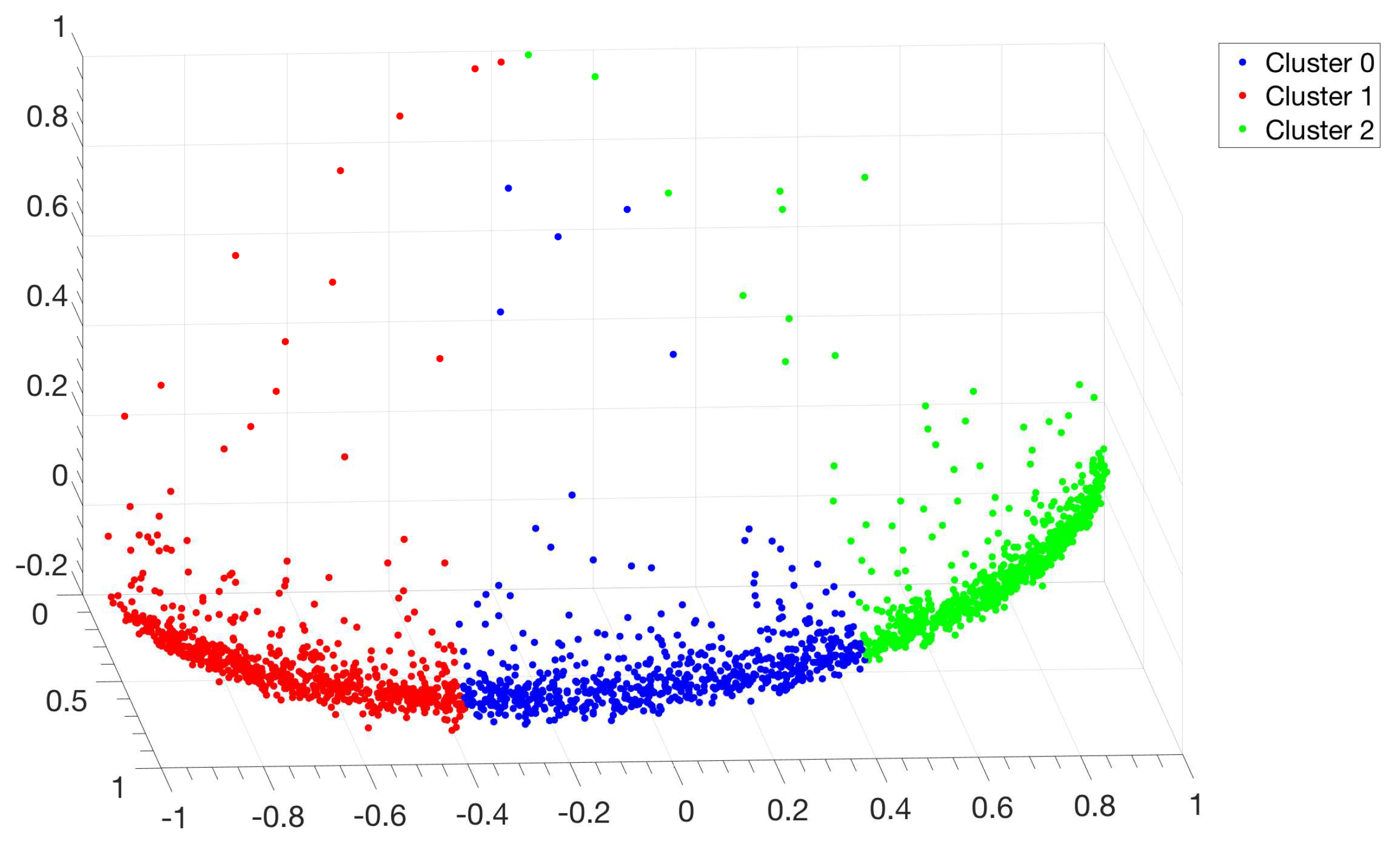
3.1. Identification and Clustering of Intergenic Sequences
3.2. Structural Features of Clusters
3.3. Correlations between Clustering and Biological Features
4. Conclusions
- First we identify the IGRs, all the noncoding portions that are upstream the TSCs annotated in the genome considered, including also the reverse complement of the genes on the opposite strand.
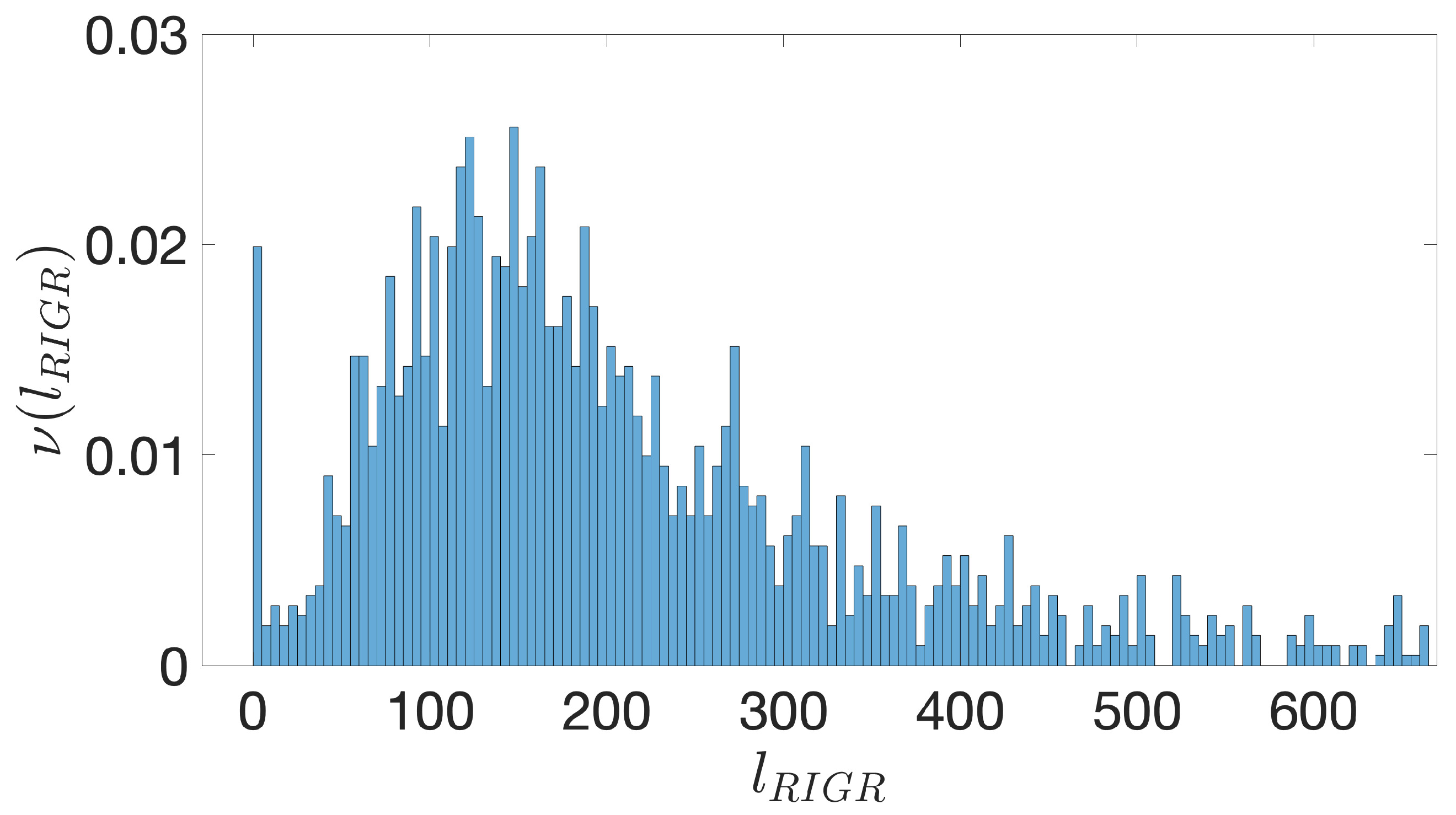
- Starting from the IGRs we can build the set of the IGSs by selecting only the noncoding part between the TSC of a gene and the end of the previous one, regardless of the strand where is located. With the help of an operon database only those that precede the TSC of a transcriptional unit (single gene or operon) are selected. We annotate the length in term of bps of each IGS. Calculating the distribution of the lengths, mean and standard deviation, we can choose a common length for all the IGSs.
- The presence of the SDSs is useful to detect approximately the position of the TSSs so as to eliminate the transcripted and not translated part for each IGS.
- These “cleaned up” noncoding sequences can be compared using alignment algorithms that provide a similarity score between them.
- Similarity matrix containing these scores is processed with a clustering algorithm and the IGSs are divided into clusters based on compositional similarities.
- the object of our research, the IGSs, are sequences of DNA upstream the TSS, charged with regulation at its very first step, since it is non-coding non-transcribed DNA (unlike RNA non-coding);
- IGSs belonging to the same organism are considered and the structural similarities are identified between sequences upstream the TSS unambiguously determined by the identification procedure, regardless of whether they are conserved or not;
- The IGSs are 175-bps long and the alignment procedure takes into consideration the whole sequence globally in its length without focusing specifically on the transcription factor binding sites allowing a correspondence between functional properties and large-scale structural features.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Supporting Information









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
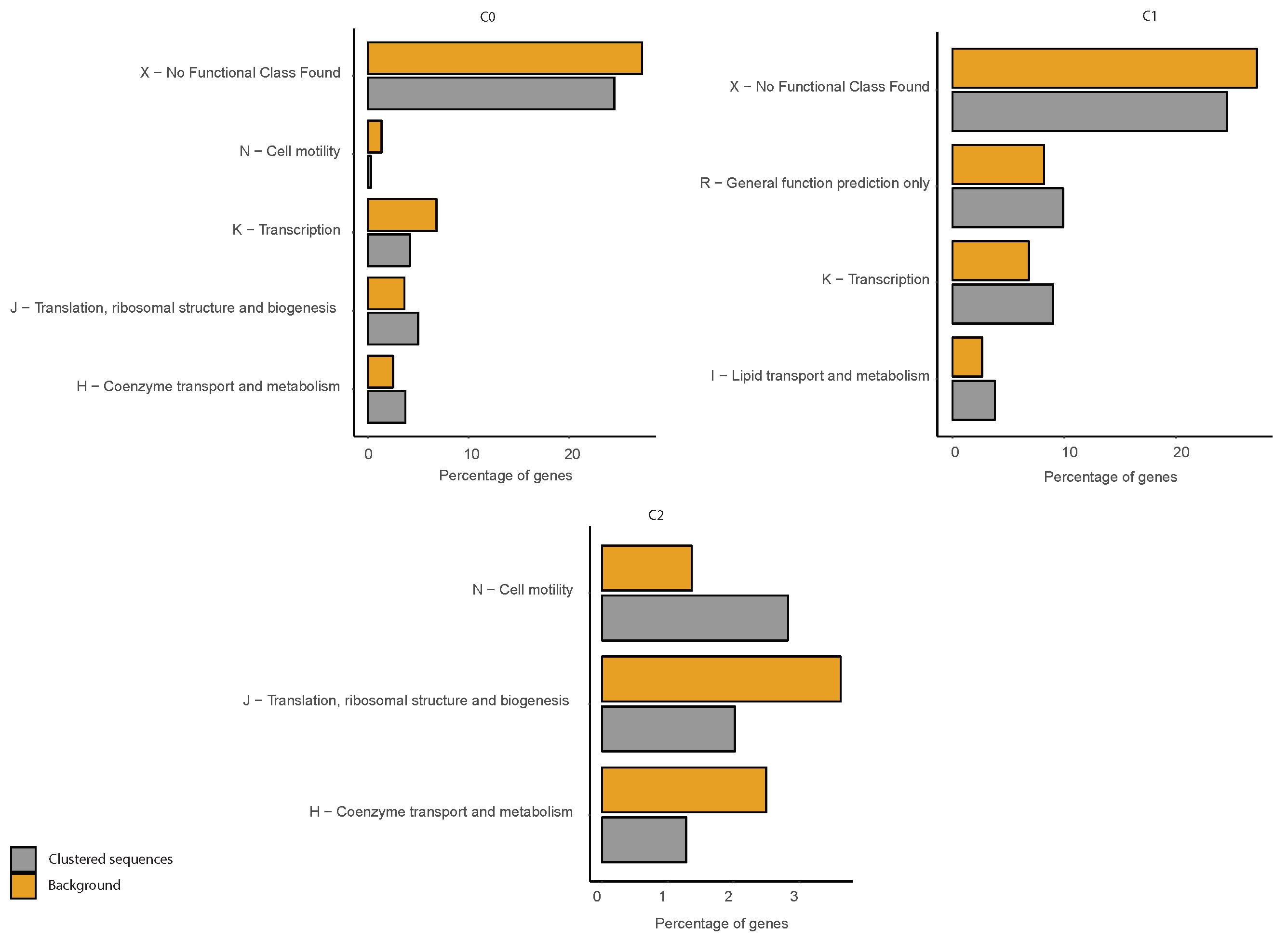{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C0 | 749 | 1341 | 25 | 25.7 | 14.4 | −0.05 | 63 | 169.6 | 189.7 | −0.56 |
| C1 | 856 | 1346 | 41 | 30.6 | 15.6 | 0.67 | 417 | 199.1 | 201.2 | 1.08 |
| C2 | 884 | 1487 | 16 | 32.3 | 16.5 | −0.99 | 28 | 217.5 | 212.2 | −0.89 |


| C0 | 749 | 1341 | 73 | 50.2 | 16.6 | 1.38 | 162 | 119.8 | 60.9 | −0.69 |
| C1 | 856 | 1346 | 71 | 63.1 | 17.4 | 0.45 | 198 | 158.3 | 77.0 | 0.52 |
| C2 | 884 | 1487 | 38 | 67.4 | 18.4 | −1.60 | 85 | 170.1 | 84.2 | −1.01 |









| C0 | 664 | 1074 | 22 | 23.0 | 12.0 | −0.08 | 36 | 102.7 | 108.8 | −0.61 |
| C1 | 718 | 1182 | 47 | 26.8 | 13.8 | 1.46 | 305 | 124.6 | 128.4 | 1.40 |
| C2 | 709 | 1079 | 24 | 26.1 | 12.0 | −0.16 | 42 | 122.1 | 121.6 | −0.66 |


| C0 | 664 | 1074 | 58 | 45.3 | 21.2 | 0.60 | 129 | 104.8 | 71.7 | 0.34 |
| C1 | 718 | 1182 | 89 | 51.4 | 21.5 | 1.75 | 441 | 120.3 | 77.1 | 4.16 |
| C2 | 709 | 1079 | 36 | 51.8 | 22.5 | −0.70 | 76 | 122.6 | 86.2 | −0.54 |

References
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- King, M.C.; Wilson, A.C. Evolution at two levels in humans and chimpanzees. Science 1975, 188, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S. Evo-Devo and the expanding evolutionary Synthesis: A genetic theory of morphological evolution. Cell 2008, 134, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Sheffield, N.C.; Fedrigo, O.; Babbitt, C.C.; Wortham, M.; Tewari, A.K.; London, D.; Song, L.; Lee, B.K.; Iyer, V.R.; et al. Extensive Evolutionary Changes in Regulatory Element Activity during Human Origins Are Associated with Altered Gene Expression and Positive Selection. PLoS Genet 2012, 8, e1002789. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Taft, R.J.; Pheasant, M.; Mattick, J.S. The relationship between non-protein-coding DNA and eukaryotic complexity. BioEssays 2007, 29, 288–299. [Google Scholar] [CrossRef]
- Ponting, C.P. The functional repertoires of metazoan genomes. Nat. Rev. Genet. 2008, 9, 689–698. [Google Scholar] [CrossRef]
- Levy, S.; Hannenhalli, S.; Workman, C. Enrichment of regulatory signals in conserved non-coding genomic sequence. Bioinformatics 2001, 17, 871–877. [Google Scholar] [CrossRef]
- Margulies, E.H.; Blanchette, M.; NISC Comparative Sequencing Program; Haussler, D.; Green, E.D. Identification and Characterization of Multi-Species Conserved Sequences. Genome Res. 2003, 13, 2507–2518. [Google Scholar] [CrossRef]
- Dermitzakis, E.T.; Reymond, A.; Scamuffa, N.; Ucla, C.; Kirkness, E.; Rossier, C.; Antonarakis, S.E. Evolutionary Discrimination of Mammalian Conserved Non-Genic Sequences (CNGs). Science 2003, 302, 1033–1035. [Google Scholar] [CrossRef]
- Bejerano, G.; Haussler, D.; Blanchette, M. Into the heart of darkness: Large-Scale clustering of human non-coding DNA. Bioinformatics 2004, 20, i40–i48. [Google Scholar] [CrossRef] [PubMed]
- Taher, L.; McGaughey, D.M.; Maragh, S.; Aneas, I.; Bessling, S.L.; Miller, W.; Nobrega, M.A.; McCallion, A.S.; Ovcharenko, I. Genome-wide identification of conserved regulatory function in diverged sequences. Genome Res. 2011, 21, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.J.; Moltke, I.; Roth, A.; Washietl, S.; Wen, J.; Kellis, M.; Breaker, R.; Pedersen, J.S. New families of human regulatory RNA structures identified by comparative analysis of vertebrate genomes. Genome Res. 2011, 21, 1929–1943. [Google Scholar] [CrossRef] [PubMed]
- Matsunami, M.; Sumiyama, K.; Saitou, N. Evolution of Conserved Non-Coding Sequences Within the Vertebrate Hox Clusters Through the Two-Round Whole Genome Duplications Revealed by Phylogenetic Footprinting Analysis. J. Mol. Evol. 2010, 71, 427–436. [Google Scholar] [CrossRef]
- Calistri, E. Variability and Constraints in Promoter Evolution. Ph.D. Thesis, Università degli Studi di Firenze, Florence, Firenze, 2008. [Google Scholar]
- Natarajan, A.; Yardımcı, G.G.; Sheffield, N.C.; Crawford, G.E.; Ohler, U. Predicting cell-type–specific gene expression from regions of open chromatin. Genome Res. 2012, 22, 1711–1722. [Google Scholar] [CrossRef]
- Neph, S.; Vierstra, J.; Stergachis, A.B.; Reynolds, A.P.; Haugen, E.; Vernot, B.; Thurman, R.E.; John, S.; Sandstrom, R.; Johnson, A.K.; et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 2012, 489, 83–90. [Google Scholar] [CrossRef]
- Landolin, J.M.; Johnson, D.S.; Trinklein, N.D.; Aldred, S.F.; Medina, C.; Shulha, H.; Weng, Z.; Myers, R.M. Sequence features that drive human promoter function and tissue specificity. Genome Res. 2010, 20, 890–898. [Google Scholar] [CrossRef]
- Hemberg, M.; Gray, J.M.; Cloonan, N.; Kuersten, S.; Grimmond, S.; Greenberg, M.E.; Kreiman, G. Integrated genome analysis suggests that most conserved non-coding sequences are regulatory factor binding sites. Nucleic Acids Res. 2012, 40, 7858–7869. [Google Scholar] [CrossRef]
- Wunderlich, Z.; Mirny, L.A. Different gene regulation strategies revealed by analysis of binding motifs. Trends Genet. 2009, 25, 434–440. [Google Scholar] [CrossRef]
- Farnham, P.J. Insights from genomic profiling of transcription factors. Nat. Rev. Genet. 2009, 10, 605–616. [Google Scholar] [CrossRef]
- Deplancke, B.; Alpern, D.; Gardeux, V. The Genetics of Transcription Factor DNA Binding Variation. Cell 2016, 166, 538–554. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Wilson, M.D.; Ballester, B.; Schwalie, P.C.; Brown, G.D.; Marshall, A.; Kutter, C.; Watt, S.; Martinez-Jimenez, C.P.; Mackay, S.; et al. Five-Vertebrate ChIP-seq Reveals the Evolutionary Dynamics of Transcription Factor Binding. Science 2010, 328, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Weirauch, M.T.; Hughes, T.R. Conserved expression without conserved regulatory sequence: The more things change, the more they stay the same. Trends Genet. 2010, 26, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Leong, B.; Vega, V.B.; Chen, X.; Lee, Y.L.; Srinivasan, K.G.; Chew, J.L.; Ruan, Y.; Wei, C.L.; Ng, H.H.; et al. Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 2008, 18, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Venkataram, S.; Fay, J.C. Is Transcription Factor Binding Site Turnover a Sufficient Explanation for Cis-Regulatory Sequence Divergence? Genome Biol. Evol. 2010, 2, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Guigo, R.; Messeguer, X. Multiple non-collinear TF-map alignments of promoter regions. BMC Bioinform. 2007, 8, 138. [Google Scholar] [CrossRef]
- Bais, A.; Grossmann, S.; Vingron, M. Incorporating evolution of transcription factor binding sites into annotated alignments. J. Biosci. 2007, 32, 841–850. [Google Scholar] [CrossRef]
- Hallikas, O.; Palin, K.; Sinjushina, N.; Rautiainen, R.; Partanen, J.; Ukkonen, E.; Taipale, J. Genome-wide Prediction of Mammalian Enhancers Based on Analysis of Transcription-Factor Binding Affinity. Cell 2006, 124, 47–59. [Google Scholar] [CrossRef]
- Parker, S.C.J.; Hansen, L.; Abaan, H.O.; Tullius, T.D.; Margulies, E.H. Local DNA Topography Correlates with Functional Noncoding Regions of the Human Genome. Science 2009, 324, 389–392. [Google Scholar] [CrossRef]
- Abeel, T.; Saeys, Y.; Bonnet, E.; Rouzé, P.; Van de Peer, Y. Generic eukaryotic core promoter prediction using structural features of DNA. Genome Res. 2008, 18, 310–323. [Google Scholar] [CrossRef]
- Reese, M.G. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem. 2001, 26, 51–56. [Google Scholar] [CrossRef]
- Demeler, B.; Zhou, G. Neural network optimization for E. coli promoter prediction. Nucleic Acids Res. 1991, 19, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Tayara, H.; Tahir, M.; Chong, K.T. Identification of prokaryotic promoters and their strength by integrating heterogeneous features. Genomics 2019. [Google Scholar] [CrossRef]
- de Avila e Silva, S.; Echeverrigaray, S.; Gerhardt, G.J. BacPP: Bacterial promoter prediction—A tool for accurate sigma-factor specific assignment in enterobacteria. J. Theor. Biol. 2011, 287, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Calistri, E.; Livi, R.; Buiatti, M. Evolutionary trends of GC/AT distribution patterns in promoters. Mol. Phylogenetics Evol. 2011, 60, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Sandelin, A.; Carninci, P.; Lenhard, B.; Ponjavic, J.; Hayashizaki, Y.; Hume, D.A. Mammalian RNA polymerase II core promoters: Insights from genome-wide studies. Nat. Rev. Genet. 2007, 8, 424–436. [Google Scholar] [CrossRef]
- Lenhard, B.; Sandelin, A.; Carninci, P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat. Rev. Genet. 2012, 13, 233–245. [Google Scholar] [CrossRef]
- Pettinato, L.; Calistri, E.; Di Patti, F.; Livi, R.; Luccioli, S. Genome-Wide Analysis of Promoters: Clustering by Alignment and Analysis of Regular Patterns. PLoS ONE 2014, 9, e85260. [Google Scholar] [CrossRef]
- van Hijum, S.A.F.T.; Medema, M.H.; Kuipers, O.P. Mechanisms and Evolution of Control Logic in Prokaryotic Transcriptional Regulation. Microbiol. Mol. Biol. Rev. 2009, 73, 481–509. [Google Scholar] [CrossRef]
- Lässig, M. From biophysics to evolutionary genetics: Statistical aspects of gene regulation. BMC Bioinform. 2007, 8, S7. [Google Scholar] [CrossRef]
- Galardini, M.; Brilli, M.; Spini, G.; Rossi, M.; Roncaglia, B.; Bani, A.; Chiancianesi, M.; Moretto, M.; Engelen, K.; Bacci, G.; et al. Evolution of Intra-specific Regulatory Networks in a Multipartite Bacterial Genome. PLoS Comput. Biol. 2015, 11, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Lipman, D.J.; Benson, D.A.; Karsch-Mizrachi, I.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2010, 39, D32–D37. [Google Scholar] [CrossRef]
- Zhou, C.; Mao, F.; Zhang, H.; Yang, J.; Ma, Q.; Lai, W.; Chen, X.; Mao, X.; Xu, Y. DOOR 2.0: Presenting operons and their functions through dynamic and integrated views. Nucleic Acids Res. 2013, 42, D654–D659. [Google Scholar] [CrossRef]
- Mao, F.; Chou, J.; Dam, P.; Olman, V.; Xu, Y. DOOR: A database for prokaryotic operons. Nucleic Acids Res. 2008, 37, D459–D463. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.; Dam, P.; Olman, V.; Xu, Y.; Su, Z. Operon prediction using both genome-specific and general genomic information. Nucleic Acids Res. 2006, 35, 288–298. [Google Scholar] [CrossRef]
- Omotajo, D.; Tate, T.; Cho, H.; Choudhary, M. Distribution and diversity of ribosome binding sites in prokaryotic genomes. BMC Genom. 2015, 16, 604. [Google Scholar] [CrossRef]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of Common Molecular Subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- von Luxburg, U. A tutorial on spectral clustering. Stat. Comput. 2007, 17, 395–416. [Google Scholar] [CrossRef]
- Stewart, G.; Sun, J. Matrix Perturbation Theory; Academic Press: New York, NY, USA, 1990. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Buchler, N.E.; Gerland, U.; Hwa, T. On schemes of combinatorial transcription logic. Proc. Natl. Acad. Sci. USA 2003, 100, 5136–5141. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Liu, J.S. De novo cis-regulatory module elicitation for eukaryotic genomes. Proc. Natl. Acad. Sci. USA 2005, 102, 7079–7084. [Google Scholar] [CrossRef] [PubMed]
- Shine, J.; Dalgarno, L. Determinant of cistron specificity in bacterial ribosomes. Nature 1975, 254, 34–38. [Google Scholar] [CrossRef]
- Sela, I.; Lukatsky, D.B. DNA Sequence Correlations Shape Nonspecific Transcription Factor-DNA Binding Affinity. Biophys. J. 2011, 101, 160–166. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]













| C0 | 930 | 1543 | 32 | 53.6 | 21.4 | −1.01 | 82 | 341.9 | 238.8 | −1.09 |
| C1 | 812 | 1451 | 62 | 43.3 | 17.6 | 1.06 | 707 | 269.4 | 194.0 | 2.26 |
| C2 | 811 | 1325 | 59 | 42.7 | 17.8 | 0.92 | 261 | 263.8 | 197.2 | −0.01 |
| C0 | 930 | 1543 | 338 | 267.7 | 35.4 | 1.99 | 1022 | 665.5 | 123.2 | 2.89 |
| C1 | 812 | 1451 | 146 | 214.4 | 35.4 | −1.93 | 326 | 503.8 | 110.5 | −1.61 |
| C2 | 811 | 1325 | 179 | 213.8 | 35.2 | −0.99 | 447 | 497.1 | 111.1 | −0.45 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenzini, L.; Di Patti, F.; Livi, R.; Fondi, M.; Fani, R.; Mengoni, A. A Method for the Structure-Based, Genome-Wide Analysis of Bacterial Intergenic Sequences Identifies Shared Compositional and Functional Features. Genes 2019, 10, 834. https://doi.org/10.3390/genes10100834
Lenzini L, Di Patti F, Livi R, Fondi M, Fani R, Mengoni A. A Method for the Structure-Based, Genome-Wide Analysis of Bacterial Intergenic Sequences Identifies Shared Compositional and Functional Features. Genes. 2019; 10(10):834. https://doi.org/10.3390/genes10100834
Chicago/Turabian StyleLenzini, Leonardo, Francesca Di Patti, Roberto Livi, Marco Fondi, Renato Fani, and Alessio Mengoni. 2019. "A Method for the Structure-Based, Genome-Wide Analysis of Bacterial Intergenic Sequences Identifies Shared Compositional and Functional Features" Genes 10, no. 10: 834. https://doi.org/10.3390/genes10100834
APA StyleLenzini, L., Di Patti, F., Livi, R., Fondi, M., Fani, R., & Mengoni, A. (2019). A Method for the Structure-Based, Genome-Wide Analysis of Bacterial Intergenic Sequences Identifies Shared Compositional and Functional Features. Genes, 10(10), 834. https://doi.org/10.3390/genes10100834