A TAC3 Missense Variant in a Domestic Shorthair Cat with Testicular Hypoplasia and Persistent Primary Dentition


1
Institute of Genetics, Vetsuisse Faculty, University of Bern, 3001 Bern, Switzerland
2
Tierarztpraxis Spiegelberg AG, 4566 Halten, Switzerland
*
Author to whom correspondence should be addressed.
Genes 2019, 10(10), 806; https://doi.org/10.3390/genes10100806
Submission received: 18 September 2019
/
Revised: 10 October 2019
/
Accepted: 12 October 2019
/
Published: 14 October 2019
(This article belongs to the Special Issue Genomics of Sexual Development and Reproduction in Mammals)
Abstract
:A single male domestic shorthair cat that did not complete puberty was reported. At four years of age, it still had primary dentition, testicular hypoplasia, and was relatively small for its age. We hypothesized that the phenotype might have been due to an inherited form of hypogonadotropic hypogonadism (HH). We sequenced the genome of the affected cat and compared the data to 38 genomes from control cats. A search for private variants in 40 candidate genes associated with human HH revealed a single protein-changing variant in the affected cat. It was located in the TAC3 gene encoding tachykinin 3, a precursor protein of the signaling molecule neurokinin B, which is known to play a role in sexual development. TAC3 variants have been reported in human patients with HH. The identified feline variant, TAC3:c.220G>A or p.(Val74Met), affects a moderately conserved region of the precursor protein, 11 residues away from the mature neurokinin B sequence. The affected cat was homozygous for the mutant allele. In a cohort of 171 randomly sampled cats, 169 were homozygous for the wildtype allele and 2 were heterozygous. These data tentatively suggest that the identified TAC3 variant might have caused the suppression of puberty in the affected cat.
1. Introduction
Gonadotropin releasing hormone (GnRH) is a main regulator of the reproductive endocrine system. Its secretion determines the pattern of secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). LH activates the release of testosterone in Leydig cells in the testicle. Testosterone is responsible for the male phenotype, body growth, and sperm production [1]. Coordinated and pulsatile GnRH secretion is induced due to the synergistic action of neurokinin B, kisspeptin, and dynorphin [2]. Neurokinin B stimulates kisspeptin neurons, which leads to GnRH secretion [3]. Dysfunctional GnRH release leads to low blood testosterone and pituitary hormone levels, resulting in hypogonadotropic hypogonadism (HH) [4]. Impaired testicular function may occur due to a primary testicular disorder or secondary to hypothalamic–pituitary dysfunction (hypogonadotropic). HH may be inherited or caused by non-genetic factors, such as drugs, encephalic trauma, or infiltrative or inflammatory pituitary lesions. Congenital hypogonadotropic hypogonadism (cHH) is a rare genetic disorder characterized by a delayed or absent pubertal development, micropenis, cryptorchidism, androgen/estrogen deficiency, and infertility due to an inadequate secretion of gonadotrophin-releasing hormone (GnRH), with an otherwise structurally and functionally normal hypothalamus and pituitary gland. Low testosterone and pituitary hormone levels confirm the diagnosis [4]. Variants in 40 genes have been shown to cause different inherited forms of HH in humans (Table 1).
This study was initiated after an owner reported a male cat with missing puberty characteristics, such as normal testicle growth and change in primary dentition. The goal of this study was to identify a possible underlying causative genetic defect.
2. Materials and Methods
2.1. Ethics Statement
All animal experiments were performed according to local regulations. The cat in this study is privately owned and was examined with the consent of the owner. The “Cantonal Committee for Animal Experiments” approved the collection of blood samples (Canton of Bern; permit 75/16).
2.2. Animal Selection
A 3-year-old male domestic shorthair cat with testicular hypoplasia and a persistent primary dentition was investigated. An EDTA blood sample was collected for genomic DNA isolation. Additionally, we used 171 blood samples from cats of various breeds which had been donated to the Vetsuisse Biobank (Table S1). These samples represented population controls without reports of testicular hypoplasia or a persistent primary dentition.
2.3. Hormone Measurement
The testosterone level of the affected cat was measured from an EDTA blood sample by labor-zentral.ch (Geuensee, Switzerland).
2.4. DNA Extraction
Genomic DNA was isolated from EDTA blood with the Maxwell RSC Whole Blood Kit using a Maxwell RSC instrument (Promega). Additionally we used DNA from EDTA blood of 171 non-affected control cats of various breeds that had been stored in our biobank.
2.5. Whole Genome Sequencing
An Illumina TruSeq PCR-free DNA library with 350 bp insert size of the affected cat (K532) was prepared. We collected 168 million 2 × 150 bp paired-end reads on a NovaSeq 6000 instrument (18x coverage). Mapping and alignment were performed as described [35]. The sequence data were deposited under the study accession PRJEB7401 and the sample accession SAMEA5885924 at the European Nucleotide Archive.
2.6. Variant Calling
Variant calling and filtering was performed as described [35]. To predict the functional effects of the called variants, SnpEFF [36] software, together with NCBI annotation release 104 for the Felis_catus_9.0 assembly, was used. For variant filtering we used 38 control genomes, which were produced during other projects of our group (Table S2).
2.7. Gene Analysis
We used the Felis_catus_9.0 cat reference genome assembly for all analyses. Numbering within the feline TAC3 gene corresponded to the NCBI RefSeq accessions XM_003988924.5 (mRNA) and XP_003988973.1 (protein).
2.8. Sanger Sequencing
The TAC3:c.220G>A variant was genotyped by direct Sanger sequencing of PCR amplicons. A 400 bp PCR product was amplified from genomic DNA using AmpliTaqGold360Mastermix together with primers 5‘-AGC CCA CTT CTC TTC CAG TG -3‘ (Primer F) and 5’-AGA GGG GAT TCA GGT CAC AA-3’ (Primer R). After treatment with exonuclease I and alkaline phosphatase, amplicons were sequenced on an ABI 3730 DNA Analyzer. Sanger sequences were analyzed using Sequencher 5.1 software.
3. Results
3.1. Clinical Examination
A 3-year-old male domestic shorthair cat was presented with persistent primary dentition consisting of one primary maxillary canine (Figure 1). Upon examination there was one small right testicle located in the scrotum. The left testicle could not be located. It was neither scrotal, nor palpable in the inguinal area. The external genitalia, including the urethral orifice, were in the normal position, although with a juvenile appearance because of their small size. The hair coat had an unkempt appearance. The cat had small body size but proportional growth. It had reportedly displayed mounting behavior toward another female cat in the household and showed an increasingly dominant–aggressive behavior toward other cats outside. The cat was presented for a follow-up examination at 4 years of age. No changes in behavior or the stage of adolescence were noticed.
3.2. Laboratory Findings
The blood testosterone level of the cat at the age of 4 years was 0.28 ng/mL, which was below the reference range for cats (0.92 ng/mL–9.17 ng/mL).
3.3. Genetic Analysis
We sequenced the genome of the affected cat and searched for homozygous and heterozygous variants in known candidate genes that were not present in 38 control cats of different breeds (Table 2).
This analysis identified a single homozygous private protein-changing variant in TAC3, a known HH candidate gene [32]. The variant was designated as ChrB4:85,517,451C>T (Felis_catus_9.0 assembly). It is a missense variant, XM_003988924.5:c.220G>A, predicted to result in the amino acid change XP_003988973.1:p.(Val74Met). This amino acid residue is moderately conserved across mammals. Primates, including humans, have a threonine at this position, while all other investigated mammalian TAC3 sequences have a valine (Figure 2). In silico predictions of the functional effect yielded conflicting results. PolyPhen-2 predicted the variant as benign with a score of 0.147 [37]. SIFT predicted that the variant affected protein function, with a score of 0.03 [38]. The SIFT prediction had low confidence due to the limited diversity of the available related sequences.
We confirmed the presence of the TAC3 variant by Sanger sequencing and genotyped 171 control cats (Figure 3). The case was homozygous for the mutant allele. Two domestic shorthair cats without reports of testicular hypoplasia or persistent primary dentition carried the alternative allele in a heterozygous state (Table 3; Table S1).
4. Discussion
In this study, we identified a TAC3:c.220G>A (p.Val74Met) missense variant in a domestic shorthair cat with absent puberty characteristics, such as normal testicle growth and change in primary dentition. The phenotype closely resembled HH, which belongs to the larger group of disorders of sexual development (DSD). The predicted amino acid substitution affects a moderately conserved region of the TAC3 precursor protein, which is 11 residues away from the mature neurokinin B sequence. The affected cat was homozygous for the mutant allele, which was found to be rare in the normal cat population. It should also be noted that the variant is close to the exon/intron boundary and might have an effect on splicing. Due to a lack of suitable RNA samples, we could not experimentally assess the TAC3 mRNA splicing pattern in the affected cat.
Cases of human HH have been reported to be caused by variants in either the TACR3 gene encoding the neurokinin B receptor or the TAC3 gene itself [32,39,40,41,42,43]. The phenotype of these patients is termed normosmic congenital hypogonadotropic hypogonadism (ncHH; OMIM #614839). ncHH is clinically characterized by a failure to enter puberty. Female patients have amenorrhea, absent breast development, and hypoplastic ovaries and uteri. Male patients have small testicles and micropenis. Body hair growth in ncHH patients resembles a juvenile pre-puberty state. ncHH in humans can be successfully treated by pulsatile administration of GnRH [42] or steroid hormones [43]. The known human pathogenic TAC3 variants consist of frameshift [43] and splice site [41,42] variants and a single missense variant, p.M90T, which affects the C-terminal residue of the mature neurokinin B peptide [32]. Additional likely pathogenic missense variants affecting the mature neurokinin B sequence were deposited in the ClinVar database.
In felines, efforts are underway to develop a method to permanently sterilize cats by RNAi-mediated silencing of KISS1 and TAC3. This method is predicted to lead to a reduction in the stray animal population and therefore decrease animal suffering and vectors for human disease [44].
5. Conclusions
Our genetic data and existing knowledge regarding the physiological function of TAC3 suggest that the TAC3:c.220G>A (p.Val74Met) missense variant may be considered a candidate causative variant for the observed HH phenotype in the studied cat. Given that this is a single case investigation and that we have no functional confirmation of neurokinin B deficiency, this result must be considered preliminary and should be interpreted with caution.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4425/10/10/806/s1, Table S1: TAC3:c.220G>A genotypes of 172 cats from 13 different cat breeds; Table S2: Accession numbers of 39 cat genome sequences.
Author Contributions
Conceptualization, T.L.; methodology, V.J.; investigation, P.H., P.K., V.J., and T.L.; resources, P.K.; data curation, V.J.; writing—original draft preparation, P.H. and T.L.; writing—review and editing, P.H., P.K., V.J., and T.L.; supervision, T.L.
Funding
This research received no external funding.
Acknowledgments
The authors are grateful to the cat owner who donated samples and participated in the study. We thank Eva Andrist, Nathalie Besuchet Schmutz, Sabrina Schenk, and Daniela Steiner for their expert technical assistance, the Next Generation Sequencing Platform of the University of Bern for performing the high-throughput sequencing experiments, and the Interfaculty Bioinformatics Unit of the University of Bern for providing high performance computing infrastructure.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Marques, P.; Skorupskaite, K.; George, J.T.; Anderson, R.A. Physiology of GNRH and Gonadotropin Secretion. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., et al., Eds.; South Dartmouth (MA): MDText.com. Available online: https://www.ncbi.nlm.nih.gov/books/NBK279070/ (accessed on 27 August 2019).
- Ikegami, K.; Minabe, S.; Ieda, N.; Goto, T.; Sugimoto, A.; Nakamura, S.; Inoue, N.; Oishi, S.; Maturana, A.D.; Sanbo, M.; et al. Evidence of involvement of neurone-glia/neurone-neurone communications via gap junctions in synchronised activity of KNDy neurones. J. Neuroendocrinol. 2017, 29. [Google Scholar] [CrossRef]
- Skorupskaite, K.; George, J.T.; Anderson, R.A. The kisspeptin-GnRH pathway in human reproductive health and disease. Hum. Reprod. Update 2014, 20, 485–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraietta, R.; Zylbergstejn, D.S.; Eseves, S.C. Hypogonadotropic Hypogonadism Revisited. Clinics 2013, 68, 81–88. [Google Scholar] [CrossRef]
- Kim, H.-G.; Kurth, I.; Lan, F.; Meliciani, I.; Wenzel, W.; Eom, S.H.; Kang, G.B.; Rosenberger, G.; Tekin, M.; Ozata, M.; et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet 2008, 83, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Achermann, J.C.; Gu, W.X.; Kotlar, T.J.; Meeks, J.J.; Sabacan, L.P.; Seminara, S.B.; Habiby, R.L.; Hindmarsh, P.C.; Bick, D.P.; Sherins, R.J.; et al. Mutational analysis of DAX1 in patients with hypogonadotropic hypogonadism or pubertal delay. J. Clin. Endocr. Metab. 1999, 84, 4497–4500. [Google Scholar] [CrossRef] [PubMed]
- Miraoui, H.; Dwyer, A.A.; Sykiotis, G.P.; Plummer, L.; Chung, W.; Feng, B.; Beenken, A.; Clarke, J.; Pers, T.H.; Dworzynski, P.; et al. Mutations in FGF17, IL17RD, DUPS6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am. J. Hum. Genet. 2013, 92, 725–743. [Google Scholar] [CrossRef] [PubMed]
- Kotan, L.D.; Hutchins, B.I.; Ozkan, Y.; Demirel, F.; Stoner, H.; Cheng, P.J.; Esen, I.; Gurbuz, F.; Bicakci, Y.K.; Mengen, E.; et al. Mutations in FEZF1 cause Kallmann syndrome. Am. J. Hum. Genet. 2014, 95, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Falardeau, J.; Chung, W.C.J.; Beenken, A.; Raivio, T.; Plummer, L.; Sidis, Y.; Jacobson-Dickman, E.E.; Eliseenkova, A.V.; Ma, J.; Dwyer, A.; et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J. Clin. Investig. 2008, 118, 2822–2831. [Google Scholar] [CrossRef] [Green Version]
- Dode, C.; Levilliers, J.; Dupont, J.M.; De Paepe, A.; Le Du, N.; Soussi-Yanicostas, N.; Coimbra, R.S.; Delmaghani, S.; Compain-Nouaille, S.; Baverel, F. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003, 33, 463–465. [Google Scholar] [CrossRef] [Green Version]
- Layman, L.C.; Lee, E.J.; Peak, D.B.; Namnoum, A.B.; Vu, K.V.; van Lingen, B.L.; Gray, M.R.; McDonough, P.G.; Reindollar, R.H.; Jameson, J.L. Delayed puberty and hypogonadism caused by mutations in the follicle-stimulating hormone beta-subunit gene. N. Eng. J. Med. 1997, 337, 607–611. [Google Scholar] [CrossRef]
- Bouligand, J.; Ghervan, C.; Tello, J.A.; Brailly-Tabard, S.; Salenave, S.; Chanson, P.; Lombes, M.; Millar, R.P.; Guiochon-Mantel, A.; Young, J. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N. Eng. J. Med. 2009, 360, 2742–2748. [Google Scholar] [CrossRef] [PubMed]
- Pralong, F.P.; Gomez, F.; Castillo, E.; Cotecchia, S.; Abuin, L.; Aubert, M.L.; Portmann, L.; Gaillard, R.C. Complete hypogonadotropic hypogonadism associated with a novel inactivating mutation of the gonadotropin-releasing hormone receptor. J. Clin. Endocr. Metab. 1999, 84, 3811–3816. [Google Scholar] [CrossRef] [PubMed]
- Tajima, T.; Hattorri, T.; Nakajima, T.; Okuhara, K.; Sato, K.; Abe, S.; Nakae, J.; Fujieda, K. Sporadic heterozygous frameshift mutation of HESX1 causing pituitary and optic nerve hypoplasia and combined pituitary hormone deficiency in a Japanese patient. J. Clin. Endocr. Metab. 2008, 88, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Bick, D.; Franco, B.; Sherins, R.J.; Heye, B.; Pike, L.; Crawford, J.; Maddalena, A.; Incerti, B.; Pragliola, A.; Meitinger, T.; et al. Brief report: intragenic deletion of the KALIG-1 gene in Kallmann’s syndrome. N. Engl. J. Med. 1992, 326, 1752–1755. [Google Scholar] [CrossRef] [PubMed]
- Topaloglu, A.K.; Tello, J.A.; Kotan, L.D.; Ozbek, M.N.; Yilmaz, M.B.; Erdogan, S.; Gurbuz, F.; Temiz, F.; Millar, R.P.; Yuksel, B. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. N. Eng. J. Med. 2012, 366, 629–635. [Google Scholar] [CrossRef]
- Brioude, F.; Bouligand, J.; Francou, B.; Fagart, J.; Roussel, R.; Viengchareun, S.; Combettes, L.; Brailly-Tabard, S.; Lombes, M.; Young, J.; et al. Two families with normosmic congenital hypogonadotropic hypogonadism and biallelic mutations in KISS1R (KISS1 receptor): clinical evaluation and molecular characterization of a novel mutation. PLoS ONE 2013, 8, e53896. [Google Scholar] [CrossRef]
- Licinio, J.; Caglayan, S.; Ozata, M.; Yildiz, B.O.; de Miranda, P.B.; O’Kirwan, F.; Whitby, R.; Liang, L.; Cohen, P.; Bhasin, S.; et al. Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behavior in leptin-deficient adults. Proc. Natl. Acad. Sci. USA 2004, 101, 4531–4536. [Google Scholar] [CrossRef] [Green Version]
- Basciani, S.; Watanabe, M.; Mariani, S.; Passeri, M.; Persichetti, A.; Fiore, D.; Scotto d’Abusco, A.; Caprio, M.; Lenzi, A.; Fabbri, A.; et al. Hypogonadism in a patient with two novel mutations of the luteinizing hormone beta-subunit gene expressed in a compound heterozygous form. J. Clin. Endocr. Metab. 2012, 97, 3031–3038. [Google Scholar] [CrossRef]
- Rajab, A.; Kelberman, D.; de Castro, S.C.P.; Biebermann, H.; Shaikh, H.; Pearce, K.; Hall, C.M.; Shaikh, G.; Gerrelli, D.; Grueters, A. Novel mutations in LHX3 are associated with hypopituitarism and sensorineural hearing loss. Hum. Mol. Genet. 2008, 17, 2150–2159. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Acierno, J.S.; Seminara, S.B. Characterization of the human nasal embryonic LHRH factor gene, NELF, and a mutation screening among 65 patients with idiopathic hypogonadotropic hypogonadism (IHH). J. Hum. Genet. 2004, 49, 265–268. [Google Scholar] [CrossRef] [Green Version]
- Margolin, D.H.; Kousi, M.; Chan, Y.M.; Lim, E.T.; Schmahmann, J.D.; Hadjivassiliou, M.; Hall, J.E.; Adam, I.; Dwyer, A.; Plummer, L. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N. Eng. J. Med. 2013, 368, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Gonzalez, M.A.; Lourenco, C.M.; Coutelier, M.; Haack, T.B.; Rebelo, A.; Hannequin, D.; Strom, T.M.; Prokisch, H.; Kernstock, C.; et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 2014, 137, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Osaka, H.; Sasaki, M.; Takanashi, J.; Hamada, K.; Yamashita, A.; Shibayama, H.; Shiina, M.; Kondo, Y.; Nishiyama, K.; et al. Mutations in POLR3A and POLR3B encoding RNA polymerase III subunits cause an autosomal-recessive hypomyelinating leukoencephalopathy. Am. J. Hum. Genet. 2011, 89, 644–651. [Google Scholar] [CrossRef]
- Dode, C.; Teixeira, L.; Levilliers, J.; Fouveaut, C.; Bouchard, P.; Kottler, M.L.; Lespinasse, J.; Lienhardt-Roussie, A.; Mathieu, M.; Moerman, A. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006, 2, e175. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, G.; Bhatia, V.; Cook, S.; Thomas, P.Q. Adrenocorticotropin deficiency in combined pituitary hormone deficiency patients homozygous for a novel PROP1 deletion. J. Clin. Endocr. Metab. 2000, 85, 4556–4561. [Google Scholar] [CrossRef] [PubMed]
- Handley, M.T.; Morris-Rosendahl, D.J.; Brown, S.; Macdonald, F.; Hardy, C.; Bem, D.; Carpanini, S.M.; Borck, G.; Martorell, L.; Izzi, C.; et al. Mutation spectrum in RAB3GAP1, RAB3GAP2, and RAB18 and genotype-phenotype correlations in Warburg Micro syndrome and Martsolf syndrome. Hum. Mutat. 2013, 34, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Seminara, S.B.; Acierno, J.S.; Abdulwahid, N.A.; Crowley, W.F.; Margolin, D.H. Hypogonadotropic hypogonadism and cerebellar ataxia: detailed phenotypic characterization of a large, extended kindred. J. Clin. Endocr. Metab. 2002, 87, 1607–1612. [Google Scholar] [CrossRef]
- Young, J.; Metay, C.; Bouligand, J.; Tou, B.; Francou, B.; Maione, L.; Tosca, L.; Sarfati, J.; Brioude, F.; Esteva, B.; et al. SEMA3A deletion in a family with Kallmann syndrome validates the role of semaphorin 3A in human puberty and olfactory system development. Hum. Reprod. 2012, 27, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Kelberman, D.; Rizzoti, K.; Avilion, A.; Bitner-Glindicz, M.; Cianfarani, S.; Collins, J.; Chong, W.K.; Kirk, J.M.W.; Achermann, J.C.; Ross, R. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J. Clin. Investig. 2006, 116, 2442–2455. [Google Scholar] [CrossRef]
- Shi, C.H.; Schisler, J.C.; Rubel, C.E.; Tan, S.; Song, B.; McDonough, H.; Xu, L.; Portbury, A.L.; Mao, C.Y.; True, C.; et al. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum. Mol. Genet. 2014, 23, 1013–1024. [Google Scholar] [CrossRef]
- Topaloglu, A.K.; Reimann, F.; Guclu, M.; Yalin, A.S.; Kotan, L.D.; Porter, K.M.; Serin, A.; Mungan, N.O.; Cook, J.R.; Ozbek, M.N.; et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for neurokinin B in the central control of reproduction. Nature Genet. 2009, 41, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Liegel, R.P.; Handley, M.T.; Ronchetti, A.; Brown, S.; Langemeyer, L.; Linford, A.; Chang, B.; Morris-Rosendahl, D.J.; Carpanini, S.; Posmyk, R.; et al. Loss-of-function mutations in TBC1D20 cause cataracts and male infertility in blind sterile mice and Warburg Micro syndrome in humans. Am. J. Hum. Genet. 2013, 93, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Ahn, J.W.; Kurth, I.; Ullmann, R.; Kim, H.T.; Kulharya, A.; Ha, K.S.; Itokawa, Y.; Meliciani, I.; Wenzel, W.; et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 2010, 87, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Dog Biomedical Variant Database Consortium (DBVDC). A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and 8 wolves. Anim Genet. 2019. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 4, 248–249. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Fukami, M.; Maruyama, T.; Dateki, S.; Sato, N.; Yoshimura, Y.; Ogata, T. Hypothalamic dysfunction in a female with isolated hypogonadotropic hypogonadism and compound heterozygous TACR3 mutations and clinical manifestation in her heterozygous mother. Horm. Res. Paediatr. 2010, 73, 477–481. [Google Scholar] [CrossRef]
- Guran, T.; Tolhurst, G.; Bereket, A.; Rocha, N.; Porter, K.; Turan, S.; Gribble, F.M.; Kotan, L.D.; Akcay, T.; Atay, Z. Hypogonadotropic hypogonadism due to a novel missense mutation in the first extracellular loop of the neurokinin B receptor. J. Clin. Endocrinol. Metab. 2009, 94, 3633–3639. [Google Scholar] [CrossRef]
- Francou, B.; Bouligand, J.; Voican, A.; Amazit, L.; Trabado, S.; Fagart, J.; Meduri, G.; Brailly-Tabard, S.; Chanson, P.; Lecomte, P. Normosmic congenital hypogonadotropic hypogonadism due to TAC3/TACR3 mutations: characterization of neuroendocrine phenotypes and novel mutations. PLoS ONE 2011, 10, e25614. [Google Scholar]
- Young, J.; Bouligand, J.; Francou, B.; Raffin-Sanson, M.L.; Gaillez, S.; Jeanpierre, M.; Grynberg, M.; Kamenicky, P.; Chanson, P.; Brailly-Tabard, S.; et al. TAC3 and TACR3 defects cause hypothalamic congenital hypogonadotropic hypogonadism in humans. J. Clin. Endocrinol. Metab. 2010, 95, 2287–2295. [Google Scholar] [CrossRef] [PubMed]
- Gianetti, E.; Tusset, C.; Noel, S.D.; Au, M.G.; Dwyer, A.A.; Hughes, V.A.; Abreu, A.P.; Carroll, J.; Trarbach, E.; Silveira, L.F.; et al. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J. Clin. Endocrinol. Metab. 2010, 95, 2857–2867. [Google Scholar] [CrossRef] [PubMed]
- Dissen, G.A.; Adachi, K.; Lomniczi, A.; Chatkupt, T.; Davidson, B.L.; Nakai, H.; Ojeda, S.R. Engineering a Gene Silencing Viral Construct that Targets the Cat Hypothalamus to Induce Permanent Sterility: An Update. Reprod. Domest. Anim. 2017, 52, 354–358. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Clinical phenotype characterized by persistent primary dentition and an “unkempt” coat appearance.
Figure 1.
Clinical phenotype characterized by persistent primary dentition and an “unkempt” coat appearance.

Figure 2.
Multi-species protein alignment of the TAC3 precursor protein in the region of the missense variant. The valine at position 74 of the feline TAC3 protein is conserved in most mammals, except primates. The 10 amino acid sequence of the mature neurokinin B signaling peptide is indicated in red. Amino acid sequences were derived from XP_003988973.1 (cat); NP_037383.1 (human); XP_005625588.1 (dog); NP_851360.1 (cattle); NP_033338.2 (mouse); NP_062035.1 (rat).
Figure 2.
Multi-species protein alignment of the TAC3 precursor protein in the region of the missense variant. The valine at position 74 of the feline TAC3 protein is conserved in most mammals, except primates. The 10 amino acid sequence of the mature neurokinin B signaling peptide is indicated in red. Amino acid sequences were derived from XP_003988973.1 (cat); NP_037383.1 (human); XP_005625588.1 (dog); NP_851360.1 (cattle); NP_033338.2 (mouse); NP_062035.1 (rat).
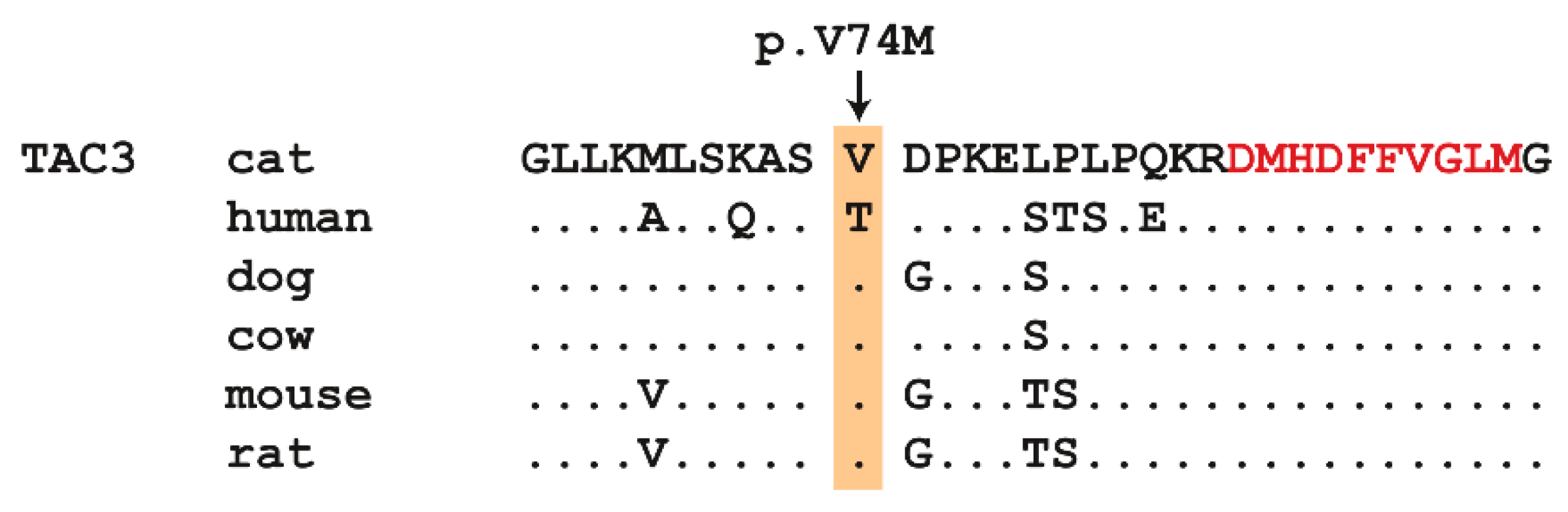
Figure 3.
Details of the TAC3:c.220G>A variant. Representative electropherograms of three cats with different genotypes are shown. Exonic bases are shown in capital letters and intronic bases in lowercase letters.
Figure 3.
Details of the TAC3:c.220G>A variant. Representative electropherograms of three cats with different genotypes are shown. Exonic bases are shown in capital letters and intronic bases in lowercase letters.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Overview on genetic causes of human HH.
| Gene | Phenotype | Inheritance | Ref. |
|---|---|---|---|
| CHD7 | Hypogonadotropic hypogonadism 5 with or without anosmia | AD | [5] |
| DAX1 | Adrenal hypoplasia, congenital | XLR | [6] |
| DUSP6 | Hypogonadotropic hypogonadism 19 with or without anosmia | AD | [7] |
| FEZF1 | Hypogonadotropic hypogonadism 22 with or without anosmia | AR | [8] |
| FGF8 | Hypogonadotropic hypogonadism 6 with or without anosmia | AD | [9] |
| FGF17 | Hypogonadotropic hypogonadism 20 with or without anosmia | AD | [7] |
| FGFR1 | Hypogonadotropic hypogonadism 2 with or without anosmia | AD | [10] |
| FLRT3 | Hypogonadotropic hypogonadism 21 with anosmia | AD | [7] |
| FSHB | Hypogonadotropic hypogonadism 24 without anosmia | AR | [11] |
| GNRH1 | Hypogonadotropic hypogonadism 12 with or without anosmia | AR | [12] |
| GNRHR | Hypogonadotropic hypogonadism 7 without anosmia | AR | [13] |
| HESX-1 | Growth hormone deficiency with pituitary anomalies | AR, AD | [14] |
| HS6ST1 | Hypogonadotropic hypogonadism 15 with or without anosmia | AD | [7] |
| IL17RD | Hypogonadotropic hypogonadism 18 with or without anosmia | AR, AD | [7] |
| KAL1 | Hypogonadotropic hypogonadism 1 with or without anosmia | XLR | [15] |
| KISS1 | Hypogonadotropic hypogonadism 13 with or without anosmia | AR | [16] |
| KISS1R | Hypogonadotropic hypogonadism 8 with or without anosmia | AR | [17] |
| LEP | Obesity, morbid, due to leptin deficiency | AR | [18] |
| LEPR | Obesity, morbid, due to leptin deficiency | AR | [18] |
| LHB | Hypogonadotropic hypogonadism 23 with or without anosmia | AR | [19] |
| LHX3 | Pituitary hormone deficiency, combined, 3 | AR | [20] |
| NELF | Hypogonadotropic hypogonadism 9 with or without anosmia | AD | [21] |
| OTUD4 | Gordon Holmes syndrome | AR | [22] |
| PNPLA6 | Boucher–Neuhauser Syndrome | AR | [23] |
| POLR3B | hypogonadotropic hypogonadism | AR | [24] |
| PROK2 | Hypogonadotropic hypogonadism 4 with or without anosmia | AD | [25] |
| PROKR2 | Hypogonadotropic hypogonadism 3 with or without anosmia | AD | [25] |
| PROP-1 | Pituitary hormone deficiency, combined, 2 | AR | [26] |
| RAB18 | Warburg micro syndrome 3 | AR | [27] |
| RAB3GAP1 | Warburg micro syndrome 1 | AR | [27] |
| RAB3GAP2 | Warburg micro syndrome 2 | AR | [27] |
| RNF216 | Cerebellar ataxia and hypogonadotropic hypogonadism | AR | [28] |
| SEMA3A | Hypogonadotropic hypogonadism 16 with or without anosmia | AD | [29] |
| SOX2 | Abnormalities of the central nervous system | AD | [30] |
| SPRY4 | Hypogonadotropic hypogonadism 17 with or without anosmia | AD | [7] |
| STUB1 | Spinocerebellar ataxia, autosomal recessive 16 | AR | [31] |
| TAC3 | Hypogonadotropic hypogonadism 10 with or without anosmia | AR | [32] |
| TACR3 | Hypogonadotropic hypogonadism 11 with or without anosmia | AR | [32] |
| TBC1D20 | Warburg micro syndrome 4 | AR | [33] |
| WDR11 | Hypogonadotropic hypogonadism 14 with or without anosmia | AD | [34] |
Table 2.
Results of variant filtering.
| Filtering Step | Homozygous Variants | Heterozygous Variant |
|---|---|---|
| Private variants | 25,355 | 209,967 |
| Protein-changing private variants | 111 | 756 |
| Private variants in known candidate genes | 1 | 0 |
Table 3.
Genotype phenotype association of the TAC3:c.220G>A variant.
| Cats | G/G | G/A | A/A |
|---|---|---|---|
| Cases (n = 1) | 0 | 0 | 1 |
| Control cats (n = 171) | 169 | 2 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hug, P.; Kern, P.; Jagannathan, V.; Leeb, T. A TAC3 Missense Variant in a Domestic Shorthair Cat with Testicular Hypoplasia and Persistent Primary Dentition. Genes 2019, 10, 806. https://doi.org/10.3390/genes10100806
AMA Style
Hug P, Kern P, Jagannathan V, Leeb T. A TAC3 Missense Variant in a Domestic Shorthair Cat with Testicular Hypoplasia and Persistent Primary Dentition. Genes. 2019; 10(10):806. https://doi.org/10.3390/genes10100806
Chicago/Turabian StyleHug, Petra, Patricia Kern, Vidhya Jagannathan, and Tosso Leeb. 2019. "A TAC3 Missense Variant in a Domestic Shorthair Cat with Testicular Hypoplasia and Persistent Primary Dentition" Genes 10, no. 10: 806. https://doi.org/10.3390/genes10100806
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.