Broken by the Cut: A Journey into the Role of Topoisomerase II in DNA Fragility


{kind=link}
{kind=link}
Abstract
1. Introduction
2. TOP2 Cleavage in Multiple Biological Processes:
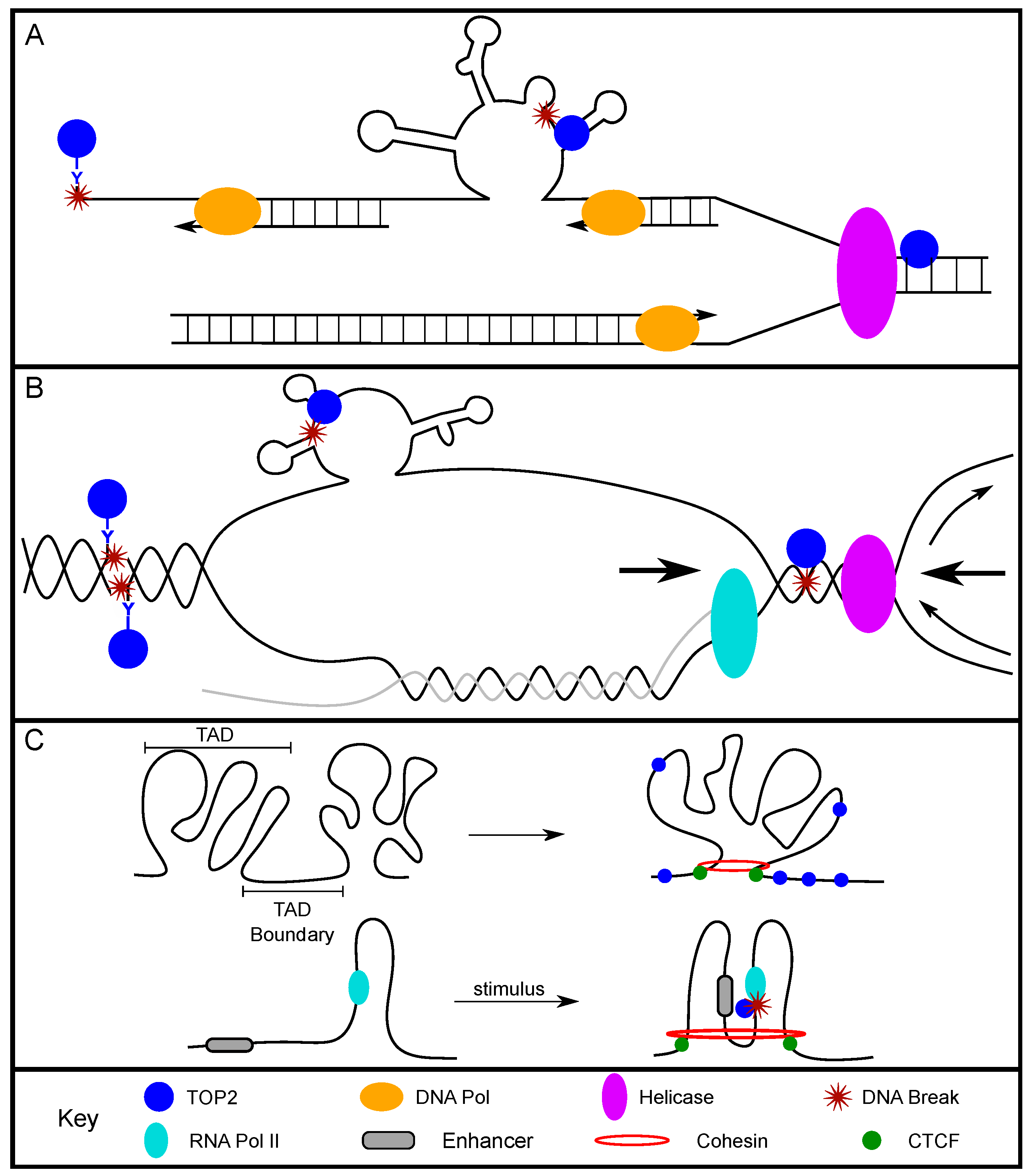
2.1. TOP2-Induced Breaks Contribute to Replication-Associated DNA Fragility
2.2. TOP2-Induced DNA Breaks and Transcription Activation
2.3. TOP2 and Chromatin Organization
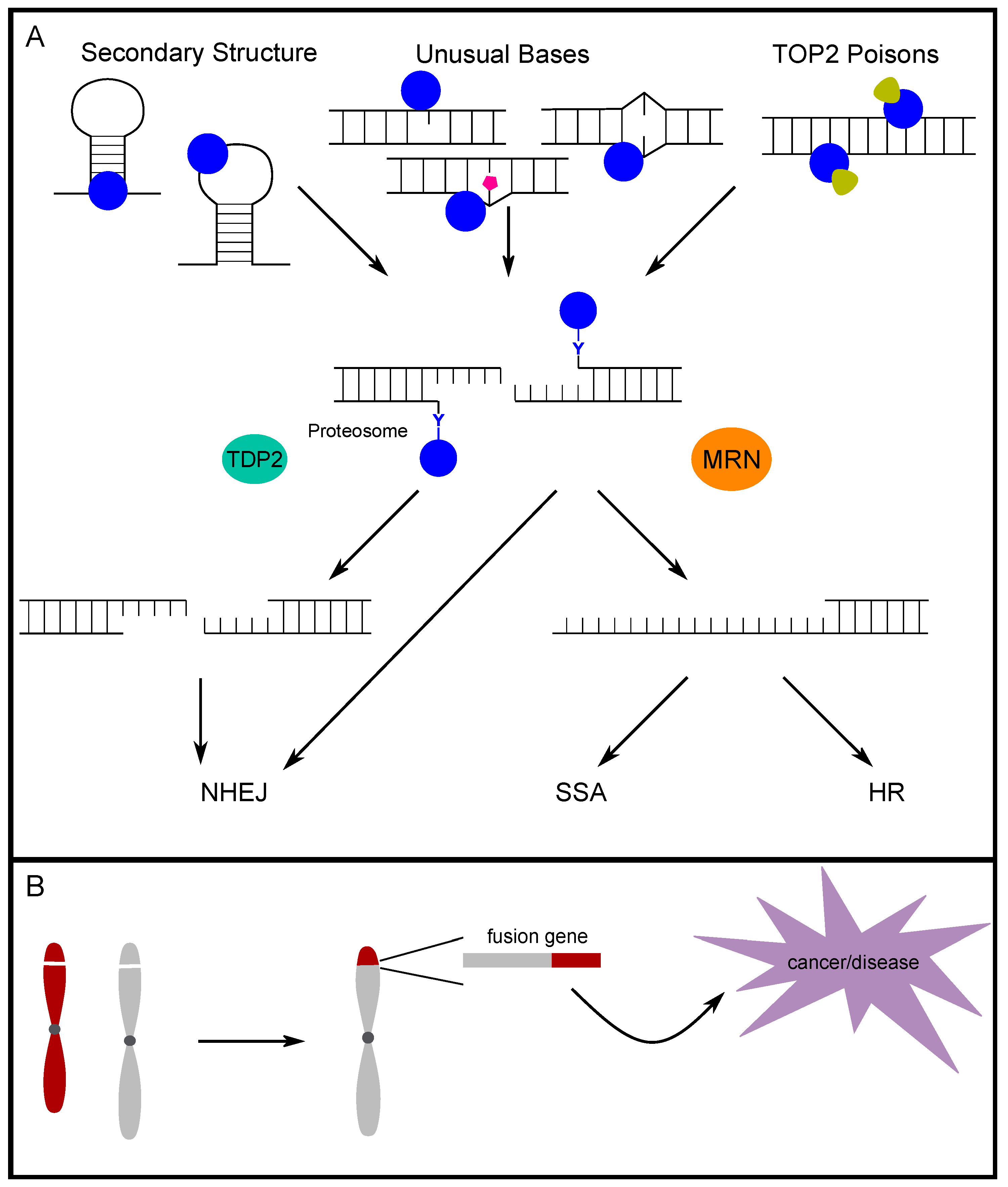
3. Alternative DNA Secondary Structures and Unusual Bases Promote TOP2-Induced DSBs
4. The Fate of TOP2-Induced Breaks
4.1. TOP2-Adduct Removal and End Processing
4.2. Determining End Structures and Repair Intermediates
4.3. Repair Pathway Choice for TOP2-Induced Breaks
5. The Role of TOP2-Induced Breaks in Disease
5.1. Therapy-Related Acute Myeloid Leukemia
5.2. Pediatric MLL-Rearranged AML
5.3. RET-Driven Papillary Thyroid Cancer
5.4. Other Diseases
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Roca, J. The mechanisms of DNA topoisomerases. Trends Biochem. Sci. 1995, 20, 156–160. [Google Scholar] [CrossRef]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Hoa, N.N.; Shimizu, T.; Zhou, Z.W.; Wang, Z.Q.; Deshpande, R.A.; Paull, T.T.; Akter, S.; Tsuda, M.; Furuta, R.; Tsutsui, K.; et al. Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol. Cell 2016, 64, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Drake, F.H.; Hofmann, G.A.; Mong, S.M.; Bartus, J.O.; Hertzberg, R.P.; Johnson, R.K.; Mattern, M.R.; Mirabelli, C.K. In vitro and intracellular inhibition of topoisomerase II by the antitumor agent merbarone. Cancer Res. 1989, 49, 2578–2583. [Google Scholar]
- Fortune, J.M.; Osheroff, N. Merbarone inhibits the catalytic activity of human topoisomerase IIalpha by blocking DNA cleavage. J. Biol. Chem. 1998, 273, 17643–17650. [Google Scholar] [CrossRef] [PubMed]
- Loike, J.D. VP16-213 and podophyllotoxin. A study on the relationship between chemical structure and biological activity. Cancer Chemother. Pharmacol. 1982, 7, 103–111. [Google Scholar]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef]
- Chen, H.; Eastmond, D.A. Topoisomerase inhibition by phenolic metabolites: A potential mechanism for benzene’s clastogenic effects. Carcinogenesis 1995, 16, 2301–2307. [Google Scholar] [CrossRef]
- Eastmond, D.A.; Mondrala, S.T.; Hasegawa, L. Topoisomerase II inhibition by myeloperoxidase-activated hydroquinone: A potential mechanism underlying the genotoxic and carcinogenic effects of benzene. Chem. Biol. Interact. 2005, 153, 207–216. [Google Scholar] [CrossRef]
- Frantz, C.E.; Chen, H.; Eastmond, D.A. Inhibition of human topoisomerase II in vitro by bioactive benzene metabolites. Environ. Health Perspect. 1996, 104, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Strick, R.; Strissel, P.L.; Borgers, S.; Smith, S.L.; Rowley, J.D. Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc. Natl. Acad. Sci. USA 2000, 97, 4790–4795. [Google Scholar] [CrossRef] [PubMed]
- Kingma, P.S.; Greider, C.A.; Osheroff, N. Spontaneous DNA lesions poison human topoisomerase IIalpha and stimulate cleavage proximal to leukemic 11q23 chromosomal breakpoints. Biochemistry 1997, 36, 5934–5939. [Google Scholar] [CrossRef] [PubMed]
- Cline, S.D.; Jones, W.R.; Stone, M.P.; Osheroff, N. DNA abasic lesions in a different light: Solution structure of an endogenous topoisomerase II poison. Biochemistry 1999, 38, 15500–15507. [Google Scholar] [CrossRef] [PubMed]
- Bigioni, M.; Zunino, F.; Tinelli, S.; Austin, C.A.; Willmore, E.; Capranico, G. Position-specific effects of base mismatch on mammalian topoisomerase II DNA cleaving activity. Biochemistry 1996, 35, 153–159. [Google Scholar] [CrossRef]
- Sabourin, M.; Osheroff, N. Sensitivity of human type II topoisomerases to DNA damage: Stimulation of enzyme-mediated DNA cleavage by abasic, oxidized and alkylated lesions. Nucleic Acids Res. 2000, 28, 1947–1954. [Google Scholar] [CrossRef]
- Long, B.H.; Stringfellow, D.A. Inhibitors of topoisomerase II: Structure-activity relationships and mechanism of action of podophyllin congeners. Adv. Enzyme Regul. 1988, 27, 223–256. [Google Scholar] [CrossRef]
- Gothe, H.J.; Bouwman, B.A.M.; Gusmao, E.G.; Piccinno, R.; Petrosino, G.; Sayols, S.; Drechsel, O.; Minneker, V.; Josipovic, N.; Mizi, A.; et al. Spatial Chromosome Folding and Active Transcription Drive DNA Fragility and Formation of Oncogenic MLL Translocations. Mol. Cell 2019, 75, 267–283. [Google Scholar] [CrossRef]
- Canela, A.; Maman, Y.; Huang, S.N.; Wutz, G.; Tang, W.; Zagnoli-Vieira, G.; Callen, E.; Wong, N.; Day, A.; Peters, J.M.; et al. Topoisomerase II-Induced Chromosome Breakage and Translocation Is Determined by Chromosome Architecture and Transcriptional Activity. Mol. Cell 2019, 75, 252–266. [Google Scholar] [CrossRef]
- Gaggioli, V.; Le Viet, B.; Germe, T.; Hyrien, O. DNA topoisomerase IIalpha controls replication origin cluster licensing and firing time in Xenopus egg extracts. Nucleic Acids Res. 2013, 41, 7313–7331. [Google Scholar] [CrossRef]
- Gonzalez, R.E.; Lim, C.U.; Cole, K.; Bianchini, C.H.; Schools, G.P.; Davis, B.E.; Wada, I.; Roninson, I.B.; Broude, E.V. Effects of conditional depletion of topoisomerase II on cell cycle progression in mammalian cells. Cell Cycle. 2011, 10, 3505–3514. [Google Scholar] [CrossRef] [PubMed]
- Abdurashidova, G.; Radulescu, S.; Sandoval, O.; Zahariev, S.; Danailov, M.B.; Demidovich, A.; Santamaria, L.; Biamonti, G.; Riva, S.; Falaschi, A. Functional interactions of DNA topoisomerases with a human replication origin. EMBO J. 2007, 26, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.G.; Baack, M.; Knippers, R. Proteins of the origin recognition complex (ORC) and DNA topoisomerases on mammalian chromatin. BMC Mol. Biol. 2009, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Catapano, C.V.; Carbone, G.M.; Pisani, F.; Qiu, J.; Fernandes, D.J. Arrest of replication fork progression at sites of topoisomerase II-mediated DNA cleavage in human leukemia CEM cells incubated with VM-26. Biochemistry 1997, 36, 5739–5748. [Google Scholar] [CrossRef] [PubMed]
- McClendon, A.K.; Rodriguez, A.C.; Osheroff, N. Human topoisomerase IIalpha rapidly relaxes positively supercoiled DNA: Implications for enzyme action ahead of replication forks. J. Biol. Chem. 2005, 280, 39337–39345. [Google Scholar] [CrossRef] [PubMed]
- Lucas, I.; Germe, T.; Chevrier-Miller, M.; Hyrien, O. Topoisomerase II can unlink replicating DNA by precatenane removal. EMBO J. 2001, 20, 6509–6519. [Google Scholar] [CrossRef] [PubMed]
- Sundin, O.; Varshavsky, A. Arrest of segregation leads to accumulation of highly intertwined catenated dimers: Dissection of the final stages of SV40 DNA replication. Cell 1981, 25, 659–669. [Google Scholar] [CrossRef]
- Fachinetti, D.; Bermejo, R.; Cocito, A.; Minardi, S.; Katou, Y.; Kanoh, Y.; Shirahige, K.; Azvolinsky, A.; Zakian, V.A.; Foiani, M. Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol. Cell 2010, 39, 595–605. [Google Scholar] [CrossRef]
- D’Arpa, P.; Beardmore, C.; Liu, L.F. Involvement of nucleic acid synthesis in cell killing mechanisms of topoisomerase poisons. Cancer Res. 1990, 50, 6919–6924. [Google Scholar]
- Holm, C.; Covey, J.M.; Kerrigan, D.; Pommier, Y. Differential requirement of DNA replication for the cytotoxicity of DNA topoisomerase I and II inhibitors in Chinese hamster DC3F cells. Cancer Res. 1989, 49, 6365–6368. [Google Scholar]
- Tammaro, M.; Barr, P.; Ricci, B.; Yan, H. Replication-dependent and transcription-dependent mechanisms of DNA double-strand break induction by the topoisomerase 2-targeting drug etoposide. PLoS ONE 2013, 8, e79202. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Jackson, S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.W.; Pierce, L.C.; Lehman, C.E.; Nikiforov, Y.E.; Wang, Y.H. DNA topoisomerases participate in fragility of the oncogene RET. PLoS ONE 2013, 8, e75741. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef]
- Gartenberg, M.R.; Wang, J.C. Positive supercoiling of DNA greatly diminishes mRNA synthesis in yeast. Proc. Natl. Acad. Sci. USA 1992, 89, 11461–11465. [Google Scholar] [CrossRef]
- Tabuchi, H.; Handa, H.; Hirose, S. Underwinding of DNA on binding of yeast TFIID to the TATA element. Biochem. Biophys. Res. Commun. 1993, 192, 1432–1438. [Google Scholar] [CrossRef]
- Sperling, A.S.; Jeong, K.S.; Kitada, T.; Grunstein, M. Topoisomerase II binds nucleosome-free DNA and acts redundantly with topoisomerase I to enhance recruitment of RNA Pol II in budding yeast. Proc. Natl. Acad. Sci. USA 2011, 108, 12693–12698. [Google Scholar] [CrossRef]
- Mizutani, M.; Ohta, T.; Watanabe, H.; Handa, H.; Hirose, S. Negative supercoiling of DNA facilitates an interaction between transcription factor IID and the fibroin gene promoter. Proc. Natl. Acad. Sci. USA 1991, 88, 718–722. [Google Scholar] [CrossRef]
- Sainsbury, S.; Bernecky, C.; Cramer, P. Structural basis of transcription initiation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 129–143. [Google Scholar] [CrossRef]
- Manville, C.M.; Smith, K.; Sondka, Z.; Rance, H.; Cockell, S.; Cowell, I.G.; Lee, K.C.; Morris, N.J.; Padget, K.; Jackson, G.H.; et al. Genome-wide ChIP-seq analysis of human TOP2B occupancy in MCF7 breast cancer epithelial cells. Biol. Open 2015, 4, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Williamson, L.M.; Lees-Miller, S.P. Estrogen receptor α-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis 2011, 32, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Bunch, H.; Lawney, B.P.; Lin, Y.F.; Asaithamby, A.; Murshid, A.; Wang, Y.E.; Chen, B.P.; Calderwood, S.K. Transcriptional elongation requires DNA break-induced signalling. Nat. Commun. 2015, 6, 10191. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Trotter, K.W.; King, H.A.; Archer, T.K. Glucocorticoid Receptor Transcriptional Activation via the BRG1-Dependent Recruitment of TOP2 β and Ku70/86. Mol. Cell Biol. 2015, 35, 2799–2817. [Google Scholar] [CrossRef]
- Tamamori-Adachi, M.; Koga, A.; Susa, T.; Fujii, H.; Tsuchiya, M.; Okinaga, H.; Hisaki, H.; Iizuka, M.; Kitajima, S.; Okazaki, T. DNA damage response induced by Etoposide promotes steroidogenesis via GADD45A in cultured adrenal cells. Sci. Rep. 2018, 8, 9636. [Google Scholar] [CrossRef]
- Vitelli, V.; Galbiati, A.; Iannelli, F.; Pessina, F.; Sharma, S.; d’Adda di Fagagna, F. Recent Advancements in DNA Damage-Transcription Crosstalk and High-Resolution Mapping of DNA Breaks. Annu. Rev. Genom. Hum. Genet. 2017, 18, 87–113. [Google Scholar] [CrossRef]
- Schwer, B.; Wei, P.C.; Chang, A.N.; Kao, J.; Du, Z.; Meyers, R.M.; Alt, F.W. Transcription-associated processes cause DNA double-strand breaks and translocations in neural stem/progenitor cells. Proc. Natl. Acad. Sci. USA 2016, 113, 2258–2263. [Google Scholar] [CrossRef]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Capra, T.; Gonzalez-Huici, V.; Fachinetti, D.; Cocito, A.; Natoli, G.; Katou, Y.; Mori, H.; Kurokawa, K.; Shirahige, K.; et al. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell 2009, 138, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.S.; Pina, B.; Roca, J. Topoisomerase II is required for the production of long Pol II gene transcripts in yeast. Nucleic Acids Res. 2012, 40, 7907–7915. [Google Scholar] [CrossRef] [PubMed]
- King, I.F.; Yandava, C.N.; Mabb, A.M.; Hsiao, J.S.; Huang, H.S.; Pearson, B.L.; Calabrese, J.M.; Starmer, J.; Parker, J.S.; Magnuson, T.; et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature 2013, 501, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, A.K.; Laflin, K.J.; Daniel, R.L.; Liao, Q.; Hawkins, K.A.; Aldaz, C.M. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3-24.1, a region frequently affected in breast cancer. Cancer Res. 2000, 60, 2140–2145. [Google Scholar] [PubMed]
- Fong, K.M.; Biesterveld, E.J.; Virmani, A.; Wistuba, I.; Sekido, Y.; Bader, S.A.; Ahmadian, M.; Ong, S.T.; Rassool, F.V.; Zimmerman, P.V.; et al. FHIT and FRA3B 3p14.2 allele loss are common in lung cancer and preneoplastic bronchial lesions and are associated with cancer-related FHIT cDNA splicing aberrations. Cancer Res. 1997, 57, 2256–2267. [Google Scholar] [PubMed]
- Inoue, H.; Ishii, H.; Alder, H.; Snyder, E.; Druck, T.; Huebner, K.; Croce, C.M. Sequence of the FRA3B common fragile region: Implications for the mechanism of FHIT deletion. Proc. Natl. Acad. Sci. USA 1997, 94, 14584–14589. [Google Scholar] [CrossRef] [PubMed]
- Ried, K.; Finnis, M.; Hobson, L.; Mangelsdorf, M.; Dayan, S.; Nancarrow, J.K.; Woollatt, E.; Kremmidiotis, G.; Gardner, A.; Venter, D.; et al. Common chromosomal fragile site FRA16D sequence: Identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum. Mol. Genet. 2000, 9, 1651–1663. [Google Scholar] [CrossRef]
- Alsop, A.E.; Taylor, K.; Zhang, J.; Gabra, H.; Paige, A.J.; Edwards, P.A. Homozygous deletions may be markers of nearby heterozygous mutations: The complex deletion at FRA16D in the HCT116 colon cancer cell line removes exons of WWOX. Genes Chromosomes Cancer 2008, 47, 437–447. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Naughton, C.; Avlonitis, N.; Corless, S.; Prendergast, J.G.; Mati, I.K.; Eijk, P.P.; Cockroft, S.L.; Bradley, M.; Ylstra, B.; Gilbert, N. Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat. Struct. Mol. Biol. 2013, 20, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Miyaji-Yamaguchi, M.; Tsutsui, K.M.; Tsutsui, K. Topoisomerase IIbeta activates a subset of neuronal genes that are repressed in AT-rich genomic environment. PLoS ONE 2008, 3, e4103. [Google Scholar] [CrossRef] [PubMed]
- Uuskula-Reimand, L.; Hou, H.; Samavarchi-Tehrani, P.; Rudan, M.V.; Liang, M.; Medina-Rivera, A.; Mohammed, H.; Schmidt, D.; Schwalie, P.; Young, E.J.; et al. Topoisomerase II β interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016, 17, 182. [Google Scholar] [CrossRef] [PubMed]
- Canela, A.; Maman, Y.; Jung, S.; Wong, N.; Callen, E.; Day, A.; Kieffer-Kwon, K.R.; Pekowska, A.; Zhang, H.; Rao, S.S.P.; et al. Genome Organization Drives Chromosome Fragility. Cell 2017, 170, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Cowell, I.G.; Sondka, Z.; Smith, K.; Lee, K.C.; Manville, C.M.; Sidorczuk-Lesthuruge, M.; Rance, H.A.; Padget, K.; Jackson, G.H.; Adachi, N.; et al. Model for MLL translocations in therapy-related leukemia involving topoisomerase IIbeta-mediated DNA strand breaks and gene proximity. Proc. Natl. Acad. Sci. USA 2012, 109, 8989–8994. [Google Scholar] [CrossRef] [PubMed]
- Capranico, G.; Tinelli, S.; Zunino, F.; Kohn, K.W.; Pommier, Y. Effects of base mutations on topoisomerase II DNA cleavage stimulated by mAMSA in short DNA oligomers. Biochemistry 1993, 32, 145–152. [Google Scholar] [CrossRef]
- Spitzner, J.R.; Muller, M.T. A consensus sequence for cleavage by vertebrate DNA topoisomerase II. Nucleic Acids Res. 1988, 16, 5533–5556. [Google Scholar] [CrossRef]
- Froelich-Ammon, S.J.; Gale, K.C.; Osheroff, N. Site-specific cleavage of a DNA hairpin by topoisomerase II. DNA secondary structure as a determinant of enzyme recognition/cleavage. J. Biol. Chem. 1994, 269, 7719–7725. [Google Scholar]
- Jonstrup, A.T.; Thomsen, T.; Wang, Y.; Knudsen, B.R.; Koch, J.; Andersen, A.H. Hairpin structures formed by α satellite DNA of human centromeres are cleaved by human topoisomerase IIalpha. Nucleic Acids Res. 2008, 36, 6165–6174. [Google Scholar] [CrossRef]
- Mills, W.E.; Spence, J.M.; Fukagawa, T.; Farr, C.J. Site-Specific Cleavage by Topoisomerase 2: A Mark of the Core Centromere. Int. J. Mol. Sci. 2018, 19, 534. [Google Scholar] [CrossRef]
- West, K.L.; Austin, C.A. Human DNA topoisomerase IIbeta binds and cleaves four-way junction DNA in vitro. Nucleic Acids Res. 1999, 27, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.W.; Pierce, L.C.; Ng, M.C.; Wang, Y.H. Role of DNA secondary structures in fragile site breakage along human chromosome 10. Hum. Mol. Genet. 2013, 22, 1443–1456. [Google Scholar] [CrossRef]
- Szlachta, K.; Thys, R.G.; Atkin, N.D.; Pierce, L.C.T.; Bekiranov, S.; Wang, Y.H. Alternative DNA secondary structure formation affects RNA polymerase II promoter-proximal pausing in human. Genome Biol. 2018, 19, 89. [Google Scholar] [CrossRef]
- Thys, R.G.; Lehman, C.E.; Pierce, L.C.; Wang, Y.H. DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genom. 2015, 16, 60–70. [Google Scholar] [CrossRef]
- Le, H.; Singh, S.; Shih, S.J.; Du, N.; Schnyder, S.; Loredo, G.A.; Bien, C.; Michaelis, L.; Toor, A.; Diaz, M.O.; et al. Rearrangements of the MLL gene are influenced by DNA secondary structure, potentially mediated by topoisomerase II binding. Genes Chromosomes Cancer 2009, 48, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Thys, R.G.; Lehman, C.E.; Pierce, L.C.; Wang, Y.H. Environmental and chemotherapeutic agents induce breakage at genes involved in leukemia-causing gene rearrangements in human hematopoietic stem/progenitor cells. Mutat. Res. 2015, 779, 86–95. [Google Scholar] [CrossRef]
- Wang, G.; Vasquez, K.M. Effects of Replication and Transcription on DNA Structure-Related Genetic Instability. Genes Basel 2017, 8, 17. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Dellino, G.I.; Palluzzi, F.; Chiariello, A.M.; Piccioni, R.; Bianco, S.; Furia, L.; De Conti, G.; Bouwman, B.A.M.; Melloni, G.; Guido, D.; et al. Release of paused RNA polymerase II at specific loci favors DNA double-strand-break formation and promotes cancer translocations. Nat. Genet. 2019, 51, 1011–1023. [Google Scholar] [CrossRef]
- Lensing, S.V.; Marsico, G.; Hansel-Hertsch, R.; Lam, E.Y.; Tannahill, D.; Balasubramanian, S. DSBCapture: In situ capture and sequencing of DNA breaks. Nat. Methods 2016, 13, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.X.; Mirzazadeh, R.; Garnerone, S.; Scott, D.; Schneider, M.W.; Kallas, T.; Custodio, J.; Wernersson, E.; Li, Y.; Gao, L.; et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017, 8, 15058. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Schellenberg, M.J.; Huang, S.Y.; Abdelmalak, M.; Marchand, C.; Nitiss, K.C.; Nitiss, J.L.; Williams, R.S.; Pommier, Y. Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2 DNA and Top2 RNA covalent complexes by tyrosyl-DNA-phosphodiesterase 2 (TDP2). J. Biol. Chem. 2014, 289, 17960–17969. [Google Scholar] [CrossRef] [PubMed]
- Cortes Ledesma, F.; El Khamisy, S.F.; Zuma, M.C.; Osborn, K.; Caldecott, K.W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 2009, 461, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Cortes-Ledesma, F.; El Khamisy, S.F.; Caldecott, K.W. TDP2/TTRAP is the major 5′-tyrosyl DNA phosphodiesterase activity in vertebrate cells and is critical for cellular resistance to topoisomerase II-induced DNA damage. J. Biol. Chem. 2011, 286, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Herreros, F.; Zagnoli-Vieira, G.; Ntai, I.; Martinez-Macias, M.I.; Anderson, R.M.; Herrero-Ruiz, A.; Caldecott, K.W. TDP2 suppresses chromosomal translocations induced by DNA topoisomerase II during gene transcription. Nat. Commun. 2017, 8, 233. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Herreros, F.; Romero-Granados, R.; Zeng, Z.; Alvarez-Quilon, A.; Quintero, C.; Ju, L.; Umans, L.; Vermeire, L.; Huylebroeck, D.; Caldecott, K.W.; et al. TDP2-dependent non-homologous end-joining protects against topoisomerase II-induced DNA breaks and genome instability in cells and in vivo. PLoS Genet. 2013, 9, e1003226. [Google Scholar] [CrossRef]
- Schellenberg, M.J.; Perera, L.; Strom, C.N.; Waters, C.A.; Monian, B.; Appel, C.D.; Vilas, C.K.; Williams, J.G.; Ramsden, D.A.; Williams, R.S. Reversal of DNA damage induced Topoisomerase 2 DNA-protein crosslinks by Tdp2. Nucleic Acids Res. 2016, 44, 3829–3844. [Google Scholar] [CrossRef]
- Lee, K.C.; Padget, K.; Curtis, H.; Cowell, I.G.; Moiani, D.; Sondka, Z.; Morris, N.J.; Jackson, G.H.; Cockell, S.J.; Tainer, J.A.; et al. MRE11 facilitates the removal of human topoisomerase II complexes from genomic DNA. Biol. Open 2012, 1, 863–873. [Google Scholar] [CrossRef]
- Deshpande, R.A.; Lee, J.H.; Arora, S.; Paull, T.T. Nbs1 Converts the Human Mre11/Rad50 Nuclease Complex into an Endo/Exonuclease Machine Specific for Protein-DNA Adducts. Mol. Cell 2016, 64, 593–606. [Google Scholar] [CrossRef]
- Aparicio, T.; Baer, R.; Gottesman, M.; Gautier, J. MRN, CtIP, and BRCA1 mediate repair of topoisomerase II-DNA adducts. J. Cell Biol. 2016, 212, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Jasrotia, A.; Bundschuh, D.; Howard, S.M.; Ranjha, L.; Stucki, M.; Cejka, P. NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Quennet, V.; Beucher, A.; Barton, O.; Takeda, S.; Lobrich, M. CtIP and MRN promote non-homologous end-joining of etoposide-induced DNA double-strand breaks in G1. Nucleic Acids Res. 2011, 39, 2144–2152. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef]
- Canela, A.; Sridharan, S.; Sciascia, N.; Tubbs, A.; Meltzer, P.; Sleckman, B.P.; Nussenzweig, A. DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Mol. Cell 2016, 63, 898–911. [Google Scholar] [CrossRef] [PubMed]
- Crosetto, N.; Mitra, A.; Silva, M.J.; Bienko, M.; Dojer, N.; Wang, Q.; Karaca, E.; Chiarle, R.; Skrzypczak, M.; Ginalski, K.; et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 2013, 10, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, B.A.M.; Crosetto, N. Endogenous DNA Double-Strand Breaks during DNA Transactions: Emerging Insights and Methods for Genome-Wide Profiling. Genes 2018, 9, 632. [Google Scholar] [CrossRef]
- Szlachta, K.; Raimer, H.M.; Comeau, L.D.; Wang, Y.-H. CNCC: An analysis tool to determine genome-wide DNA break end structure at single-nucleotide resolution. bioRxiv 2019, 611756. [Google Scholar] [CrossRef]
- Zhou, Y.; Caron, P.; Legube, G.; Paull, T.T. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res. 2014, 42, e19. [Google Scholar] [CrossRef]
- Chang, H.H.; Watanabe, G.; Gerodimos, C.A.; Ochi, T.; Blundell, T.L.; Jackson, S.P.; Lieber, M.R. Different DNA End Configurations Dictate Which NHEJ Components Are Most Important for Joining Efficiency. J. Biol. Chem. 2016, 291, 24377–24389. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Tomimatsu, N.; Mukherjee, B.; Catherine Hardebeck, M.; Ilcheva, M.; Vanessa Camacho, C.; Louise Harris, J.; Porteus, M.; Llorente, B.; Khanna, K.K.; Burma, S. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat. Commun. 2014, 5, 3561. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.K.; Lavin, M.F.; Jackson, S.P.; Mulhern, T.D. ATM, a central controller of cellular responses to DNA damage. Cell Death Differ. 2001, 8, 1052–1065. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 1. [Google Scholar] [CrossRef] [PubMed]
- Bracker, T.U.; Giebel, B.; Spanholtz, J.; Sorg, U.R.; Klein-Hitpass, L.; Moritz, T.; Thomale, J. Stringent regulation of DNA repair during human hematopoietic differentiation: A gene expression and functional analysis. Stem. Cells 2006, 24, 722–730. [Google Scholar] [CrossRef]
- Milyavsky, M.; Gan, O.I.; Trottier, M.; Komosa, M.; Tabach, O.; Notta, F.; Lechman, E.; Hermans, K.G.; Eppert, K.; Konovalova, Z.; et al. A distinctive DNA damage response in human hematopoietic stem cells reveals an apoptosis-independent role for p53 in self-renewal. Cell Stem. Cell 2010, 7, 186–197. [Google Scholar] [CrossRef]
- Beadle, G.; Baade, P.; Fritschi, L. Acute myeloid leukemia after breast cancer: A population-based comparison with hematological malignancies and other cancers. Ann. Oncol. 2009, 20, 103–109. [Google Scholar] [CrossRef]
- Morton, L.M.; Dores, G.M.; Tucker, M.A.; Kim, C.J.; Onel, K.; Gilbert, E.S.; Fraumeni, J.F.; Curtis, R.E. Evolving risk of therapy-related acute myeloid leukemia following cancer chemotherapy among adults in the United States, 1975–2008. Blood 2013, 121, 2996–3004. [Google Scholar] [CrossRef]
- Leone, G.; Mele, L.; Pulsoni, A.; Equitani, F.; Pagano, L. The incidence of secondary leukemias. Haematologica 1999, 84, 937–945. [Google Scholar]
- Pirani, M.; Marcheselli, R.; Marcheselli, L.; Bari, A.; Federico, M.; Sacchi, S. Risk for second malignancies in non-Hodgkin’s lymphoma survivors: A meta-analysis. Ann. Oncol. 2011, 22, 1845–1858. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S. Therapy-related myelodysplasia and acute myeloid leukemia. Semin. Oncol. 2013, 40, 666–675. [Google Scholar] [CrossRef]
- Zahid, M.F.; Parnes, A.; Savani, B.N.; Litzow, M.R.; Hashmi, S.K. Therapy-related myeloid neoplasms—What have we learned so far? World J. Stem. Cells 2016, 8, 231–242. [Google Scholar] [CrossRef] [PubMed]
- McNerney, M.E.; Godley, L.A.; Le Beau, M.M. Therapy-related myeloid neoplasms: When genetics and environment collide. Nat. Rev. Cancer 2017, 17, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Howe, R.; Micallef, I.N.; Inwards, D.J.; Ansell, S.M.; Dewald, G.W.; Dispenzieri, A.; Gastineau, D.A.; Gertz, M.A.; Geyer, S.M.; Hanson, C.A.; et al. Secondary myelodysplastic syndrome and acute myelogenous leukemia are significant complications following autologous stem cell transplantation for lymphoma. Bone Marrow Transpl. 2003, 32, 317–324. [Google Scholar] [CrossRef]
- Krishnan, A.; Bhatia, S.; Slovak, M.L.; Arber, D.A.; Niland, J.C.; Nademanee, A.; Fung, H.; Bhatia, R.; Kashyap, A.; Molina, A.; et al. Predictors of therapy-related leukemia and myelodysplasia following autologous transplantation for lymphoma: An assessment of risk factors. Blood 2000, 95, 1588–1593. [Google Scholar]
- Pedersen-Bjergaard, J.; Philip, P. Balanced translocations involving chromosome bands 11q23 and 21q22 are highly characteristic of myelodysplasia and leukemia following therapy with cytostatic agents targeting at DNA-topoisomerase II. Blood 1991, 78, 1147–1148. [Google Scholar]
- Broeker, P.L.; Super, H.G.; Thirman, M.J.; Pomykala, H.; Yonebayashi, Y.; Tanabe, S.; Zeleznik-Le, N.; Rowley, J.D. Distribution of 11q23 breakpoints within the MLL breakpoint cluster region in de novo acute leukemia and in treatment-related acute myeloid leukemia: Correlation with scaffold attachment regions and topoisomerase II consensus binding sites. Blood 1996, 87, 1912–1922. [Google Scholar]
- Felix, C.A. Secondary leukemias induced by topoisomerase-targeted drugs. Biochim. Biophys. Acta 1998, 1400, 233–255. [Google Scholar] [CrossRef]
- Meyer, C.; Burmeister, T.; Groger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-de-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Kersting, G.; Willmann, S.; Wurthwein, G.; Lippert, J.; Boos, J.; Hempel, G. Physiologically based pharmacokinetic modelling of high- and low-dose etoposide: From adults to children. Cancer Chemother. Pharmacol. 2012, 69, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Kontny, N.E.; Wurthwein, G.; Joachim, B.; Boddy, A.V.; Krischke, M.; Fuhr, U.; Thompson, P.A.; Jorger, M.; Schellens, J.H.; Hempel, G. Population pharmacokinetics of doxorubicin: Establishment of a NONMEM model for adults and children older than 3 years. Cancer Chemother. Pharmacol. 2013, 71, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Aplan, P.D.; Chervinsky, D.S.; Stanulla, M.; Burhans, W.C. Site-specific DNA cleavage within the MLL breakpoint cluster region induced by topoisomerase II inhibitors. Blood 1996, 87, 2649–2658. [Google Scholar] [CrossRef] [PubMed]
- Felix, C.A.; Lange, B.J.; Hosler, M.R.; Fertala, J.; Bjornsti, M.A. Chromosome band 11q23 translocation breakpoints are DNA topoisomerase II cleavage sites. Cancer Res. 1995, 55, 4287–4292. [Google Scholar] [PubMed]
- Strissel, P.L.; Strick, R.; Rowley, J.D.; Zeleznik-Le, N.J. An in vivo topoisomerase II cleavage site and a DNase I hypersensitive site colocalize near exon 9 in the MLL breakpoint cluster region. Blood 1998, 92, 3793–3803. [Google Scholar] [PubMed]
- Cimino, G.; Rapanotti, M.C.; Biondi, A.; Elia, L.; Lo Coco, F.; Price, C.; Rossi, V.; Rivolta, A.; Canaani, E.; Croce, C.M.; et al. Infant acute leukemias show the same biased distribution of ALL1 gene breaks as topoisomerase II related secondary acute leukemias. Cancer Res. 1997, 57, 2879–2883. [Google Scholar]
- Ross, J.A.; Davies, S.M.; Potter, J.D.; Robison, L.L. Epidemiology of childhood leukemia, with a focus on infants. Epidemiol. Rev. 1994, 16, 243–272. [Google Scholar] [CrossRef]
- Ross, J.A.; Potter, J.D.; Reaman, G.H.; Pendergrass, T.W.; Robison, L.L. Maternal exposure to potential inhibitors of DNA topoisomerase II and infant leukemia (United States): A report from the Children’s Cancer Group. Cancer Causes Control 1996, 7, 581–590. [Google Scholar] [CrossRef]
- Alexander, F.E.; Patheal, S.L.; Biondi, A.; Brandalise, S.; Cabrera, M.E.; Chan, L.C.; Chen, Z.; Cimino, G.; Cordoba, J.C.; Gu, L.J.; et al. Transplacental chemical exposure and risk of infant leukemia with MLL gene fusion. Cancer Res. 2001, 61, 2542–2546. [Google Scholar]
- Schroder-van der Elst, J.P.; van der Heide, D.; Rokos, H.; Morreale de Escobar, G.; Kohrle, J. Synthetic flavonoids cross the placenta in the rat and are found in fetal brain. Am. J. Physiol. 1998, 274, 253–256. [Google Scholar] [CrossRef]
- Gale, K.B.; Ford, A.M.; Repp, R.; Borkhardt, A.; Keller, C.; Eden, O.B.; Greaves, M.F. Backtracking leukemia to birth: Identification of clonotypic gene fusion sequences in neonatal blood spots. Proc. Natl. Acad. Sci. USA 1997, 94, 13950–13954. [Google Scholar] [CrossRef]
- Ford, A.M.; Bennett, C.A.; Price, C.M.; Bruin, M.C.; Van Wering, E.R.; Greaves, M. Fetal origins of the TEL-AML1 fusion gene in identical twins with leukemia. Proc. Natl. Acad. Sci. USA 1998, 95, 4584–4588. [Google Scholar] [CrossRef] [PubMed]
- Gill Super, H.J.; Rothberg, P.G.; Kobayashi, H.; Freeman, A.I.; Diaz, M.O.; Rowley, J.D. Clonal, nonconstitutional rearrangements of the MLL gene in infant twins with acute lymphoblastic leukemia: In utero chromosome rearrangement of 11q23. Blood 1994, 83, 641–644. [Google Scholar] [PubMed]
- Baan, R.; Grosse, Y.; Straif, K.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part F: Chemical agents and related occupations. Lancet. Oncol. 2009, 10, 1143–1144. [Google Scholar] [CrossRef]
- Carlos-Wallace, F.M.; Zhang, L.; Smith, M.T.; Rader, G.; Steinmaus, C. Parental, In Utero, and Early-Life Exposure to Benzene and the Risk of Childhood Leukemia: A Meta-Analysis. Am. J. Epidemiol. 2016, 183, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.A.; Patel, S.; Ono, K.; Nakane, H.; Fisher, L.M. Site-specific DNA cleavage by mammalian DNA topoisomerase II induced by novel flavone and catechin derivatives. Biochem. J. 1992, 282, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Bandele, O.J.; Osheroff, N. Bioflavonoids as poisons of human topoisomerase II α and II β. Biochemistry 2007, 46, 6097–6108. [Google Scholar] [CrossRef] [PubMed]
- Constantinou, A.; Mehta, R.; Runyan, C.; Rao, K.; Vaughan, A.; Moon, R. Flavonoids as DNA topoisomerase antagonists and poisons: Structure-activity relationships. J. Nat. Prod. 1995, 58, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Jensen, C.D.; Block, G.; Buffler, P.; Ma, X.; Selvin, S.; Month, S. Maternal dietary risk factors in childhood acute lymphoblastic leukemia (United States). Cancer Causes Control 2004, 15, 559–570. [Google Scholar] [CrossRef]
- Latino-Martel, P.; Chan, D.S.; Druesne-Pecollo, N.; Barrandon, E.; Hercberg, S.; Norat, T. Maternal alcohol consumption during pregnancy and risk of childhood leukemia: Systematic review and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1238–1260. [Google Scholar] [CrossRef]
- Spector, L.G.; Xie, Y.; Robison, L.L.; Heerema, N.A.; Hilden, J.M.; Lange, B.; Felix, C.A.; Davies, S.M.; Slavin, J.; Potter, J.D.; et al. Maternal diet and infant leukemia: The DNA topoisomerase II inhibitor hypothesis: A report from the children’s oncology group. Cancer Epidemiol. Biomark. Prev. 2005, 14, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Lanoue, L.; Green, K.K.; Kwik-Uribe, C.; Keen, C.L. Dietary factors and the risk for acute infant leukemia: Evaluating the effects of cocoa-derived flavanols on DNA topoisomerase activity. Exp. Biol. Med. Maywood 2010, 235, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Bariar, B.; Vestal, C.G.; Deem, B.; Goodenow, D.; Ughetta, M.; Engledove, R.W.; Sahyouni, M.; Richardson, C. Bioflavonoids promote stable translocations between MLL-AF9 breakpoint cluster regions independent of normal chromosomal context: Model system to screen environmental risks. Environ. Mol. Mutagen. 2019, 60, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Jemal, A.; Ward, E.M. Increasing incidence of differentiated thyroid cancer in the United States, 1988–2005. Cancer 2009, 115, 3801–3807. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.; Welch, H.G. Increasing incidence of thyroid cancer in the United States, 1973-2002. JAMA 2006, 295, 2164–2167. [Google Scholar] [CrossRef] [PubMed]
- Enewold, L.; Zhu, K.; Ron, E.; Marrogi, A.J.; Stojadinovic, A.; Peoples, G.E.; Devesa, S.S. Rising thyroid cancer incidence in the United States by demographic and tumor characteristics, 1980–2005. Cancer Epidemiol. Biomark. Prev. 2009, 18, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Ron, E.; Lubin, J.H.; Shore, R.E.; Mabuchi, K.; Modan, B.; Pottern, L.M.; Schneider, A.B.; Tucker, M.A.; Boice, J.D. Thyroid cancer after exposure to external radiation: A pooled analysis of seven studies. Radiat. Res. 1995, 141, 259–277. [Google Scholar] [CrossRef] [PubMed]
- Wartofsky, L. Increasing world incidence of thyroid cancer: Increased detection or higher radiation exposure? Hormones Athens 2010, 9, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Rowland, J.M.; Bove, K.E.; Monforte-Munoz, H.; Fagin, J.A. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res. 1997, 57, 1690–1694. [Google Scholar] [PubMed]
- Finn, S.P.; Smyth, P.; O’Leary, J.; Sweeney, E.C.; Sheils, O. Ret/PTC chimeric transcripts in an Irish cohort of sporadic papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 938–941. [Google Scholar] [CrossRef][Green Version]
- Fenton, C.L.; Lukes, Y.; Nicholson, D.; Dinauer, C.A.; Francis, G.L.; Tuttle, R.M. The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults. J. Clin. Endocrinol. Metab. 2000, 85, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Fugazzola, L.; Pilotti, S.; Pinchera, A.; Vorontsova, T.V.; Mondellini, P.; Bongarzone, I.; Greco, A.; Astakhova, L.; Butti, M.G.; Demidchik, E.P.; et al. Oncogenic rearrangements of the RET proto-oncogene in papillary thyroid carcinomas from children exposed to the Chernobyl nuclear accident. Cancer Res. 1995, 55, 5617–5620. [Google Scholar] [PubMed]
- Klugbauer, S.; Lengfelder, E.; Demidchik, E.P.; Rabes, H.M. High prevalence of RET rearrangement in thyroid tumors of children from Belarus after the Chernobyl reactor accident. Oncogene 1995, 11, 2459–2467. [Google Scholar] [PubMed]
- Burrow, A.A.; Williams, L.E.; Pierce, L.C.; Wang, Y.H. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genom. 2009, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.; Dillon, L.W.; Pramanik, S.; Nikiforov, Y.E.; Wang, Y.H. DNA breaks at fragile sites generate oncogenic RET/PTC rearrangements in human thyroid cells. Oncogene 2010, 29, 2272–2280. [Google Scholar] [CrossRef] [PubMed]
- National Toxicology Program. NTP 12th Report on Carcinogens. Rep Carcinog. 2011, 12, 499. [Google Scholar]
- Yunis, J.J.; Soreng, A.L.; Bowe, A.E. Fragile sites are targets of diverse mutagens and carcinogens. Oncogene 1987, 1, 59–69. [Google Scholar]
- Dillon, L.W.; Lehman, C.E.; Wang, Y.H. The role of fragile sites in sporadic papillary thyroid carcinoma. J. Thyroid. Res. 2012, 2012, 927683. [Google Scholar] [CrossRef]
- Pellegriti, G.; De Vathaire, F.; Scollo, C.; Attard, M.; Giordano, C.; Arena, S.; Dardanoni, G.; Frasca, F.; Malandrino, P.; Vermiglio, F.; et al. Papillary thyroid cancer incidence in the volcanic area of Sicily. J. Natl. Cancer Inst. 2009, 101, 1575–1583. [Google Scholar] [CrossRef]
- Pecoraino, G.; Scalici, L.; Avellone, G.; Ceraulo, L.; Favara, R.; Candela, E.G.; Provenzano, M.C.; Scaletta, C. Distribution of volatile organic compounds in Sicilian groundwaters analysed by head space-solid phase micro extraction coupled with gas chromatography mass spectrometry (SPME/GC/MS). Water Res. 2008, 42, 3563–3577. [Google Scholar] [CrossRef]
- Boffetta, P.; Kaldor, J.M. Secondary malignancies following cancer chemotherapy. Acta Oncol. 1994, 33, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, A.J.; Douglas, A.J.; Hudson, G.V.; Hudson, B.V.; Bennett, M.H.; MacLennan, K.A. Risk of second primary cancers after Hodgkin’s disease by type of treatment: Analysis of 2846 patients in the British National Lymphoma Investigation. BMJ 1992, 304, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Miller, R.W.; Ishikawa, Y.; Sugano, H. Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol. Biomark. Prev. 1996, 5, 239–246. [Google Scholar]
- Jimenez, M.; Leon, P.; Castro, L.; Azcona, C.; Sierrasesumaga, L. Second tumors in pediatric oncologic patients. Report of 5 cases. Rev. Med. Univ. Navar. 1995, 40, 72–77. [Google Scholar]
- Tsuchiya, H.; Tomita, K.; Ohno, M.; Inaoki, M.; Kawashima, A. Werner’s syndrome combined with quintuplicate malignant tumors: A case report and review of literature data. Jpn. J. Clin. Oncol. 1991, 21, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Verneris, M.; McDougall, I.R.; Becton, D.; Link, M.P. Thyroid carcinoma after successful treatment of osteosarcoma: A report of three patients. J. Pediatr. Hematol. Oncol. 2001, 23, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Yen, B.C.; Kahn, H.; Schiller, A.L.; Klein, M.J.; Phelps, R.G.; Lebwohl, M.G. Multiple hamartoma syndrome with osteosarcoma. Arch. Pathol. Lab. Med. 1993, 117, 1252–1254. [Google Scholar]
- Venkitaraman, R.; Affolter, A.; Ahmed, M.; Thomas, V.; Pritchard-Jones, K.; Sharma, A.K.; Marais, R.; Nutting, C.M. Childhood papillary thyroid cancer as second malignancy after successful treatment of rhabdomyosarcoma. Acta Oncol. 2008, 47, 469–472. [Google Scholar] [CrossRef]
- De Vathaire, F.; Hawkins, M.; Campbell, S.; Oberlin, O.; Raquin, M.A.; Schlienger, J.Y.; Shamsaldin, A.; Diallo, I.; Bell, J.; Grimaud, E.; et al. Second malignant neoplasms after a first cancer in childhood: Temporal pattern of risk according to type of treatment. Br. J. Cancer 1999, 79, 1884–1893. [Google Scholar] [CrossRef]
- Gow, K.W.; Lensing, S.; Hill, D.A.; Krasin, M.J.; McCarville, M.B.; Rai, S.N.; Zacher, M.; Spunt, S.L.; Strickland, D.K.; Hudson, M.M. Thyroid carcinoma presenting in childhood or after treatment of childhood malignancies: An institutional experience and review of the literature. J. Pediatr. Surg. 2003, 38, 1574–1580. [Google Scholar] [CrossRef]
- Vane, D.; King, D.R.; Boles, E.T. Secondary thyroid neoplasms in pediatric cancer patients: Increased risk with improved survival. J. Pediatr. Surg. 1984, 19, 855–860. [Google Scholar] [CrossRef]
- Smith, M.B.; Xue, H.; Strong, L.; Takahashi, H.; Jaffe, N.; Ried, H.; Zietz, H.; Andrassy, R.J. Forty-year experience with second malignancies after treatment of childhood cancer: Analysis of outcome following the development of the second malignancy. J. Pediatr. Surg. 1993, 28, 1342–1348. [Google Scholar] [CrossRef]
- Lehman, C.E.; Dillon, L.W.; Nikiforov, Y.E.; Wang, Y.H. DNA fragile site breakage as a measure of chemical exposure and predictor of individual susceptibility to form oncogenic rearrangements. Carcinogenesis 2017, 38, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Broderick, L.; Yost, S.; Li, D.; McGeough, M.D.; Booshehri, L.M.; Guaderrama, M.; Brydges, S.D.; Kucharova, K.; Patel, N.C.; Harr, M.; et al. Mutations in topoisomerase IIbeta result in a B cell immunodeficiency. Nat. Commun. 2019, 10, 3644. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.H.; Hwang, J.; Shang, H.F.; Tsai, S.T. Characterization of human DNA topoisomerase II as an autoantigen recognized by patients with IDDM. Diabetes 1996, 45, 408–414. [Google Scholar] [CrossRef]
- Gomez-Herreros, F.; Schuurs-Hoeijmakers, J.H.; McCormack, M.; Greally, M.T.; Rulten, S.; Romero-Granados, R.; Counihan, T.J.; Chaila, E.; Conroy, J.; Ennis, S.; et al. TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat. Genet. 2014, 46, 516–521. [Google Scholar] [CrossRef]
- Miyamoto, R.; Morino, H.; Yoshizawa, A.; Miyazaki, Y.; Maruyama, H.; Murakami, N.; Fukada, K.; Izumi, Y.; Matsuura, S.; Kaji, R.; et al. Exome sequencing reveals a novel MRE11 mutation in a patient with progressive myoclonic ataxia. J. Neurol. Sci. 2014, 337, 219–223. [Google Scholar] [CrossRef]
- Fernet, M.; Gribaa, M.; Salih, M.A.; Seidahmed, M.Z.; Hall, J.; Koenig, M. Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Hum. Mol. Genet. 2005, 14, 307–318. [Google Scholar] [CrossRef]
- Delia, D.; Piane, M.; Buscemi, G.; Savio, C.; Palmeri, S.; Lulli, P.; Carlessi, L.; Fontanella, E.; Chessa, L. MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. Hum. Mol. Genet. 2004, 13, 2155–2163. [Google Scholar] [CrossRef]
- Dovey, M.; Patton, E.E.; Bowman, T.; North, T.; Goessling, W.; Zhou, Y.; Zon, L.I. Topoisomerase II α is required for embryonic development and liver regeneration in zebrafish. Mol. Cell Biol. 2009, 29, 3746–3753. [Google Scholar] [CrossRef]
- Sapetto-Rebow, B.; McLoughlin, S.C.; O’Shea, L.C.; O’Leary, O.; Willer, J.R.; Alvarez, Y.; Collery, R.; O’Sullivan, J.; Van Eeden, F.; Hensey, C.; et al. Maternal topoisomerase II α, not topoisomerase II β, enables embryonic development of zebrafish top2a-/-mutants. BMC Dev. Biol. 2011, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Akimitsu, N.; Adachi, N.; Hirai, H.; Hossain, M.S.; Hamamoto, H.; Kobayashi, M.; Aratani, Y.; Koyama, H.; Sekimizu, K. Enforced cytokinesis without complete nuclear division in embryonic cells depleting the activity of DNA topoisomerase IIalpha. Genes Cells 2003, 8, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.L.; Wang, J.C. Aberrant lamination in the cerebral cortex of mouse embryos lacking DNA topoisomerase IIbeta. Proc. Natl. Acad. Sci. USA 2003, 100, 7123–7128. [Google Scholar] [CrossRef] [PubMed]
- Nur, E.K.A.; Meiners, S.; Ahmed, I.; Azarova, A.; Lin, C.P.; Lyu, Y.L.; Liu, L.F. Role of DNA topoisomerase IIbeta in neurite outgrowth. Brain Res. 2007, 1154, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, K.; Tsutsui, K.; Sano, K.; Kikuchi, A.; Tokunaga, A. Involvement of DNA topoisomerase IIbeta in neuronal differentiation. J. Biol. Chem. 2001, 276, 5769–5778. [Google Scholar] [CrossRef]
- Yang, X.; Li, W.; Prescott, E.D.; Burden, S.J.; Wang, J.C. DNA topoisomerase IIbeta and neural development. Science 2000, 287, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Meguid, N.A.; Dardir, A.A.; Abdel-Raouf, E.R.; Hashish, A. Evaluation of oxidative stress in autism: Defective antioxidant enzymes and increased lipid peroxidation. Biol. Trace Elem. Res. 2011, 143, 58–65. [Google Scholar] [CrossRef]
- Kern, J.K.; Jones, A.M. Evidence of toxicity, oxidative stress, and neuronal insult in autism. J. Toxicol. Environ. Health B Crit. Rev. 2006, 9, 485–499. [Google Scholar] [CrossRef]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef]
- Chauhan, A.; Chauhan, V.; Brown, W.T.; Cohen, I. Oxidative stress in autism: Increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin—The antioxidant proteins. Life Sci. 2004, 75, 2539–2549. [Google Scholar] [CrossRef]
- Fehsel, K.; Jalowy, A.; Qi, S.; Burkart, V.; Hartmann, B.; Kolb, H. Islet cell DNA is a target of inflammatory attack by nitric oxide. Diabetes 1993, 42, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Chou, H.Y.; Shen, T.L.; Chang, W.J.; Tai, P.H.; Li, T.K. Topoisomerase II-mediated DNA cleavage and mutagenesis activated by nitric oxide underlie the inflammation-associated tumorigenesis. Antioxid. Redox. Signal. 2013, 18, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Quilon, A.; Serrano-Benitez, A.; Lieberman, J.A.; Quintero, C.; Sanchez-Gutierrez, D.; Escudero, L.M.; Cortes-Ledesma, F. ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Nat. Commun. 2014, 5, 3347. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J. ATM and ataxia telangiectasia. EMBO Rep. 2004, 5, 772–776. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atkin, N.D.; Raimer, H.M.; Wang, Y.-H. Broken by the Cut: A Journey into the Role of Topoisomerase II in DNA Fragility. Genes 2019, 10, 791. https://doi.org/10.3390/genes10100791
Atkin ND, Raimer HM, Wang Y-H. Broken by the Cut: A Journey into the Role of Topoisomerase II in DNA Fragility. Genes. 2019; 10(10):791. https://doi.org/10.3390/genes10100791
Chicago/Turabian StyleAtkin, Naomi D., Heather M. Raimer, and Yuh-Hwa Wang. 2019. "Broken by the Cut: A Journey into the Role of Topoisomerase II in DNA Fragility" Genes 10, no. 10: 791. https://doi.org/10.3390/genes10100791
APA StyleAtkin, N. D., Raimer, H. M., & Wang, Y.-H. (2019). Broken by the Cut: A Journey into the Role of Topoisomerase II in DNA Fragility. Genes, 10(10), 791. https://doi.org/10.3390/genes10100791