Conservation Genetic Assessment of Savannah Elephants (Loxodonta africana) in the Greater Kruger Biosphere, South Africa


 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Area and Sample Collection
2.2. Laboratory Procedures and Microsatellite Quality Control
2.3. Genetic Diversity and Structure
2.4. Effective Population Size (Ne) and Demographic History
3. Results
3.1. Microsatellite Data Validation
3.2. Genetic Diversity and Structure
3.3. Effective Population Size and Demographic History
4. Discussion
4.1. Genetic Diversity and Structure
4.2. Demographic History and Effective Population Size
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fahrig, L. Relative effects of habitat loss and fragmentation on population extinction. J. Wildl. Manag. 1997, 61, 603–610. [Google Scholar] [CrossRef]
- Hoare, R.E. Determinants of human-elephant conflict in a land-use mosaic. J. Appl. Ecol. 1999, 36, 689–700. [Google Scholar] [CrossRef]
- Milner-Gulland, E.J.; Leader-Williams, N. A model of incentives for the illegal exploitation of black rhinos and elephants: Poaching pays in Luangwa Valley, Zambia. J. Appl. Ecol. 1992, 29, 388–401. [Google Scholar] [CrossRef]
- Winiarski, J.M.; Moorman, C.E.; Carpenter, J.P.; Hess, G.R. Reproductive consequences of habitat fragmentation for a declining resident bird of the longleaf pine ecosystem. Ecosphere 2017, 8, e01898. [Google Scholar] [CrossRef]
- Banks, S.C.; Piggott, M.P.; Stow, A.J.; Taylor, A.C. Sex and sociality in a disconnected world: A review of the impacts of habitat fragmentation on animal social interactions. Can. J. Zool. 2007, 85, 1065–1079. [Google Scholar] [CrossRef]
- Graham, M.D.; Douglas-Hamilton, I.; Adams, W.M.; Lee, P.C. The movement of African elephants in a human-dominated land-use mosaic. Anim. Conserv. 2009, 12, 445–455. [Google Scholar] [CrossRef]
- Montgomery, M.E.; Woodworth, L.M.; Nurthen, R.K.; Gilligan, D.M.; Briscoe, D.A.; Frankham, R. Relationships between population size and loss of genetic diversity: Comparisons of experimental results with theoretical predictions. Conserv. Genet. 2000, 1, 33–43. [Google Scholar] [CrossRef]
- Frankham, R. Genetics and extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Ouborg, N.J.; van Treuren, R.; van Damme, J.M.M. The significance of genetic erosion in the process of extinction. Oecologia 1991, 86, 359–367. [Google Scholar] [CrossRef]
- Haag, T.; Santos, A.S.; Sana, D.A.; Morato, R.G.; Cullen, L., Jr.; Crawshaw, P.G., Jr.; De Angelo, C.; Di Bitetti, M.S.; Salzano, F.M.; Eizirik, E. The effect of habitat fragmentation on the genetic structure of a top predator: Loss of diversity and high differentiation among remnant populations of Atlantic Forest jaguars (Panthera onca). Mol. Ecol. 2010, 19, 4906–4921. [Google Scholar] [CrossRef]
- Stockwell, C.A.; Hendry, A.P.; Kinnison, M.T. Contemporary evolution meets conservation biology. Trends Ecol. Evol. 2003, 18, 94–101. [Google Scholar] [CrossRef]
- Comer, C.E.; Kilgo, J.C.; D’Angelo, G.J.; Glenn, T.C.; Miller, K.V. Fine scale genetic structure and social organization in female white-tailed deer. J. Wildl. Manag. 2005, 69, 332–344. [Google Scholar] [CrossRef]
- Gobush, K.; Kerr, B.; Wasser, S. Genetic relatedness and disrupted social structure in a poached population of African elephants. Mol. Ecol. 2009, 18, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Chase, M.J.; Schlossberg, S.; Griffin, C.R.; Bouché, P.J.C.; Djene, S.W.; Elkan, P.W.; Ferreira, S.; Grossman, F.; Kohi, E.M.; Landen, K.; et al. Continent-wide survey reveals massive decline in African savannah elephants. PeerJ 2016, 4, e2354. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.S.; Trimble, M.J.; Purdon, A.; Young-Overton, K.D.; Pimm, S.L.; van Aarde, R.J. Savanna elephant numbers are only a quarter of their expected values. PLoS ONE 2017, 12, e0175942. [Google Scholar] [CrossRef] [PubMed]
- Blanc, J. Loxodonta Africana. The IUCN Red List of Threatened Species 2008: E.T12392A3339343. 2008. Available online: http://dx.doi.org/10.2305/IUCN.UK.2008.RLTS.T12392A3339343.en (accessed on 19 September 2017).
- Archie, E.A.; Chiyo, P.I. Elephant behaviour and conservation: Social relationships, the effects of poaching, and genetic tools for management. Mol. Ecol. 2012, 21, 765–778. [Google Scholar] [CrossRef]
- Douglas-Hamilton, I. African elephants: Population trends and their causes. Oryx 1987, 21, 11–24. [Google Scholar] [CrossRef]
- Milner-Gulland, E.J.; Mace, R. The impact of the ivory trade on the African elephant Loxodonta africana population as assessed by data from the trade. Biol. Conserv. 1991, 55, 215–229. [Google Scholar] [CrossRef]
- van Aarde, R.; Jackson, T. Megaparks for metapopulations: Addressing the causes of locally high elephant numbers in southern Africa. Biol. Conserv. 2007, 134, 289–297. [Google Scholar] [CrossRef]
- Nyakaana, S.; Arctander, P.; Siegismund, H.R. Population structure of the African savannah elephant inferred from mitochondrial control region sequences and nuclear microsatellite loci. Heredity 2002, 89, 90–98. [Google Scholar] [CrossRef]
- Comstock, K.E.; Georgiadis, N.; Pecon-Slattery, J.; Roca, A.L.; Ostrander, E.A.; O’Brien, S.J.; Wasser, S.K. Patterns of molecular genetic variation among African elephant populations. Mol. Ecol. 2002, 11, 2489–2498. [Google Scholar] [CrossRef] [PubMed]
- Roca, A.L. Genetic evidence for two species of elephant in Africa. Science 2001, 293, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Roca, A.L.; Georgiadis, N.; O’Brien, S.J. Cytonuclear genomic dissociation in African elephant species. Nat. Genet. 2005, 37, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Hall-Martin, A.J. Distribution and status of the African elephant Loxodonta africana in South Africa, 1652–1992. Koedoe 1992, 35, 65–88. [Google Scholar] [CrossRef]
- Selier, S.A.J.; Henley, M.; Pretorius, Y.; Garai, M. A conservation assessment of Loxodonta africana. In The Red List of Mammals of South Africa, Swaziland and Lesotho; Child, M.F., Roxburgh, L., Do Linh San, E., Raimondo, D., Davies-Mostert, H.T., Eds.; South African National Biodiversity Institute and Endangered Wildlife Trust: Modderfontein, South Africa, 2016. [Google Scholar]
- Lacy, R.C. Importance of genetic variation to the viability of mammalian populations. J. Mammal. 1997, 78, 320–335. [Google Scholar] [CrossRef]
- Whitehouse, A.M.; Harley, E.H. Post-bottleneck genetic diversity of elephant populations in South Africa, revealed using microsatellite analysis. Mol. Ecol. 2001, 10, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- DeSalle, R.; Amato, G. The expansion of conservation genetics. Nat. Rev. Genet. 2004, 5, 702–712. [Google Scholar] [CrossRef]
- de Flamingh, A.; Roca, A.L.; van Aarde, R.J. Origin and phylogeography of African savannah elephants (Loxodonta africana) in Kruger and nearby parks in southern Africa. Conserv. Genet. 2018, 19, 155–167. [Google Scholar] [CrossRef]
- Whyte, I.; Aarde, R.; Pimm, S.L. Managing the elephants of Kruger National Park. Anim. Conserv. 1998, 1, 77–83. [Google Scholar] [CrossRef]
- Henley, M.D. Report on Elephant Movements in Relation to Water and the Effect of the 2012 Floods within the Associated Private Nature Reserves. Unpublished Report to the Associated Private Nature Reserves. 2014. [Google Scholar]
- Moss, C.J. The demography of an African elephant (Loxodonta africana) population in Amboseli, Kenya. J. Zool. 2001, 255, 145–156. [Google Scholar] [CrossRef]
- Henley, M. Report on the Demographics of the Bull Population of the Associated Private Nature Reserves; Save the Elephants: Nairobi, Kenya, 2013; pp. 1–24. [Google Scholar]
- Comstock, K.E.; Wasser, S.K.; Ostrander, E.A. Polymorphic microsatellite DNA loci identified in the African elephant (Loxodonta africana). Mol. Ecol. 2000, 9, 1004–1006. [Google Scholar] [CrossRef] [PubMed]
- Eggert, L.S.; Ramakrishnan, U.; Mundy, N.I.; Woodruff, D.S. Polymorphic microsatellite DNA markers in the African elephant (Loxondonta africana) and their use in the Asian elephant (Elephas maximus). Mol. Ecol. 2000, 9, 2223–2225. [Google Scholar] [CrossRef] [PubMed]
- Archie, E.A.; Moss, C.J.; Alberts, S.C. Characterization of tetranucleotide microsatellite loci in the African Savannah Elephant (Loxodonta africana africana). Mol. Ecol. Notes 2003, 3, 244–246. [Google Scholar] [CrossRef]
- Ahlering, M.A.; Hailer, F.; Roberts, M.T.; Foley, C. A simple and accurate method to sex savannah, forest and Asian elephants using noninvasive sampling techniques. Mol. Ecol. Resour. 2011, 11, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Frantz, A.C.; Pope, L.C.; Carpenter, P.J.; Roper, T.J.; Wilson, G.J.; Delahay, R.J.; Burke, T. Reliable microsatellite genotyping of the Eurasian badger (Meles meles) using faecal DNA. Mol. Ecol. 2003, 12, 1649–1661. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.; Ben-David, M.; Mcdonald, D.B. TECHNICAL ADVANCES: Effects of genotyping protocols on success and errors in identifying individual river otters (Lontra canadensis) from their faeces. Mol. Ecol. Resour. 2008, 8, 282–289. [Google Scholar] [CrossRef]
- Valière, N. GIMLET: A computer program for analysing genetic individual identification data. Mol. Ecol. Resour. 2002, 2, 377–379. [Google Scholar] [CrossRef]
- Broquet, T.; Petit, E. Quantifying genotyping errors in noninvasive population genetics. Mol. Ecol. 2004, 13, 3601–3608. [Google Scholar] [CrossRef]
- Waits, L.P.; Luikart, G.; Taberlet, P. Estimating the probability of identity among genotypes in natural populations: Cautions and guidelines. Mol. Ecol. 2001, 10, 249–256. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Sawaya, M.A.; Taper, M.L. Individual identification and distribution of genotypic differences between individuals. J. Wildl. Manag. 2006, 70, 1148–1150. [Google Scholar] [CrossRef]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Rousset, F. Genepop’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Gaetano, J. Holm-Bonferroni Sequential Correction: An EXCEL Calculator (1.2) [Microsoft Excel Workbook]. Available online: https://www.researchgate.net/publication/242331583_Holm-Bonferroni_Sequential_Correction_An_EXCEL_Calculator_-_Ver_12 (accessed on 30 July 2018).
- Dąbrowski, M.J.; Bornelöv, S.; Kruczyk, M.; Baltzer, N.; Komorowski, J. ‘True’ null allele detection in microsatellite loci: A comparison of methods, assessment of difficulties and survey of possible improvements. Mol. Ecol. Resour. 2015, 15, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Summers, K.; Amos, W. Behavioral, ecological, and molecular genetic analyses of reproductive strategies in the Amazonian dart-poison frog, Dendrobates ventrimaculatus. Behav. Ecol. 1997, 8, 260–267. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L. Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv. Genet. 2006, 7, 991–995. [Google Scholar] [CrossRef]
- Chybicki, I.J.; Burczyk, J. Simultaneous estimation of null alleles and inbreeding coefficients. J. Hered. 2009, 100, 106–113. [Google Scholar] [CrossRef]
- Kalinowski, S.T. hp-rare 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 2005, 5, 187–189. [Google Scholar] [CrossRef]
- Belkhir, K.; Borsa, P.; Chikhi, L.; Bonhomme, F. Genetix 4.05: WindowsTM Software for Population Genetics; University of Montpelier II, Laboratoire Genome de Populations: Montpelier, France, 1996. [Google Scholar]
- Whyte, I. Conservation management of the Kruger National Park elephant population. Ph.D. Thesis, University of Pretoria, Tshwance, South Africa, 2001. [Google Scholar]
- Archie, E.A.; Maldonado, J.E.; Hollister-Smith, J.A.; Poole, J.H.; Moss, C.J.; Fleischer, R.C.; Alberts, S.C. Fine-scale population genetic structure in a fission-fusion society. Mol. Ecol. 2008, 17, 2666–2679. [Google Scholar] [CrossRef]
- Kioko, J.; Muruthi, P.; Omondi, P.; Chiyo, P.I. The performance of electric fences as elephant barriers in Amboseli, Kenya. S. Afr. J. Wildl. Res. 2008, 38, 52–58. [Google Scholar] [CrossRef]
- Loarie, S.R.; Aarde, R.J.V.; Pimm, S.L. Fences and artificial water affect African savannah elephant movement patterns. Biol. Conserv. 2009, 142, 3086–3098. [Google Scholar] [CrossRef]
- Osborn, F.V.; Parker, G.E. Towards an integrated approach for reducing the conflict between elephants and people: A review of current research. Oryx 2003, 37, 80–84. [Google Scholar] [CrossRef]
- Druce, H.C.; Pretorius, K.; Slotow, R. The response of an elephant population to conservation area expansion: Phinda Private Game Reserve, South Africa. Biol. Conserv. 2008, 141, 3127–3138. [Google Scholar] [CrossRef]
- Harris, G.M.; Russell, G.J.; van Aarde, R.I.; Pimm, S.L. Rules of habitat use by elephants Loxodonta africana in southern Africa: Insights for regional management. Oryx 2008, 42, 66–75. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Smouse, P.E.; Peakall, R. Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity 1999, 82, 561–573. [Google Scholar] [CrossRef]
- Moran, P.A.P. Notes on continuous stochastic phenomena. Biometrika 1950, 37, 17–23. [Google Scholar] [CrossRef]
- Peakall, R.; Ruibal, M.; Lindenmayer, D.B. Spatial autocorrelation analysis offers insights into gene flow in the Australian bush rat, Rattus fuscipes. Evolution 2003, 57, 1182–1195. [Google Scholar] [CrossRef]
- Epperson, B.K. Estimating dispersal from short distance spatial autocorrelation. Heredity 2005, 95, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.; Brown, G.P.; Madsen, T.; Shine, R. Male-biased dispersal in a tropical Australian snake (Stegonotus cucullatus, Colubridae). Mol. Ecol. 2008, 17, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Smouse, P.E.; Peakall, R.; Gonzales, E. A heterogeneity test for fine-scale genetic structure. Mol. Ecol. 2008, 17, 3389–3400. [Google Scholar] [CrossRef] [PubMed]
- Banks, S.C.; Peakall, R. Genetic spatial autocorrelation can readily detect sex-biased dispersal. Mol. Ecol. 2012, 21, 2092–2105. [Google Scholar] [CrossRef] [PubMed]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar]
- Frantz, A.C.; Hamann, J.-L.; Klein, F. Fine-scale genetic structure of red deer (Cervus elaphus) in a French temperate forest. Eur. J. Wildl. Res. 2008, 54, 44–52. [Google Scholar] [CrossRef]
- Rousset, F. Genetic differentiation between individuals. J. Evol. Biol. 2000, 13, 58–62. [Google Scholar] [CrossRef]
- Loiselle, B.A.; Sork, V.L.; Nason, J.; Graham, C. Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am. J. Bot. 1995, 82, 1420–1425. [Google Scholar] [CrossRef]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Resour. 2002, 2, 618–620. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Guillot, G.; Mortier, F.; Estoup, A. GENELAND: A computer package for landscape genetics. Mol. Ecol. Resour. 2005, 5, 712–715. [Google Scholar] [CrossRef]
- Chen, C.; Durand, E.; Forbes, F.; François, O. Bayesian clustering algorithms ascertaining spatial population structure: A new computer program and a comparison study. Mol. Ecol. Notes 2007, 7, 747–756. [Google Scholar] [CrossRef]
- Blair, C.; Weigel, D.E.; Balazik, M.; Keeley, A.T.H.; Walker, F.M.; Landguth, E.; Cushman, S.; Murphy, M.; Waits, L.; Balkenhol, N. A simulation-based evaluation of methods for inferring linear barriers to gene flow. Mol. Ecol. Resour. 2012, 12, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Latch, E.K.; Dharmarajan, G.; Glaubitz, J.C.; Rhodes, O.E. Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation. Conserv. Genet. 2006, 7, 295–302. [Google Scholar] [CrossRef]
- François, O.; Durand, E. Spatially explicit Bayesian clustering models in population genetics. Mol. Ecol. Resour. 2010, 10, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Basto, M.P.; Santos-Reis, M.; Simões, L.; Grilo, C.; Cardoso, L.; Cortes, H.; Bruford, M.W.; Fernandes, C. Assessing genetic structure in common but ecologically distinct carnivores: The stone marten and red fox. PLoS ONE 2016, 11, e0145165. [Google Scholar] [CrossRef]
- Blanchet, E.; Lecoq, M.; Sword, G.A.; Berthier, K.; Pages, C.; Billot, C.; Rivallan, R.; Foucart, A.; Vassal, J.-M.; Risterucci, A.M.; et al. A comparative analysis of fine-scale genetic structure in three closely related syntopic species of the grasshopper genus Calliptamus. Can. J. Zool. 2012, 90, 31–41. [Google Scholar] [CrossRef]
- Guillot, G. Inference of structure in subdivided populations at low levels of genetic differentiation—The correlated allele frequencies model revisited. Bioinformatics 2008, 24, 2222–2228. [Google Scholar] [CrossRef]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Rosenberg, N.A. Distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2003, 4, 137–138. [Google Scholar] [CrossRef]
- Goudet, J.; Raymond, M.; de Meeus, T.; Rousset, F. Testing differentiation in diploid populations. Genetics 1996, 144, 1933–1940. [Google Scholar] [PubMed]
- Goudet, J. FSTAT (version 1.2): A computer program to calculate F-statistics. J. Hered. 1995, 86, 485–486. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Meirmans, P.G.; Hedrick, P.W. Assessing population structure: FST and related measures. Mol. Ecol. Resour. 2011, 11, 5–18. [Google Scholar] [CrossRef]
- Jost, L. Gst and its relatives do not measure differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef]
- Meirmans, P.G.; Van Tienderen, P.H. Genotype and genodive: Two programs for the analysis of genetic diversity of asexual organisms. Mol. Ecol. Notes 2004, 4, 792–794. [Google Scholar] [CrossRef]
- Goudet, J.; Perrin, N.; Waser, P. Tests for sex-biased dispersal using bi-parentally inherited genetic markers. Mol. Ecol. 2002, 11, 1103–1114. [Google Scholar] [CrossRef]
- Wright, S. Evolution in Mendelian populations. Genetics 1931, 16, 97–159. [Google Scholar]
- Waples, R.S. A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv. Genet. 2006, 7, 167–184. [Google Scholar] [CrossRef]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size Ne from genetic data. Mol. Ecol. Resour. 2014, 14, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Peel, D.; Waples, R.S.; Macbeth, G.M.; Do, C.; Ovenden, J.R. Accounting for missing data in the estimation of contemporary genetic effective population size (Ne). Mol. Ecol. Resour. 2013, 13, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S.; Do, C. Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: A largely untapped resource for applied conservation and evolution. Evol. Appl. 2010, 3, 244–262. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.T.; Ovenden, J.R.; Wang, Y.-G. Improved confidence intervals for the linkage disequilibrium method for estimating effective population size. Heredity 2016, 117, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, K.J.; Whitlock, M.C. Evaluating methods for estimating local effective population size with and without migration. Evolution 2015, 69, 2154–2166. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S.; England, P.R. Estimating contemporary effective population size on the basis of linkage disequilibrium in the face of migration. Genetics 2011, 189, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. A comparison of single-sample estimators of effective population sizes from genetic marker data. Mol. Ecol. 2016, 25, 4692–4711. [Google Scholar] [CrossRef]
- Sved, J.A.; Cameron, E.C.; Gilchrist, A.S. Estimating effective population size from linkage disequilibrium between unlinked loci: Theory and application to fruit fly outbreak populations. PLoS ONE 2013, 8, e69078. [Google Scholar] [CrossRef]
- Holleley, C.E.; Nichols, R.A.; Whitehead, M.R.; Adamack, A.T.; Gunn, M.R.; Sherwin, W.B. Testing single-sample estimators of effective population size in genetically structured populations. Conserv. Genet. 2014, 15, 23–35. [Google Scholar] [CrossRef]
- Luikart, G.; Ryman, N.; Tallmon, D.A.; Schwartz, M.K.; Allendorf, F.W. Estimation of census and effective population sizes: The increasing usefulness of DNA-based approaches. Conserv. Genet. 2010, 11, 355–373. [Google Scholar] [CrossRef]
- Robinson, J.D.; Moyer, G.R. Linkage disequilibrium and effective population size when generations overlap. Evol. Appl. 2013, 6, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S.; Antao, T.; Luikart, G. Effects of overlapping generations on linkage disequilibrium estimates of effective population size. Genetics 2014, 197, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Sinnock, P. The Wahlund Effect for the Two-Locus Model. Am. Nat. 1975, 109, 565–570. [Google Scholar] [CrossRef]
- Wang, J.; Brekke, P.; Huchard, E.; Knapp, L.A.; Cowlishaw, G. Estimation of parameters of inbreeding and genetic drift in populations with overlapping generations. Evolution 2010, 64, 1704–1718. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Fu, Y.-X. Estimating effective population size or mutation rate with microsatellites. Genetics 2004, 166, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 2004, 5, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.L.; Wong, C. Mutation of human short tandem repeats. Hum. Mol. Genet. 1993, 2, 1123–1128. [Google Scholar] [CrossRef]
- Whittaker, J.C.; Harbord, R.M.; Boxall, N.; Mackay, I.; Dawson, G.; Sibly, R.M. Likelihood-based estimation of microsatellite mutation rates. Genetics 2003, 164, 781–787. [Google Scholar]
- Xu, X.; Peng, M.; Fang, Z.; Xu, X. The direction of microsatellite mutations is dependent upon allele length. Nat. Genet. 2000, 24, 396–399. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J.-M. BOTTLENECK: A computer program for detecting recent reductions in the effective size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar] [PubMed]
- Luikart, G.; Cornuet, J.-M. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency. Conserv. Biol. 1998, 12, 228–237. [Google Scholar] [CrossRef]
- Peery, M.Z.; Kirby, R.; Reid, B.N.; Stoelting, R.; Doucet-BëEr, E.; Robinson, S.; VáSquez-Carrillo, C.; Pauli, J.N.; PalsbøLl, P.J. Reliability of genetic bottleneck tests for detecting recent population declines. Mol. Ecol. 2012, 21, 3403–3418. [Google Scholar] [CrossRef] [PubMed]
- Luikart, G.; Allendorf, F.W.; Cornuet, J.M.; Sherwin, W.B. Distortion of allele frequency distributions provides a test for recent population bottlenecks. J. Hered. 1998, 89, 238–247. [Google Scholar] [CrossRef]
- Garza, J.C.; Williamson, E.G. Detection of reduction in population size using data from microsatellite loci. Mol. Ecol. 2001, 10, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Leblois, R.; Pudlo, P.; Néron, J.; Bertaux, F.; Reddy Beeravolu, C.; Vitalis, R.; Rousset, F. Maximum-likelihood inference of population size contractions from microsatellite data. Mol. Biol. Evol. 2014, 31, 2805–2823. [Google Scholar] [CrossRef]
- Beaumont, M.A. Detecting population expansion and decline using microsatellites. Genetics 1999, 153, 2013–2029. [Google Scholar]
- Faurby, S.; Pertoldi, C. The consequences of the unlikely but critical assumption of stepwise mutation in the population genetic software, MSVAR. Evol. Ecol. Res. 2012, 14, 859–879. [Google Scholar]
- Girod, C.; Vitalis, R.; Leblois, R.; Fréville, H. Inferring population decline and expansion from microsatellite data: A simulation-based evaluation of the Msvar method. Genetics 2011, 188, 165–179. [Google Scholar] [CrossRef]
- Nikolic, N.; Chevalet, C. Detecting past changes of effective population size. Evol. Appl. 2014, 7, 663–681. [Google Scholar] [CrossRef]
- Pompanon, F.; Bonin, A.; Bellemain, E.; Taberlet, P. Genotyping errors: Causes, consequences and solutions. Nat. Rev. Genet. 2005, 6, 847–859. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.S.; Schwartz, M.K. Genetic errors associated with population estimation using non-invasive molecular tagging: Problems and new solutions. J. Wildl. Manag. 2004, 68, 439–448. [Google Scholar] [CrossRef]
- Carlsson, J. Effects of microsatellite null alleles on assignment testing. J. Hered. 2008, 99, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, M.-P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Dakin, E.E.; Avise, J.C. Microsatellite null alleles in parentage analysis. Heredity 2004, 93, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Ritland, K.; Dunn, D.W.; Qi, X.; Guo, S.; Li, B. Estimating relatedness in the presence of null alleles. Genetics 2016, 202, 247–260. [Google Scholar] [CrossRef]
- Storz, J.F.; Ramakrishnan, U.; Alberts, S.C. Genetic effective size of a wild primate population: Influence of current and historical demography. Evolution 2002, 56, 817–829. [Google Scholar] [CrossRef][Green Version]
- Waples, R.S. Testing for Hardy–Weinberg Proportions: Have we lost the plot? J. Hered. 2015, 106, 1–19. [Google Scholar] [CrossRef]
- Kamath, P.L.; Haroldson, M.A.; Luikart, G.; Paetkau, D.; Whitman, C.; van Manen, F.T. Multiple estimates of effective population size for monitoring a long-lived vertebrate: An application to Yellowstone grizzly bears. Mol. Ecol. 2015, 24, 5507–5521. [Google Scholar] [CrossRef]
- Skrbinšek, T.; Jelenčič, M.; Waits, L.; Kos, I.; Jerina, K.; Trontelj, P. Monitoring the effective population size of a brown bear (Ursus arctos) population using new single-sample approaches. Mol. Ecol. 2012, 21, 862–875. [Google Scholar] [CrossRef]
- Neel, M.C.; McKelvey, K.; Ryman, N.; Lloyd, M.W.; Short Bull, R.; Allendorf, F.W.; Schwartz, M.K.; Waples, R.S. Estimation of effective population size in continuously distributed populations: There goes the neighborhood. Heredity 2013, 111, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Dobson, F.S.; Chesser, R.K.; Hoogland, J.L.; Sugg, D.W.; Foltz, D.W. The influence of social breeding groups on effective population size in black-tailed prairie dogs. J. Mammal. 2004, 85, 58–66. [Google Scholar] [CrossRef]
- Wang, J. Effective size and F-statistics of subdivided populations. Genetics 1997, 146, 1465–1474. [Google Scholar] [PubMed]
- Miller, M.P.; Mullins, T.D.; Haig, S.M.; Takano, L.; Garcia, K. Genetic structure, diversity, and interisland dispersal in the endangered Mariana Common Moorhen (Gallinula chloropus guami). Condor 2015, 117, 660–669. [Google Scholar] [CrossRef]
- Leblois, R.; (National Institute for Agronomical Research, Montpellier, France). Personal communication, 2018.
- Valdes, A.M.; Slatkin, M.; Freimert, N.B. Allele frequencies at microsatellite loci: The stepwise mutation model revisited. Genetics 1993, 133, 737–749. [Google Scholar]
- Di Rienzo, A.; Peterson, A.C.; Garza, J.C.; Valdes, A.M.; Slatkin, M.; Freimer, N.B. Mutational processes of simple-sequence repeat loci in human populations. Proc. Natl. Acad. Sci. USA 1994, 91, 3166–3170. [Google Scholar] [CrossRef]
- Ishida, Y.; Van Coeverden de Groot, P.J.; Leggett, K.E.A.; Putnam, A.S.; Fox, V.E.; Lai, J.; Boag, P.T.; Georgiadis, N.J.; Roca, A.L. Genetic connectivity across marginal habitats: The elephants of the Namib Desert. Ecol. Evol. 2016, 6, 6189–6201. [Google Scholar] [CrossRef]
- Ishengoma, D.R.S.; Shedlock, A.M.; Foley, C.A.H.; Foley, L.J.; Wasser, S.K.; Balthazary, S.T.; Mutayoba, B.M. Effects of poaching on bull mating success in a free ranging African elephant (Loxodonta africana) population in Tarangire National Park, Tanzania. Conserv. Genet. 2008, 9, 247–255. [Google Scholar] [CrossRef]
- Ahlering, M.A.; Eggert, L.S.; Western, D.; Estes, A.; Munishi, L.; Fleischer, R.; Roberts, M.; Maldonado, J.E. Identifying source populations and genetic structure for savannah elephants in human-dominated landscapes and protected areas in the Kenya-Tanzania borderlands. PLoS ONE 2012, 7, e52288. [Google Scholar] [CrossRef]
- Okello, J.B.A.; Masembe, C.; Rasmussen, H.B.; Wittemyer, G.; Omondi, P.; Kahindi, O.; Muwanika, V.B.; Arctander, P.; Douglas-Hamilton, I.; Nyakaana, S.; et al. Population genetic structure of savannah elephants in Kenya: Conservation and management implications. J. Hered. 2008, 99, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Okello, J.B.A.; Wittemyer, G.; Rasmussen, H.B.; Arctander, P.; Nyakaana, S.; Douglas-Hamilton, I.; Siegismund, H.R. Effective population size dynamics reveal impacts of historic climatic events and recent anthropogenic pressure in African elephants. Mol. Ecol. 2008, 17, 3788–3799. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.C.; Neigel, J.E.; Leberg, P.L. Experimental evaluation of the usefulness of microsatellite DNA for detecting demographic bottlenecks. Mol. Ecol. 2000, 9, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Nyakaana, S.; Arctander, P. Population genetic structure of the African elephant in Uganda based on variation at mitochondrial and nuclear loci: Evidence for male-biased gene flow. Mol. Ecol. 1999, 8, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Williamson-Natesan, E.G. Comparison of methods for detecting bottlenecks from microsatellite loci. Conserv. Genet. 2005, 6, 551–562. [Google Scholar] [CrossRef]
- Hundertmark, K.J.; Van Daele, L.J. Founder effect and bottleneck signatures in an introduced, insular population of elk. Conserv. Genet. 2010, 11, 139–147. [Google Scholar] [CrossRef]
- Hoban, S.M.; Gaggiotti, O.E.; Bertorelle, G. The number of markers and samples needed for detecting bottlenecks under realistic scenarios, with and without recovery: A simulation-based study. Mol. Ecol. 2013, 22, 3444–3450. [Google Scholar] [CrossRef]
- Busch, J.D.; Waser, P.M.; DeWoody, J.A. Recent demographic bottlenecks are not accompanied by a genetic signature in banner-tailed kangaroo rats (Dipodomys spectabilis). Mol. Ecol. 2007, 16, 2450–2462. [Google Scholar] [CrossRef]
- Lippé, C.; Dumont, P.; Bernatchez, L. High genetic diversity and no inbreeding in the endangered copper redhorse, Moxostoma hubbsi (Catostomidae, Pisces): The positive sides of a long generation time. Mol. Ecol. 2006, 15, 1769–1780. [Google Scholar] [CrossRef]
- Waples, R.S.; Gaggiotti, O. What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol. Ecol. 2006, 15, 1419–1439. [Google Scholar] [CrossRef]
- Temple, H.J.; Hoffman, J.I.; Amos, W. Dispersal, philopatry and intergroup relatedness: Fine-scale genetic structure in the white-breasted thrasher, Ramphocinclus brachyurus. Mol. Ecol. 2006, 15, 3449–3458. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhan, X.; Qi, D.; Wei, F. Spatial genetic structure and dispersal of giant pandas on a mountain-range scale. Conserv. Genet. 2010, 11, 2145–2155. [Google Scholar] [CrossRef]
- De Villiers, P.A.; Kok, O.B. Home range, association and related aspects of elephants in the eastern Transvaal Lowveld. Afr. J. Ecol. 1997, 35, 224–236. [Google Scholar] [CrossRef]
- Ahlering, M.A.; Maldonado, J.E.; Fleischer, R.C.; Western, D.; Eggert, L.S. Fine-scale group structure and demography of African savanna elephants recolonizing lands outside protected areas. Divers. Distrib. 2012, 18, 952–961. [Google Scholar] [CrossRef]
- Wakeley, J. Nonequilibrium migration in human history. Genetics 1999, 153, 1863–1871. [Google Scholar]
- Chikhi, L.; Sousa, V.C.; Luisi, P.; Goossens, B.; Beaumont, M.A. The confounding effects of population structure, genetic diversity and the sampling scheme on the detection and quantification of population size changes. Genetics 2010, 186, 983–995. [Google Scholar] [CrossRef]
- Charlesworth, B. Fundamental concepts in genetics: Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 2009, 10, 195–205. [Google Scholar] [CrossRef]
- Nunney, L.; Elam, D.R. Estimating the effective population size of conserved populations. Conserv. Biol. 1994, 8, 175–184. [Google Scholar] [CrossRef]
- Leberg, P. Genetic approaches for estimating the effective size of populations. J. Wildl. Manag. 2005, 69, 1385–1399. [Google Scholar] [CrossRef]
- Nunney, L.; Campbell, K.A. Assessing minimum viable population size: Demography meets population genetics. Trends Ecol. Evol. 1993, 8, 6. [Google Scholar] [CrossRef]
- Palstra, F.P.; Ruzzante, D.E. Genetic estimates of contemporary effective population size: What can they tell us about the importance of genetic stochasticity for wild population persistence? Mol. Ecol. 2008, 17, 3428–3447. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S. Genetic estimates of contemporary effective population size: To what time periods do the estimates apply? Mol. Ecol. 2005, 14, 3335–3352. [Google Scholar] [CrossRef] [PubMed]
- Waples, R.S.; Do, C.; Chopelet, J. Calculating Ne and Ne/N in age-structured populations: A hybrid Felsenstein-Hill approach. Ecology 2011, 92, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Li, W.-H. Linkage disequilibrium in subdivided populations. Genetics 1973, 75, 213–219. [Google Scholar] [PubMed]
- Waples, R.S. Making sense of genetic estimates of effective population size. Mol. Ecol. 2016, 25, 4689–4691. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.X.; Helgason, A.; Masson, G.; Ebenesersdóttir, S.S.; Li, H.; Mallick, S.; Gnerre, S.; Patterson, N.; Kong, A.; Reich, D.; et al. A direct characterization of human mutation based on microsatellites. Nat. Genet. 2012, 44, 1161–1165. [Google Scholar] [CrossRef]
- Yang, J.; Jiang, Z. Genetic diversity, population genetic structure and demographic history of Przewalski’s gazelle (Procapra przewalskii): Implications for conservation. Conserv. Genet. 2011, 12, 1457–1468. [Google Scholar] [CrossRef]
- Waples, R.S. Spatial-temporal stratifications in natural populations and how they affect understanding and estimation of effective population size. Mol. Ecol. Resour. 2010, 10, 785–796. [Google Scholar] [CrossRef]
- Lehmann, T.; Hawley, W.A.; Grebert, H.; Collins, F.H. The effective population size of Anopheles gambiae in Kenya: Implications for population structure. Mol. Biol. Evol. 1998, 15, 264–276. [Google Scholar] [CrossRef]
- Beerli, P.; Felsenstein, J. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl. Acad. Sci. USA 2001, 98, 4563–4568. [Google Scholar] [CrossRef]
- Vitalis, R.; Couvet, D. Estimation of effective population size and migration rate from one- and two-locus identity measures. Genetics 2001, 157, 911–925. [Google Scholar] [PubMed]
- Franklin, I.R. The 50/500 rule is still valid—Reply to Frankham et al. Biol. Conserv. 2014, 176, 284–285. [Google Scholar] [CrossRef]
- Franklin, I.R.; Frankham, R. How large must populations be to retain evolutionary potential? Anim. Conserv. 1998, 1, 69–70. [Google Scholar] [CrossRef]
- Lynch, M.; Lande, R. The critical effective size for a genetically secure population. Anim. Conserv. 1998, 1, 70–72. [Google Scholar] [CrossRef]
- Frankham, R.; Bradshaw, C.J.A.; Brook, B.W. 50/500 rules need upward revision to 100/1000—Response to Franklin et al. Biol. Conserv. 2014, 176, 286. [Google Scholar] [CrossRef]
- Frankham, R.; Bradshaw, C.J.A.; Brook, B.W. Genetics in conservation management: Revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol. Conserv. 2014, 170, 56–63. [Google Scholar] [CrossRef]
- Rowe, G.; Beebee, T.J.C. Reconciling genetic and demographic estimators of effective population size in the anuran amphibian Bufo calamita. Conserv. Genet. 2004, 5, 287–298. [Google Scholar] [CrossRef]
- Nunney, L. Measuring the ratio of effective population size to adult numbers using genetic and ecological data. Evolution 1995, 49, 389. [Google Scholar] [CrossRef]
- Frankham, R. Effective population size/adult population size ratios in wildlife: A review. Genet. Res. 1995, 66, 95–107. [Google Scholar] [CrossRef]
- Gaggiotti, O.E. Genetic threats to population persistence. Ann. Zool. Fenn. 2003, 40, 155–168. [Google Scholar]
- Palstra, F.P.; Fraser, D.J. Effective/census population size ratio estimation: A compendium and appraisal. Ecol. Evol. 2012, 2, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Vucetich, J.A.; Waite, T.A.; Nunney, L. Fluctuating population size and the ratio of effective to census population size. Evolution 1997, 51, 2017–2021. [Google Scholar] [CrossRef] [PubMed]
- Waite, T.A.; Parker, P.G. Dimensionless life histories and effective population size. Conserv. Biol. 1996, 10, 1456–1462. [Google Scholar] [CrossRef]
- Waples, R.S.; Luikart, G.; Faulkner, J.R.; Tallmon, D.A. Simple life-history traits explain key effective population size ratios across diverse taxa. Proc. R. Soc. B Biol. Sci. 2013, 280, 20131339. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-H.; Janzen, F.J. Genetic effects of a persistent bottleneck on a natural population of ornate box turtles (Terrapene ornata). Conserv. Genet. 2004, 5, 425–437. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Waples, R.S. Relationship of effective to census size in fluctuating populations. Conserv. Biol. 2002, 16, 129–136. [Google Scholar] [CrossRef]
- Bishop, J.M.; Leslie, A.J.; Bourquin, S.L.; O’Ryan, C. Reduced effective population size in an overexploited population of the Nile crocodile (Crocodylus niloticus). Biol. Conserv. 2009, 142, 2335–2341. [Google Scholar] [CrossRef]
- Schwartz, M.; Luikart, G.; Waples, R. Genetic monitoring as a promising tool for conservation and management. Trends Ecol. Evol. 2007, 22, 25–33. [Google Scholar] [CrossRef]
- Nunziata, S.O.; Weisrock, D.W. Estimation of contemporary effective population size and population declines using RAD sequence data. Heredity 2018, 120, 196–207. [Google Scholar] [CrossRef]
- Cook, R.M.; Henley, M.D.; Parrini, F. Elephant movement patterns in relation to human inhabitants in and around the Great Limpopo Transfrontier Park. Koedoe 2015, 57, 1–7. [Google Scholar] [CrossRef]
- Roever, C.L.; van Aarde, R.J.; Leggett, K. Functional connectivity within conservation networks: Delineating corridors for African elephants. Biol. Conserv. 2013, 157, 128–135. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
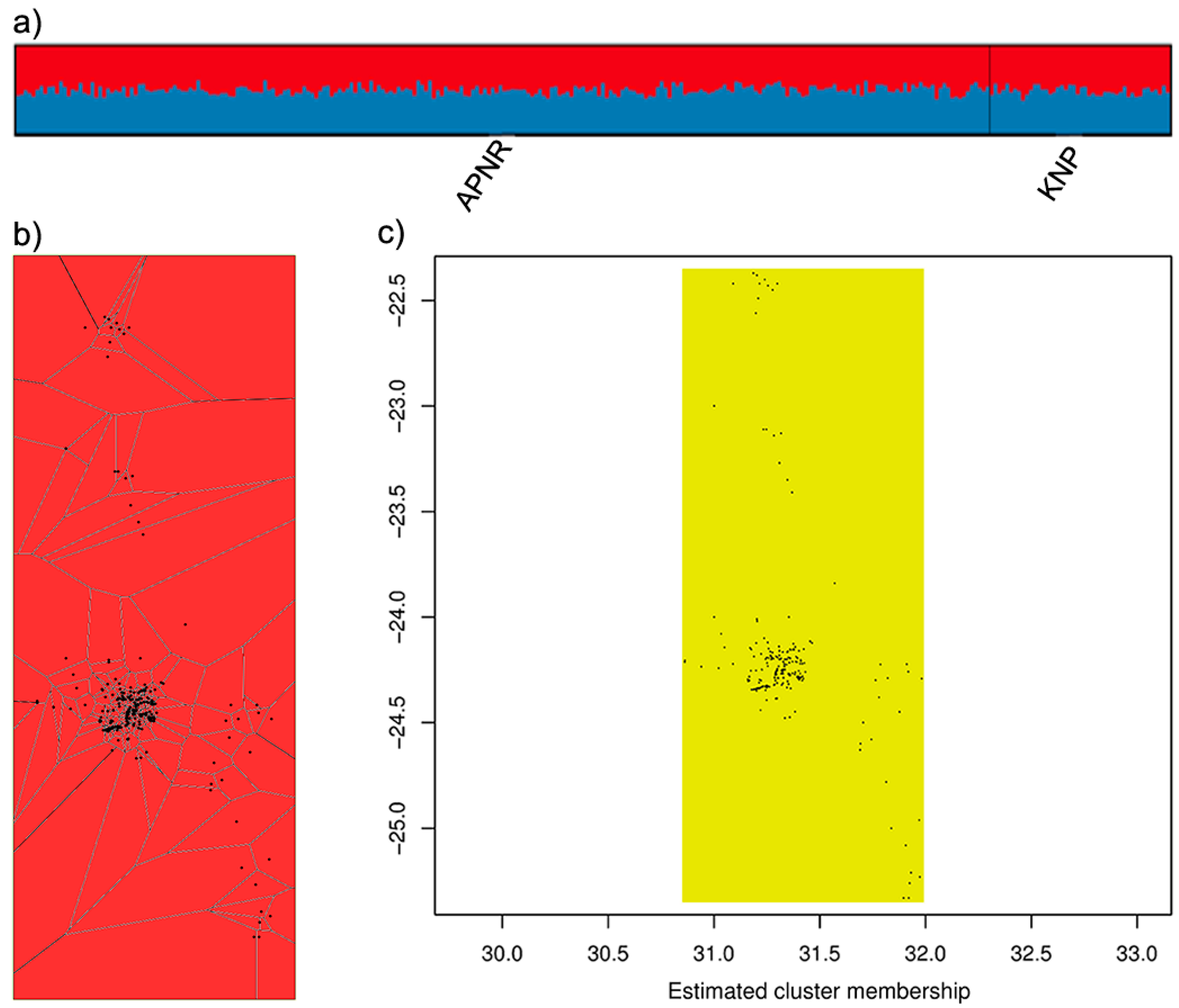
| APNR | KNP | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus | NA | He | Ho | FIS | AR | PAR | NA | He | Ho | FIS | AR | PAR |
| FH1 | 5 | 0.711 | 0.702 | 0.013 | 4.936 | 0.012 | 5 | 0.712 | 0.717 | −0.007 | 4.987 | 0.063 |
| FH19 | 8 | 0.724 | 0.673 | 0.070 | 6.796 | 0.077 | 8 | 0.759 | 0.739 | 0.026 | 7.877 | 1.157 |
| FH39 | 12 | 0.715 | 0.750 | −0.049 | 8.261 | 0.847 | 10 | 0.731 | 0.717 | 0.019 | 9.439 | 2.025 |
| FH40 | 6 | 0.512 | 0.548 | −0.070 | 4.673 | 0.676 | 4 | 0.557 | 0.533 | 0.042 | 4.000 | 0.003 |
| FH48 | 8 | 0.596 | 0.605 | −0.015 | 6.342 | 0.651 | 7 | 0.639 | 0.652 | −0.021 | 6.509 | 0.818 |
| FH60 | 4 | 0.427 | 0.444 | −0.039 | 3.113 | 0.153 | 3 | 0.369 | 0.348 | 0.058 | 2.988 | 0.027 |
| FH67 | 8 | 0.666 | 0.625 | 0.062 | 7.006 | 2.035 | 5 | 0.646 | 0.605 | 0.064 | 4.999 | 0.028 |
| FH71 | 3 | 0.564 | 0.500 | 0.114 | 2.998 | 0.000 | 3 | 0.525 | 0.478 | 0.089 | 3.000 | 0.002 |
| FH94 | 6 | 0.700 | 0.671 | 0.042 | 5.034 | 1.034 | 4 | 0.564 | 0.630 | −0.120 | 4.000 | 0.000 |
| FH103 | 5 | 0.353 | 0.339 | 0.040 | 4.896 | 0.240 | 5 | 0.328 | 0.370 | −0.127 | 4.758 | 0.102 |
| LA5 | 7 | 0.570 | 0.581 | −0.018 | 4.729 | 0.803 | 5 | 0.555 | 0.565 | −0.019 | 4.521 | 0.595 |
| LA6 | 4 | 0.463 | 0.492 | −0.062 | 2.901 | 0.094 | 5 | 0.484 | 0.478 | 0.013 | 4.510 | 1.703 |
| LaT06 | 13 | 0.649 | 0.539 | 0.171 * | 8.590 | 2.715 | 7 | 0.448 | 0.324 | 0.279 | 6.937 | 1.062 |
| LaT08 | 14 | 0.845 | 0.863 | −0.022 | 11.188 | 0.849 | 13 | 0.861 | 0.870 | −0.010 | 12.148 | 1.809 |
| LaT13 | 11 | 0.800 | 0.742 | 0.073 | 7.520 | 0.933 | 8 | 0.753 | 0.761 | −0.010 | 7.411 | 0.824 |
| LaT18 | 10 | 0.775 | 0.781 | −0.008 | 7.182 | 1.586 | 6 | 0.795 | 0.486 | 0.393 * | 6.000 | 0.404 |
| LaT24 | 13 | 0.851 | 0.834 | 0.020 | 9.947 | 2.648 | 8 | 0.827 | 0.658 | 0.206 | 7.916 | 0.617 |
| LaT25 | 8 | 0.673 | 0.558 | 0.170 * | 6.458 | 0.647 | 7 | 0.764 | 0.783 | −0.025 | 6.746 | 0.935 |
| Overall | 8.06 | 0.644 ± 0.142 | 0.625 ± 0.140 | 0.030 * | 6.25 | 0.89 | 6.28 | 0.629 ± 0.158 | 0.595 ± 0.158 | 0.054 * | 6.04 | 0.68 |
| Even Sample Classes | Even Distance Classes (5 km) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
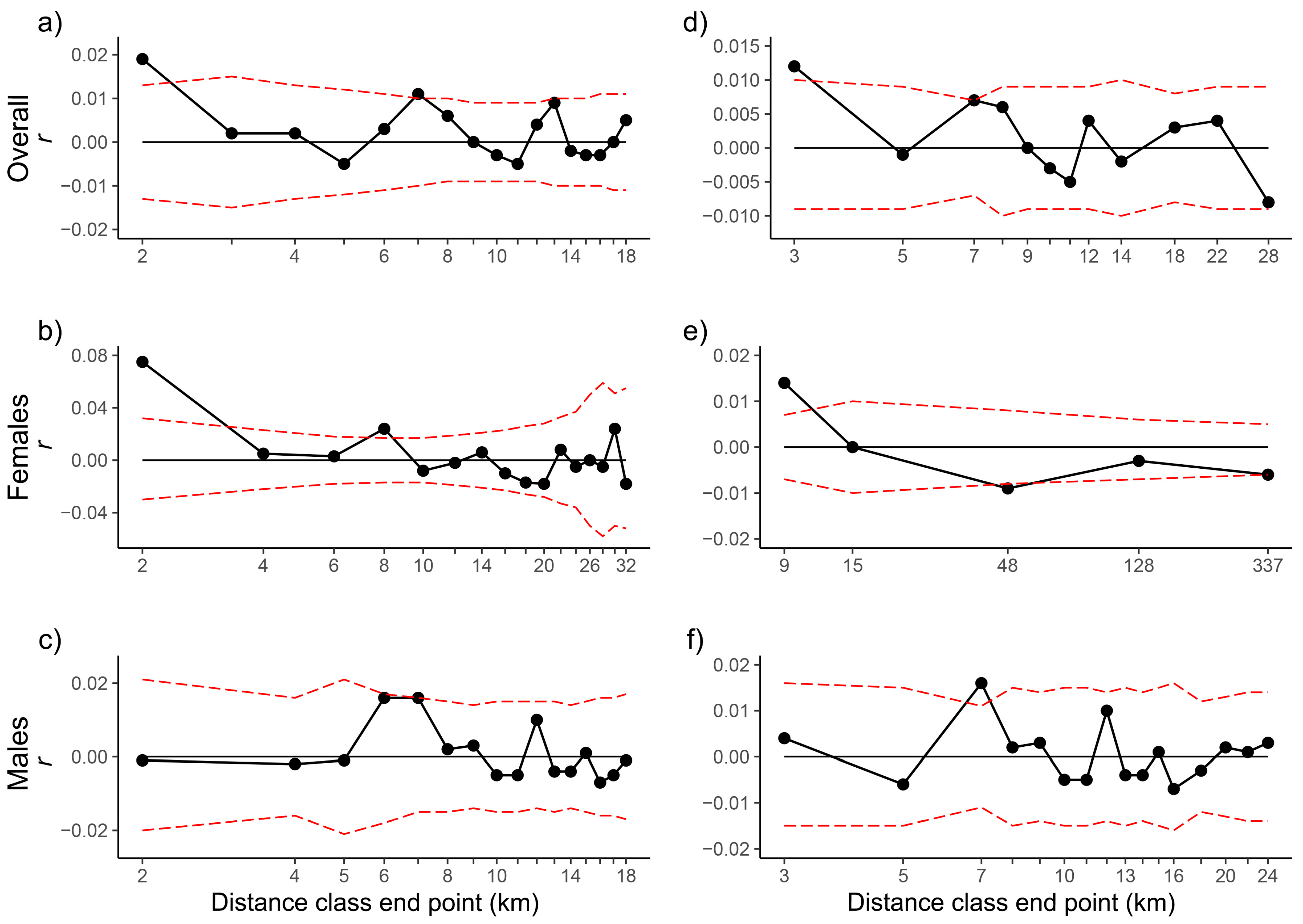
| D | r | U | L | Ur | Lr | P | ω | Pω | C | r | U | L | Ur | Lr | P | ω | Pω | |
| APNR + KNP adults | 2 | 0.019 | 0.013 | −0.013 | 0.034 | 0.004 | 0.003 | 188.700 | 0.000 | 1663 | 0.004 | 0.006 | −0.006 | 0.012 | −0.003 | 0.080 | 224.281 | 0.000 |
| 3 | 0.012 | 0.010 | −0.009 | 0.023 | 0.001 | 0.011 | 115.622 | 0.000 | - | - | - | - | - | - | - | - | - | |
| 4 | 0.008 | 0.008 | −0.007 | 0.017 | −0.001 | 0.020 | 74.778 | 0.000 | - | - | - | - | - | - | - | - | - | |
| APNR + KNP adult females | 3 | 0.040 | 0.024 | −0.021 | 0.068 | 0.013 | 0.001 | 125.857 | 0.000 | 320 | 0.024 | 0.015 | −0.013 | 0.042 | 0.007 | 0.001 | 263.182 | 0.000 |
| 9 | 0.014 | 0.007 | −0.007 | 0.025 | 0.003 | 0.000 | 37.493 | 0.000 | - | - | - | - | - | - | - | - | - | |
| APNR + KNP adult males | 2 | −0.001 | 0.020 | −0.019 | 0.022 | −0.023 | 0.533 | 180.299 | 0.000 | 604 | −0.001 | 0.011 | −0.010 | 0.011 | −0.013 | 0.583 | 212.809 | 0.000 |
| 3 | 0.004 | 0.016 | −0.015 | 0.022 | −0.013 | 0.290 | 100.978 | 0.002 | - | - | - | - | - | - | - | - | - | |
| APNR adults | 2 | 0.018 | 0.014 | −0.013 | 0.033 | 0.003 | 0.006 | 116.296 | 0.001 | 1639 | 0.004 | 0.006 | −0.006 | 0.011 | −0.003 | 0.122 | 81.399 | 0.002 |
| 3 | 0.011 | 0.010 | −0.009 | 0.022 | −0.001 | 0.023 | 75.154 | 0.001 | - | - | - | - | - | - | - | - | - | |
| APNR adult females | 3 | 0.038 | 0.023 | −0.021 | 0.065 | 0.011 | 0.001 | 58.721 | 0.000 | 316 | 0.023 | 0.014 | −0.013 | 0.040 | 0.005 | 0.002 | 40.909 | 0.004 |
| 8 | 0.017 | 0.008 | −0.007 | 0.029 | 0.005 | 0.000 | 25.058 | 0.001 | - | - | - | - | - | - | - | - | - | |
| 9 | 0.011 | 0.007 | −0.006 | 0.022 | 0.000 | 0.002 | 23.625 | 0.004 | - | - | - | - | - | - | - | - | - | |
| APNR adult males | 2 | 0.000 | 0.021 | −0.020 | 0.023 | −0.023 | 0.516 | 85.590 | 0.015 | 596 | −0.001 | 0.011 | −0.010 | 0.011 | −0.013 | 0.573 | 67.208 | 0.028 |
| 3 | 0.004 | 0.017 | −0.015 | 0.022 | −0.014 | 0.300 | 74.756 | 0.018 | - | - | - | - | - | - | - | - | - | |
| Overall | Adults | Adult Males | Adult Females | |
|---|---|---|---|---|
| Fst | 0.004 ± 0.002 * | 0.004 ± 0.003 * | −0.001 ± 0.003 | 0.014 ± 0.007 * |
| G’’st | 0.011 ± 0.007 * | 0.010 ± 0.007 * | −0.003 ± 0.007 | 0.039 ± 0.018 * |
| Jost’s Dest | 0.007 ± 0.004 * | 0.006 ± 0.004 * | −0.002 ± 0.005 | 0.025 ± 0.012 * |
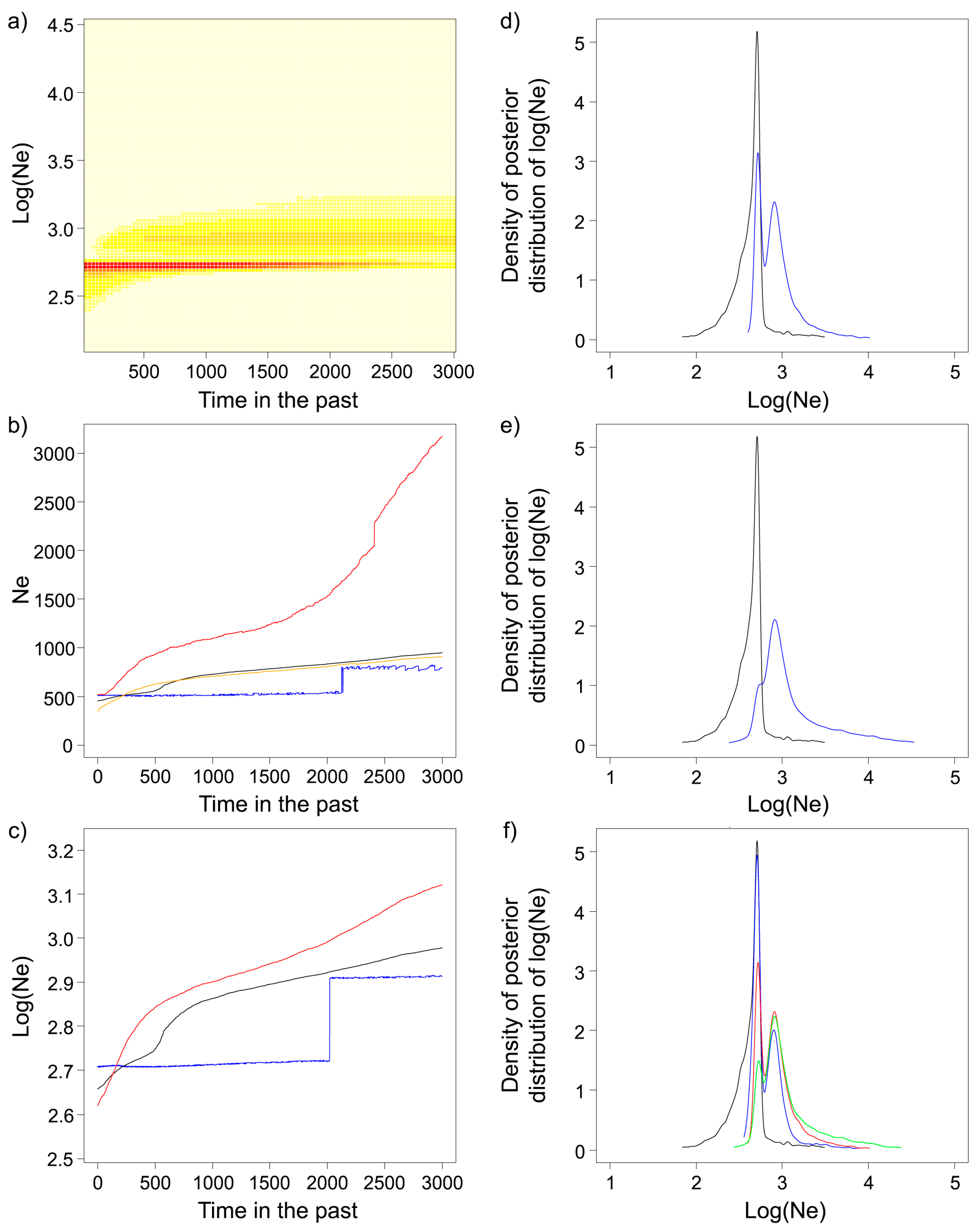
| APNR | APNR+KNP | |
|---|---|---|
| LD Method | 388.3 [245.2–809.3] | 510.2 [319.9–1091.4] |
| EPA | 197 [83–436] | 692 [109–743] |
| VarEff | ||
| Harmonic mean | 343.8 | 339.3 |
| Median | 454.7 | 443.5 |
| 90% central range | 160.9–970.9 | 157.5–920.8 |
| Migraine | 737 [590–915]; 7370 [5900–9150] | 723 [581–895]; 7230 [5810–8950] |
| ΘF | 618.5; 6185 | 614.7; 6147 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, T.L.; Fernandes, C.; Henley, M.D.; Dawson, D.A.; Mumby, H.S. Conservation Genetic Assessment of Savannah Elephants (Loxodonta africana) in the Greater Kruger Biosphere, South Africa. Genes 2019, 10, 779. https://doi.org/10.3390/genes10100779
Santos TL, Fernandes C, Henley MD, Dawson DA, Mumby HS. Conservation Genetic Assessment of Savannah Elephants (Loxodonta africana) in the Greater Kruger Biosphere, South Africa. Genes. 2019; 10(10):779. https://doi.org/10.3390/genes10100779
Chicago/Turabian StyleSantos, Teresa L., Carlos Fernandes, Michelle D. Henley, Deborah A. Dawson, and Hannah S. Mumby. 2019. "Conservation Genetic Assessment of Savannah Elephants (Loxodonta africana) in the Greater Kruger Biosphere, South Africa" Genes 10, no. 10: 779. https://doi.org/10.3390/genes10100779
APA StyleSantos, T. L., Fernandes, C., Henley, M. D., Dawson, D. A., & Mumby, H. S. (2019). Conservation Genetic Assessment of Savannah Elephants (Loxodonta africana) in the Greater Kruger Biosphere, South Africa. Genes, 10(10), 779. https://doi.org/10.3390/genes10100779