Common Nevus and Skin Cutaneous Melanoma: Prognostic Genes Identified by Gene Co-Expression Network Analysis

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Raw Data and Procession
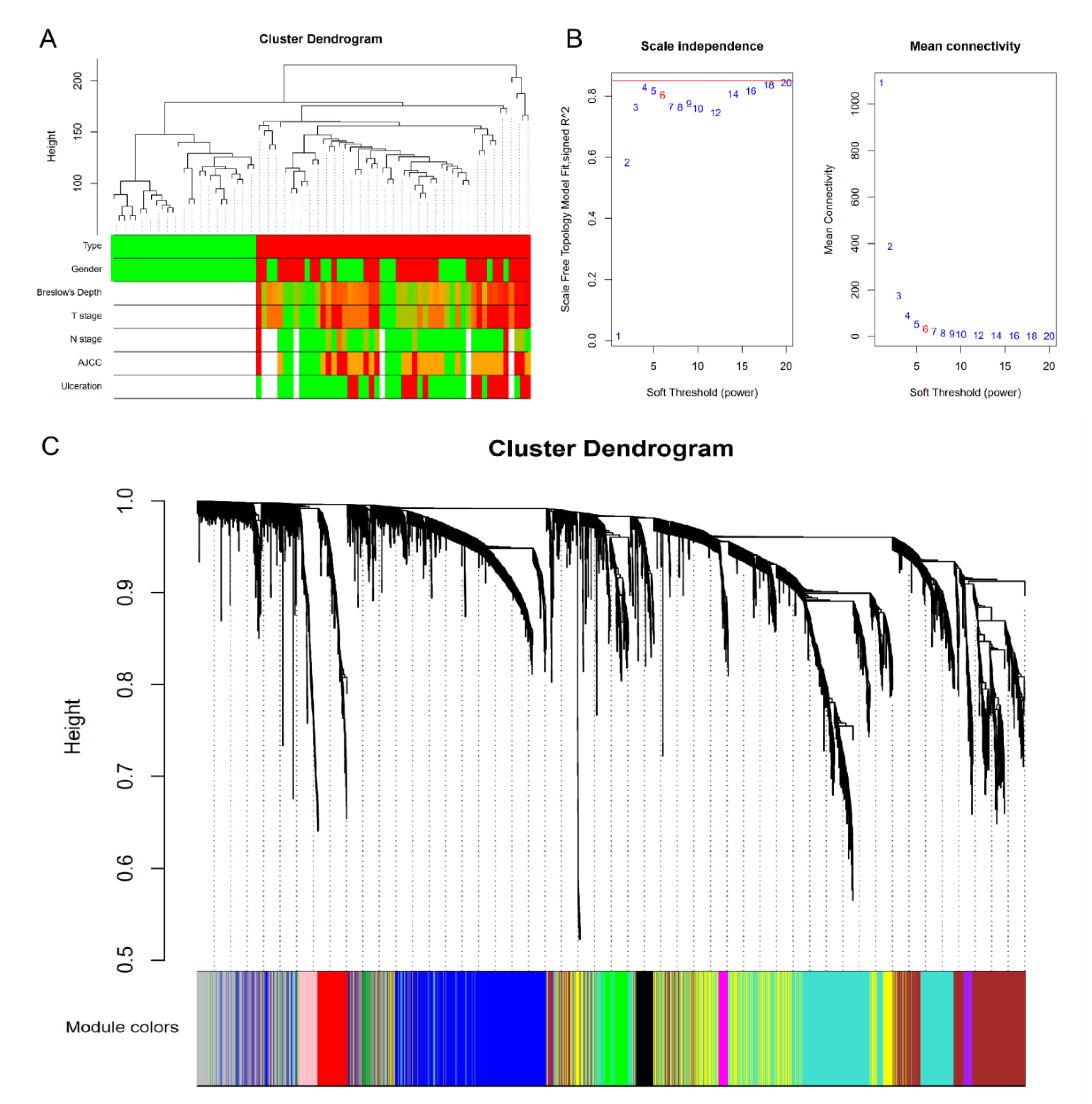
2.2. Gene Co-Expression Network and Modules
2.3. Hub Gene Identification and Validation
3. Results
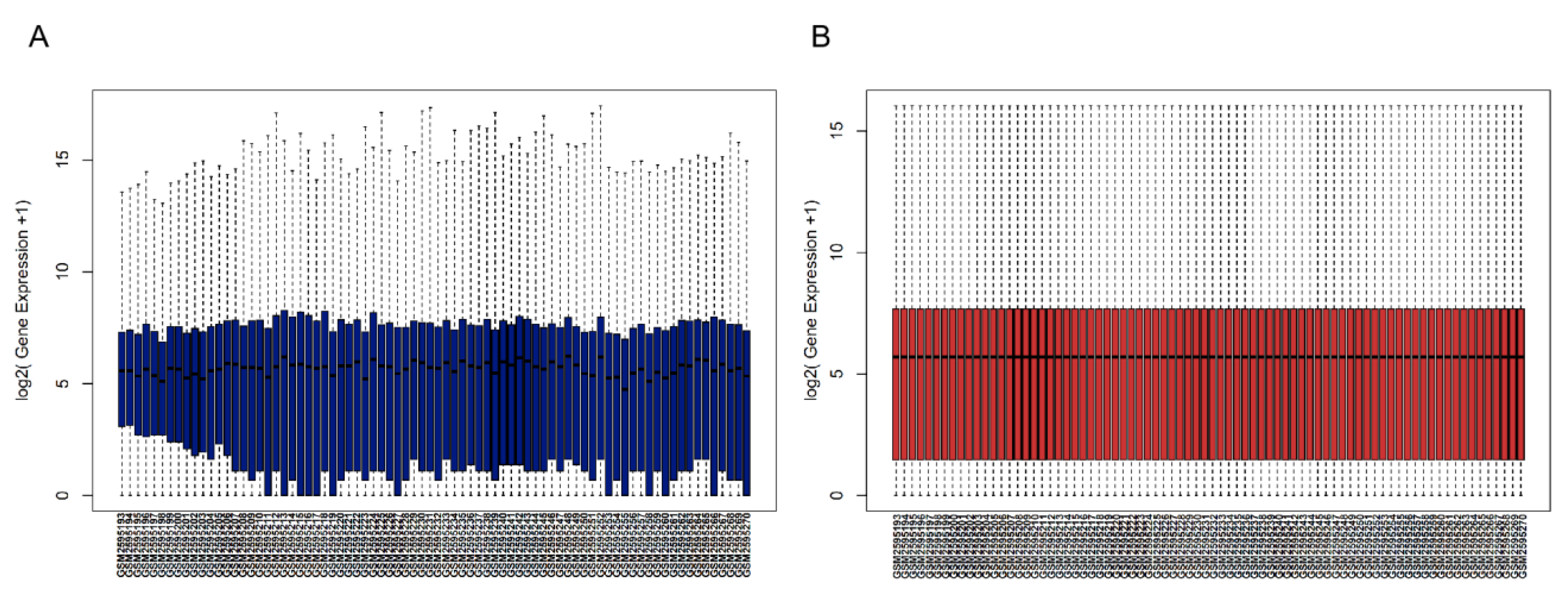
3.1. Gene Expression Data
3.2. Clinically Significant Modules
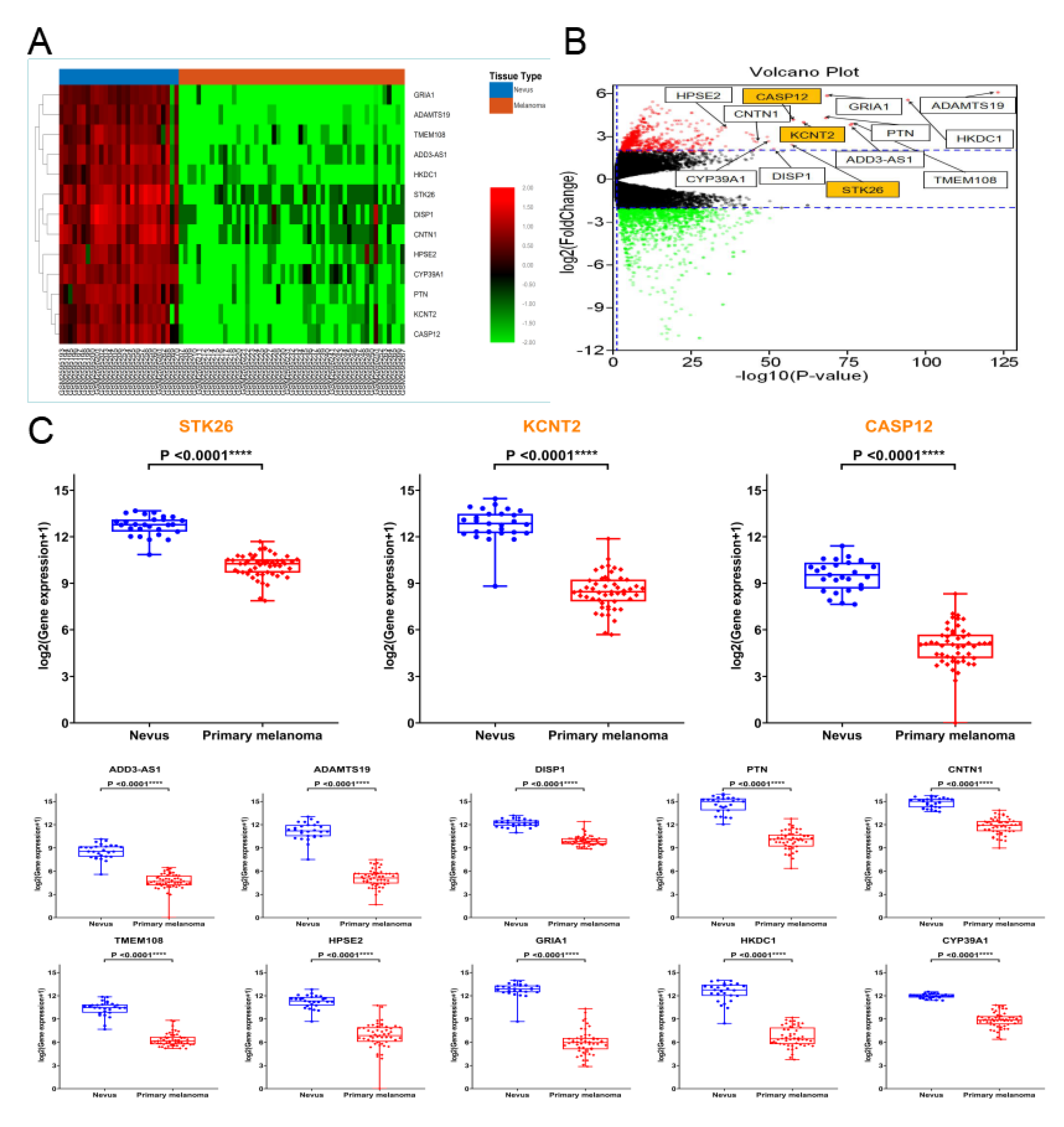
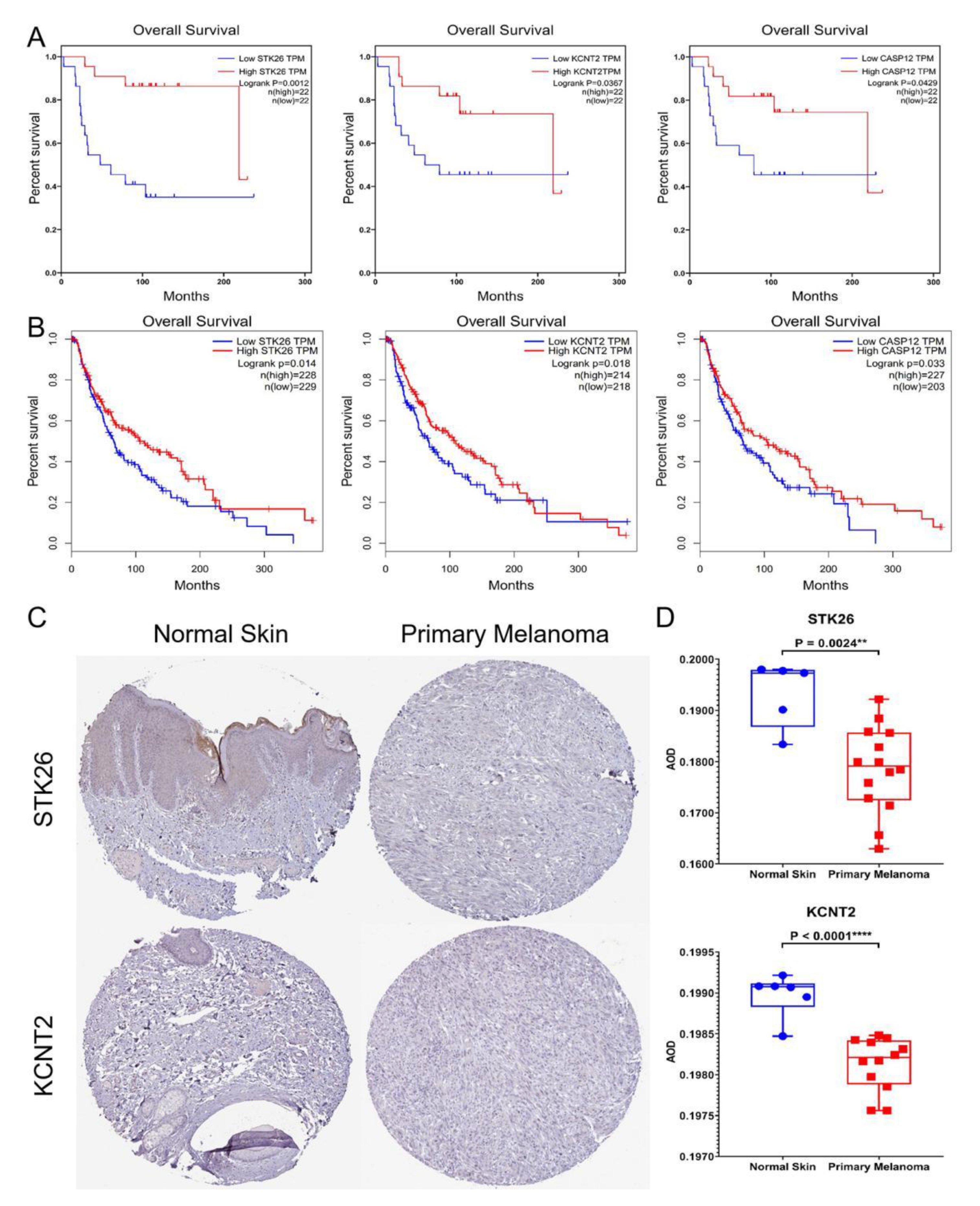
3.3. Hub Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Liu, W.; Gotlieb, V. The rapidly evolving therapies for advanced melanoma—Towards immunotherapy, molecular targeted therapy, and beyond. Crit. Rev. Oncol. Hematol. 2015, 99, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Ekwueme, D.U.; Guy, G.P.; Li, C.; Rim, S.H.; Parelkar, P.; Chen, S.C. The health burden and economic costs of cutaneous melanoma mortality by race/ethnicity—United States, 2000 to 2006. J. Am. Acad. Dermatol. 2011, 65, S131–CS133. [Google Scholar] [CrossRef] [PubMed]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Rigel, D.S.; Rivers, J.K.; Kopf, A.W.; Friedman, R.J.; Vinokur, A.F.; Heilman, E.R.; Levenstein, M. Dysplastic nevi. Markers for increased risk for melanoma. Cancer Am. Cancer Soc. 1989, 63, 386–389. [Google Scholar]
- Chen, S.T.; Geller, A.C.; Tsao, H. Update on the Epidemiology of Melanoma. Curr. Dermatol. Rep. 2013, 2, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Gargalovic, P.S.; Imura, M.; Zhang, B.; Gharavi, N.M.; Clark, M.J.; Pagnon, J.; Yang, W.; He, A.; Truong, A.; Patel, S.; et al. Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc. Nalt. Acad. Sci. USA 2006, 103, 12741–12746. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zhang, B.; Carlson, M.; Lu, K.V.; Zhu, S.; Felciano, R.M.; Laurance, M.F.; Zhao, W.; Qi, S.; Chen, Z.; et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc. Nalt. Acad. Sci. USA 2006, 103, 17402–17407. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Kong, D.; Cui, Q.; Wang, K.; Zhang, D.; Gong, Y.; Wu, G. Prognostic genes of breast cancer identified by gene co-expression network analysis. Front. Oncol. 2018, 8, 374. [Google Scholar] [CrossRef]
- Shi, H.; Zhang, L.; Qu, Y.; Hou, L.; Wang, L.; Zheng, M. Prognostic genes of breast cancer revealed by gene co-expression network analysis. Oncol. Lett. 2017, 14, 4535–4542. [Google Scholar] [CrossRef][Green Version]
- Badal, B.; Solovyov, A.; Di Cecilia, S.; Chan, J.M.; Chang, L.; Iqbal, R.; Aydin, I.T.; Rajan, G.S.; Chen, C.; Abbate, F.; et al. Transcriptional dissection of melanoma identifies a high-risk subtype underlying TP53 family genes and epigenome deregulation. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B. Preprocess core: A collection of pre-processing functions. R package version 1.46.0. Available online: https://github.com/bmbolstad/preprocessCore (accessed on 2 May 2019).
- Fernandez-Banet, J.; Esposito, A.; Coffin, S.; Horvath, I.B.; Estrella, H.; Schefzick, S.; Deng, S.; Wang, K.; AChing, K.; Ding, Y.; et al. OASIS: Web-based platform for exploring cancer multi-omics data. Nat. Methods 2016, 13, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; He, Y.; Xia, R. TBtools, a toolkit for biologists integrating various biological data handling tools with a user-friendly interface. BioRxiv 2018. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucl. Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucl. Acids Res. 2017, 45, W98–CW102. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Spatz, A.; Robert, C. Cutaneous melanoma. Lancet 2014, 383, 816–827. [Google Scholar] [CrossRef]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Warf, M.B.; Flake, D.N.; Hartman, A.R.; Tahan, S.; Shea, C.R.; Gerami, P.; Messina, J.; Florell, S.R.; Wenstrup, R.J.; et al. Clinical validation of a gene expression signature that differentiates benign nevi from malignant melanoma. J. Cutan. Pathol. 2015, 42, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.R.; Zhang, B.; Fang, Z.; Mischel, P.S.; Horvath, S.; Nelson, S.F. Gene connectivity, function, and sequence conservation: Predictions from modular yeast co-expression networks. BMC Genomics 2006, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.A.; Oldham, M.C.; Geschwind, D.H. A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. J. Neurosci. 2008, 28, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhao, H.; Shan, J.; Long, F.; Chen, Y.; Chen, Y.; Zhang, Y.; Han, X.; Ma, D. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol. Biol. Cell 2007, 18, 1965–1978. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.L.; Chen, H.C.; Fang, H.I.; Robinson, D.; Kung, H.J.; Shih, H.M. MST4, a new Ste20-related kinase that mediates cell growth and transformation via modulating ERK pathway. Oncogene 2001, 20, 6559–6569. [Google Scholar] [CrossRef][Green Version]
- Mardakheh, F.K.; Self, A.; Marshall, C.J. RHO binding to FAM65A regulates Golgi reorientation during cell migration. J. Cell Sci. 2016, 129, 4466–4479. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, X.; Peng, S.; Nan, X.; Zhao, H. Differential expression of MST4, STK25 and PDCD10 between benign prostatic hyperplasia and prostate cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 8105–8111. [Google Scholar]
- Lin, Z.H.; Wang, L.; Zhang, J.B.; Liu, Y.; Li, X.Q.; Guo, L.; Zhang, B.; Zhu, W.W.; Ye, Q.H. MST4 promotes hepatocellular carcinoma epithelial-mesenchymal transition and metastasis via activation of the p-ERK pathway. Int. J. Oncol. 2014, 45, 629–640. [Google Scholar] [CrossRef]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell 2017, 32, 840–855. [Google Scholar] [CrossRef]
- Berens, E.B.; Gilardi, M.; Moretti, M.; Riegel, A.T.; Wellstein, A. Abstract 908: A role for MST4 in organelle organization and breast cancer cell vascular invasion. Cancer Res. 2017, 77, 908. [Google Scholar]
- Hayashi, M.; Novak, I. Molecular basis of potassium channels in pancreatic duct epithelial cells. Channels 2013, 7, 432–441. [Google Scholar] [CrossRef]
- Gururaj, S.; Palmer, E.E.; Sheehan, G.D.; Kandula, T.; Macintosh, R.; Ying, K.; Morris, P.; Tao, J.; Dias, K.; Zhu, Y.; et al. A de novo mutation in the sodium-activated potassium channel kcnt2 alters ion selectivity and causes epileptic encephalopathy. Cell Rep. 2017, 21, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, R.; Dilorenzo, S.; Lundin-Strom, K.B.; Olsson, L.; Biloglav, A.; Lilljebjorn, H.; Rissler, M.; Wahlberg, P.; Lundmark, A.; Castor, A.; et al. Mutation, methylation, and gene expression profiles in dup(1q)-positive pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia 2018, 32, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.L.C.S.; Massieu, L. Caspases and their role in inflammation and ischemic neuronal death. Focus on caspase-12. Apoptosis 2016, 21, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Dupaul-Chicoine, J.; Yeretssian, G.; Doiron, K.; Bergstrom, K.S.B.; McIntire, C.R.; LeBlanc, P.M.; Meunier, C.; Turbide, C.; Gros, P.; Beauchemin, N.; et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 2010, 32, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.K.; Hsu, C.C.; Huang, S.F.; Hsu, C.C.; Chow, S.E. Caspase 12 degrades IkappaBalpha protein and enhances MMP-9 expression in human nasopharyngeal carcinoma cell invasion. Oncotarget 2017, 8, 33515–33526. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chow, S.; Kao, C.; Liu, Y.A.; Cheng, M.; Yang, Y.; Huang, Y.; Hsu, C.; Wang, J. Resveratrol induced ER expansion and ER caspase-mediated apoptosis in human nasopharyngeal carcinoma cells. Apoptosis 2014, 19, 527–541. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes Symbol | Full Name | R | P-Value |
|---|---|---|---|
| ADAMTS19 | Disintegrin and Metalloprotease Domain (ADAM) Metallopeptidase with Thrombospondin Type 1 Motif 19 | 0.96 | 6.68 × 10−40 |
| KCNT2 | Potassium Sodium-Activated Channel Subfamily T Member 2 | 0.95 | 1.50 × 10−39 |
| CASP12 | Caspase 12 | 0.94 | 4.48 × 10−39 |
| ADD3-AS1 | Adducin 3 Antisense RNA 1 | 0.93 | 2.78 × 10−33 |
| DISP1 | Dispatched RND Transporter Family Member 1 | 0.93 | 1.57 × 10−32 |
| PTN | Pleiotrophin | 0.92 | 8.81 × 10−32 |
| CNTN1 | Contactin 1 | 0.92 | 1.94 × 10−31 |
| TMEM108 | Transmembrane Protein 108 | 0.92 | 1.61 × 10−30 |
| HPSE2 | Heparanase 2 | 0.92 | 2.14 × 10−30 |
| GRIA1 | Glutamate Ionotropic Receptor AMPA Type Subunit 1 | 0.91 | 6.86 × 10−30 |
| HKDC1 | Hexokinase Domain Containing 1 | 0.91 | 1.58 × 10−29 |
| STK26 | Serine/Threonine Kinase 26 | 0.91 | 1.36 × 10−28 |
| CYP39A1 | Cytochrome P450 Family 39 Subfamily A Member 1 | 0.90 | 3.26 × 10−28 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Xu, Y.; Yan, Y.; Luo, P.; Chen, S.; Zheng, B.; Yan, W.; Chen, Y.; Wang, C. Common Nevus and Skin Cutaneous Melanoma: Prognostic Genes Identified by Gene Co-Expression Network Analysis. Genes 2019, 10, 747. https://doi.org/10.3390/genes10100747
Yang L, Xu Y, Yan Y, Luo P, Chen S, Zheng B, Yan W, Chen Y, Wang C. Common Nevus and Skin Cutaneous Melanoma: Prognostic Genes Identified by Gene Co-Expression Network Analysis. Genes. 2019; 10(10):747. https://doi.org/10.3390/genes10100747
Chicago/Turabian StyleYang, Lingge, Yu Xu, Yan Yan, Peng Luo, Shiqi Chen, Biqiang Zheng, Wangjun Yan, Yong Chen, and Chunmeng Wang. 2019. "Common Nevus and Skin Cutaneous Melanoma: Prognostic Genes Identified by Gene Co-Expression Network Analysis" Genes 10, no. 10: 747. https://doi.org/10.3390/genes10100747
APA StyleYang, L., Xu, Y., Yan, Y., Luo, P., Chen, S., Zheng, B., Yan, W., Chen, Y., & Wang, C. (2019). Common Nevus and Skin Cutaneous Melanoma: Prognostic Genes Identified by Gene Co-Expression Network Analysis. Genes, 10(10), 747. https://doi.org/10.3390/genes10100747