1. Introduction
Genomic analyses such as comparative genomics, population genomics, and transcriptomics have advanced our knowledge on diverse biological fields [
1]. For example, genome-wide data have helped identify the genetic bases of morphological change or ecological adaptation for organisms in nature [
2,
3]. Such research may require reference genomes that are often unavailable for non-model species. Thus, the demands for de novo assembling genomes from short sequence reads have been increasing in many biological fields. However, sequence reads do not always have correct signals at all their bases due to errors in sequencing procedures or base calling [
4]; low quality bases might prevent correct genome assembly. Removing low quality bases from sequence reads (i.e., read trimming) is often assumed as necessary for de novo genome assembly [
5]. Therefore, one usually executes the trimming step without evaluating its costs and benefits on genome assembly.
Different trimming strategies might lead to different genome assembly results and thus should be carefully examined. Trimming low quality bases may not only lower base error rates, but also reduce read numbers and lengths, which could increase (or decrease) assembled genome contiguity and completeness [
6,
7]. In addition, read trimming could potentially reduce computational time [
6]. As sequencing costs have decreased dramatically over the past decade [
8,
9], budgeting time for genomic analyses has become an increasingly important factor, especially for research teams with limited computational resources. Evaluation based on empirical data to explore different trimming strategies is often needed as the assembled genome quality and computation time may vary among taxa and assembly approaches. In addition, researchers should take into account the applications of assembled genomes to determine the optimal trimming strategy for genome assembly.
The rufous-capped babbler (
Cyanoderma ruficeps Blyth) is a widespread passerine in East Asia [
10]. This bird occurs in diverse bioclimatic zones and thus provides a good system to examine the genetic mechanism underlying avian adaptation to different climatic niches. Comparing genomic sequences between populations from different bioclimatic zones will facilitate the identification of candidate genes for local adaptation, and de novo assembling a reference genome is the first step to this end. In this study, we used the rufous-capped babbler as an empirical example to evaluate how different trimming strategies affect de novo genome assembly. Two assembly approaches were used to examine the effects of three trimming strategies on assembled genome quality and computational time. We used assembly summary statistics, gene prediction results, and chromosome-level genome alignments to the chicken (
Gallus gallus L.), zebra finch (
Taeniopygia guttata Vieillot), and collared flycatcher (
Ficedula albicollis Temminck) to access the quality of the assembled genomes. We also discussed the trade-offs among the trimming strategies based on the applications of the assembled genomes. We expect that the rufous-capped babbler genome will offer a good research system to study the ecological adaptation of passerines in East Asia.
4. Discussion
Trimming low quality bases from sequence reads is considered not only as the first step to improve genome assembly quality, but also a possible way to determine the computational time [
6]. As the cost of genomic sequencing decreases, one of the major concerns remaining for genomic projects is the amount of computational time needed to obtain reliable results. In this study, we examined how different trimming strategies affected the efficiency of assembling the rufous-capped babbler genome.
Researchers often need to consider three factors when assembling a genome: (1) sequence read quality, (2) assembled genome quality, and (3) computational time for assembly. These three factors could be highly correlated. Lower quality reads may result in less accurate and less complete assembled genomes and longer computational times. Thus, removing low quality bases from sequence reads may seem to be the best strategy. However, our results showed an inconsistent relationship between the three factors when using different assembly approaches (i.e., PLATANUS and DISCOVARdenovo + SOAPdenovo2), except that the lengths of the scaffolds (i.e., N50) were always longer for lower quality, untrimmed reads (i.e., the raw PE dataset) than higher quality, trimmed ones (i.e., the trimmed PE and cut off PE datasets). Among the three PLATANUS-assembled genomes, the genome assembled from the trimmed PE dataset had a medium scaffold N50 value and took a medium amount of computational time, but yielded the fewest Ns and gaps (
Table 1). In contrast, the trimmed PE genome had a medium scaffold N50 value and medium numbers of Ns and gaps, but its computation time was the longest of the three DISCOVAR+SOAPdenovo2 assembled genomes (
Table 2).
The trade-offs between computational time and genome quality (e.g., genome contiguity and assembly completeness) should be considered based on the goals of the project and assembly approach. In this study, the two assembly approaches took relatively different amounts of time among the three datasets. However, given that the PLATANUS-assembled genomes yielded much (7–10 times) larger scaffold N50 values (i.e., better genome contiguity) than the DISCOVARdenovo+SOAPdenovo2 ones, we discuss the effect of trimming strategies on assembly quality based only on the former. A previous study suggests that the effect of read trimming on genome assembly might be detrimental as more aggressive trimming might result in lower scaffold N50 values and less accurate assemble rates, although they did not conduct gene prediction for the assembled genomes [
6]. Surprisingly, we found few differences in the numbers and completeness of predicted genes and transcripts among the three PLATANUS-assembled genomes, indicating similar levels of assembly (gene prediction) completeness. That is, read-trimming had little impact on assembly completeness for coding genes.
We also found that the computational times increased 292%–383% while the N50 values only increased 8%–27% when assembling untrimmed reads compared with trimmed reads. Thus, if reference genome contiguity is not critical for focal studies such as those that aim to conduct SNP calling or RNAseq mapping, then one should at least perform a gentle trimming step to save a considerable amount of computational time, especially when using PLATANUS, as this does not appear to decrease assembly completeness. For studies focusing on genomic structural variation, one might consider assembling reference genomes based on untrimmed reads to increase genome contiguity; nevertheless, the computational time needed to assemble untrimmed reads might be too long and not affordable for every team.
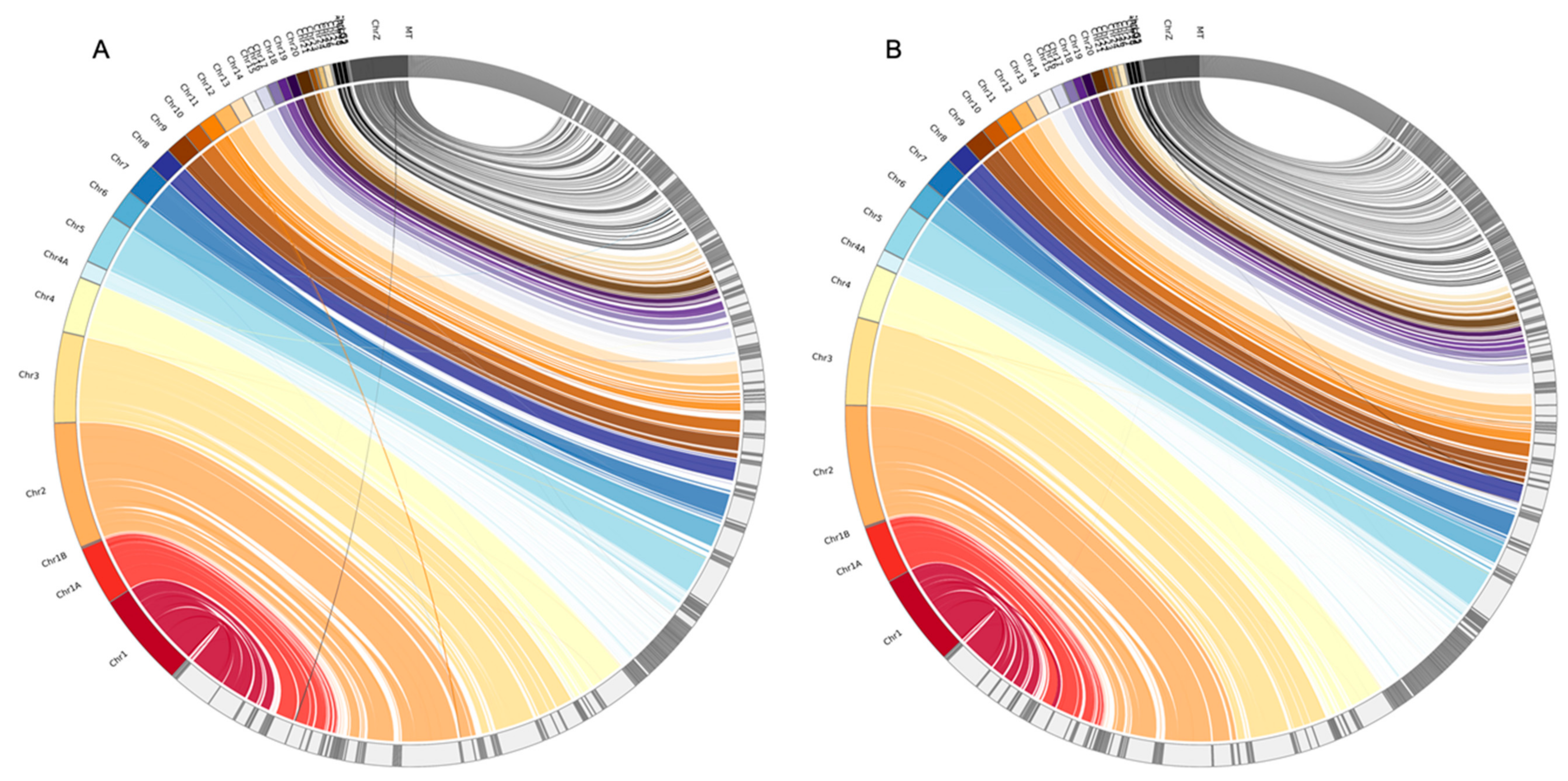
Longer assembled scaffolds, however, do not always guarantee a more correct assembly. Although the PLATANUS-assembled genome based on the raw PE dataset with the longest scaffolds (scaffold N50 = 16.9 MB) revealed more cases of reversion than that based on the trimmed PE dataset (scaffold N50 = 15.6 MB), that based on the cut off PE dataset with the shortest scaffolds (scaffold N50 = 13.3 MB) also showed a higher reversion rate than that based on the trimmed PE dataset (
Figure S1). The results imply that assembled genomes with longer scaffolds do not guarantee more accurate estimates of genome structure changes, especially if mis-assembly such as incorrect joining between mis-oriented segments occurs [
34]. Raw reads might generate longer scaffolds with more mis-assemblies than trimmed reads because the former have higher levels of sequencing errors or adapter contamination than the latter. The read-trimming practice could reduce these error resources and thus lower the frequencies of mis-assemblies.
The scaffolds corresponding to the chromosome Z, W, and 4A were poorly assembled in all of the three read datasets. Interestingly, parts of chromosome 4A have been found translocated into the W and Z chromosomes, forming neo-sex chromosomes in Sylvioidea, an avian superfamily that includes babblers [
35,
36]. The complex genomic structure caused by the fusion history might make the neo-sex chromosomes difficult to assemble. In addition, a richness of repeats and the haploid nature of sex chromosomes may also increase assembly difficulties [
37,
38]. Overall, all of the PLATANUS-assembled genomes based on the three datasets with different trimming strategies performed similarly when being used to detect such genomic structure changes, while their computational times for assembly varied considerably.





{kind=link}
{kind=link}