Reference Genomes from Distantly Related Species Can Be Used for Discovery of Single Nucleotide Polymorphisms to Inform Conservation Management

 ,
,  , and
, and
Abstract
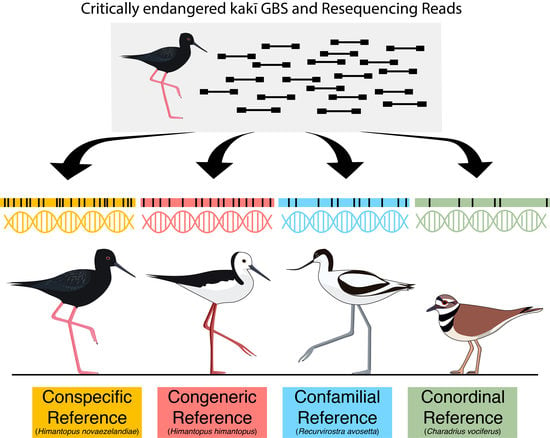
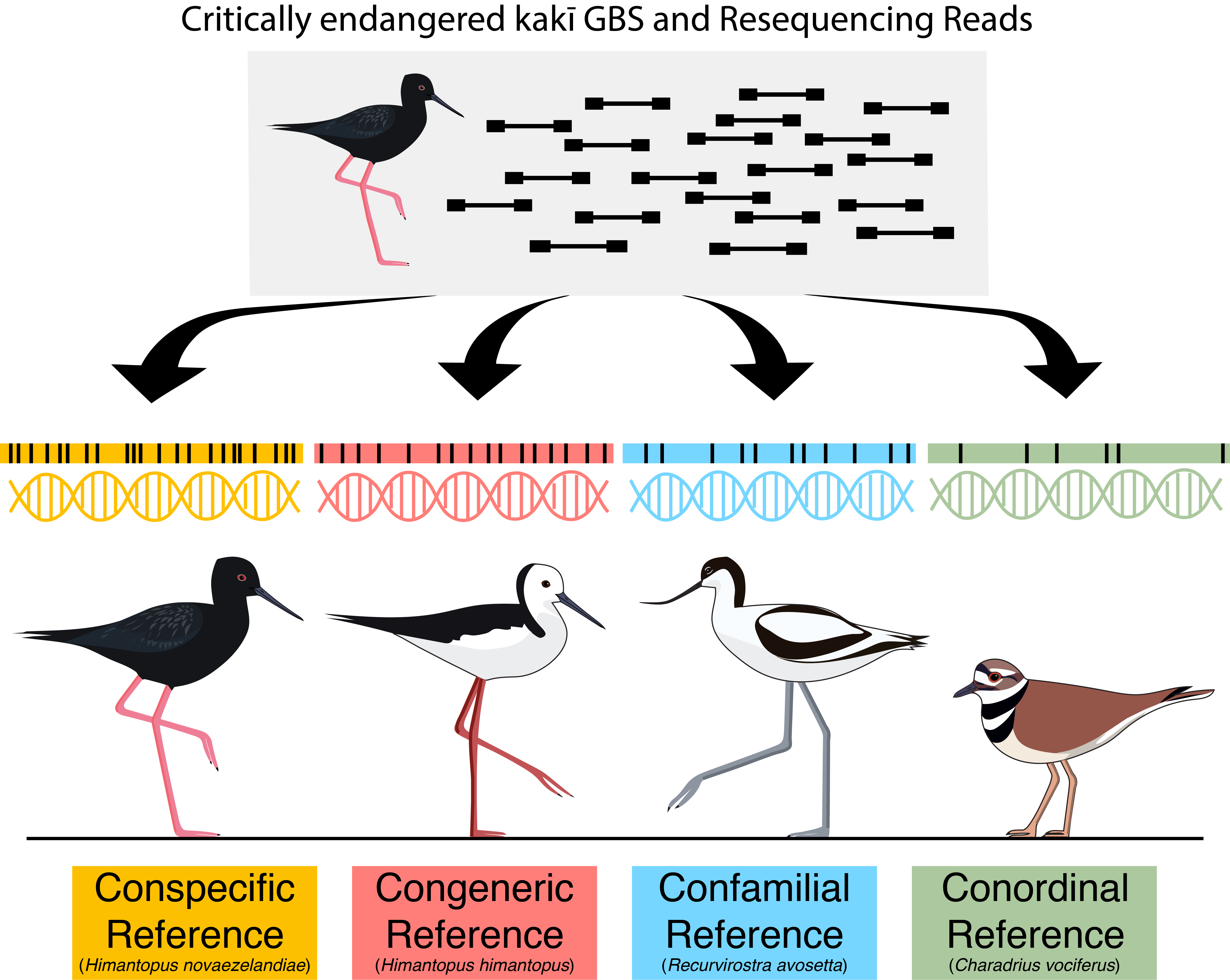
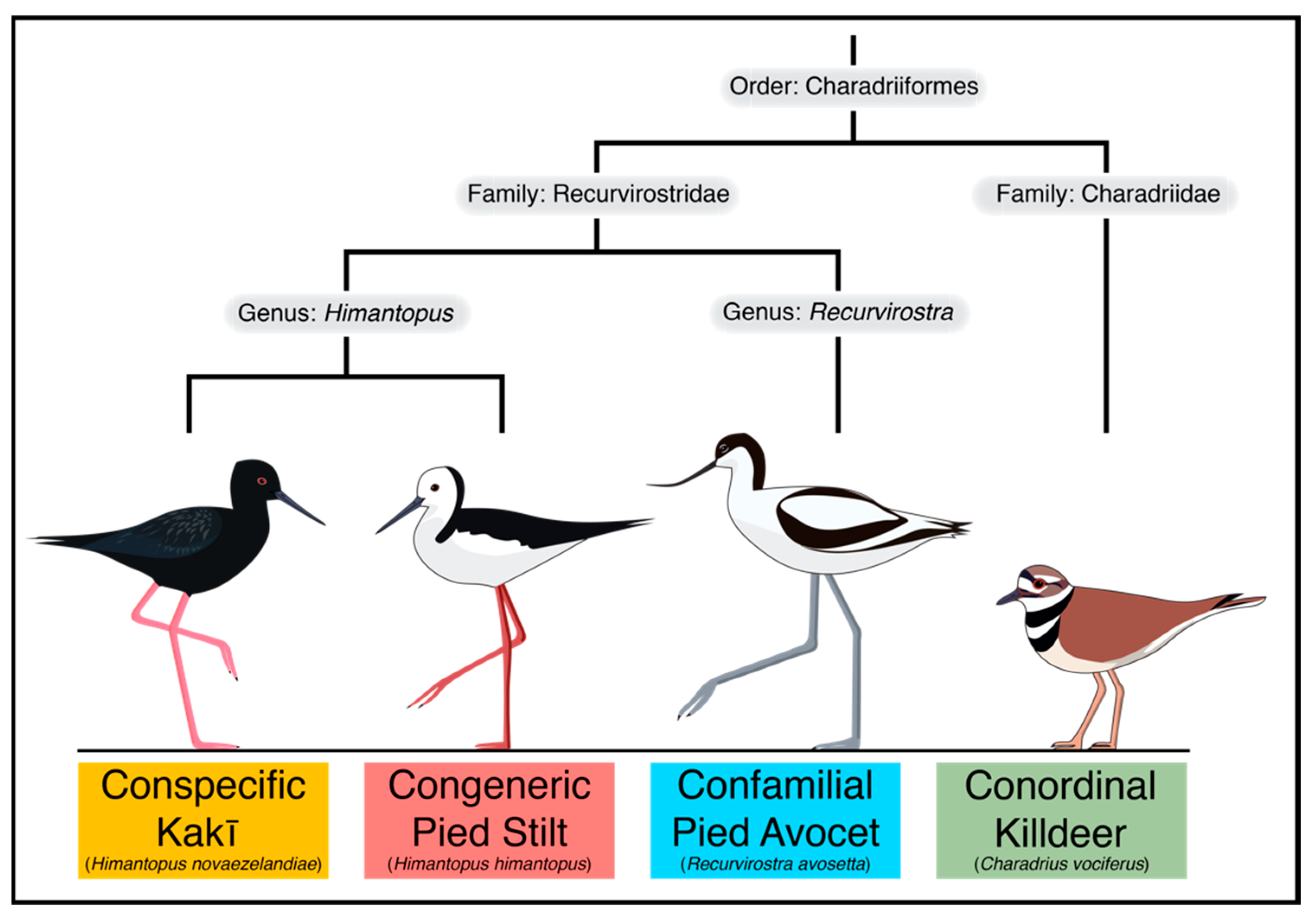
1. Introduction
2. Materials and Methods
2.1. Tissue Sampling and DNA Extractions
2.2. Reference Genome Library Preparation and Sequencing
2.3. Reference Genome Sequence Processing and Assembly
2.3.1. Kakī and Australian Pied Stilt
2.3.2. Pied Avocet
2.3.3. Killdeer
2.4. Genotyping-by-Sequencing
2.5. Resequencing
2.6. Diversity Estimates
3. Results
3.1. Reference Genome Sequencing and Assembly
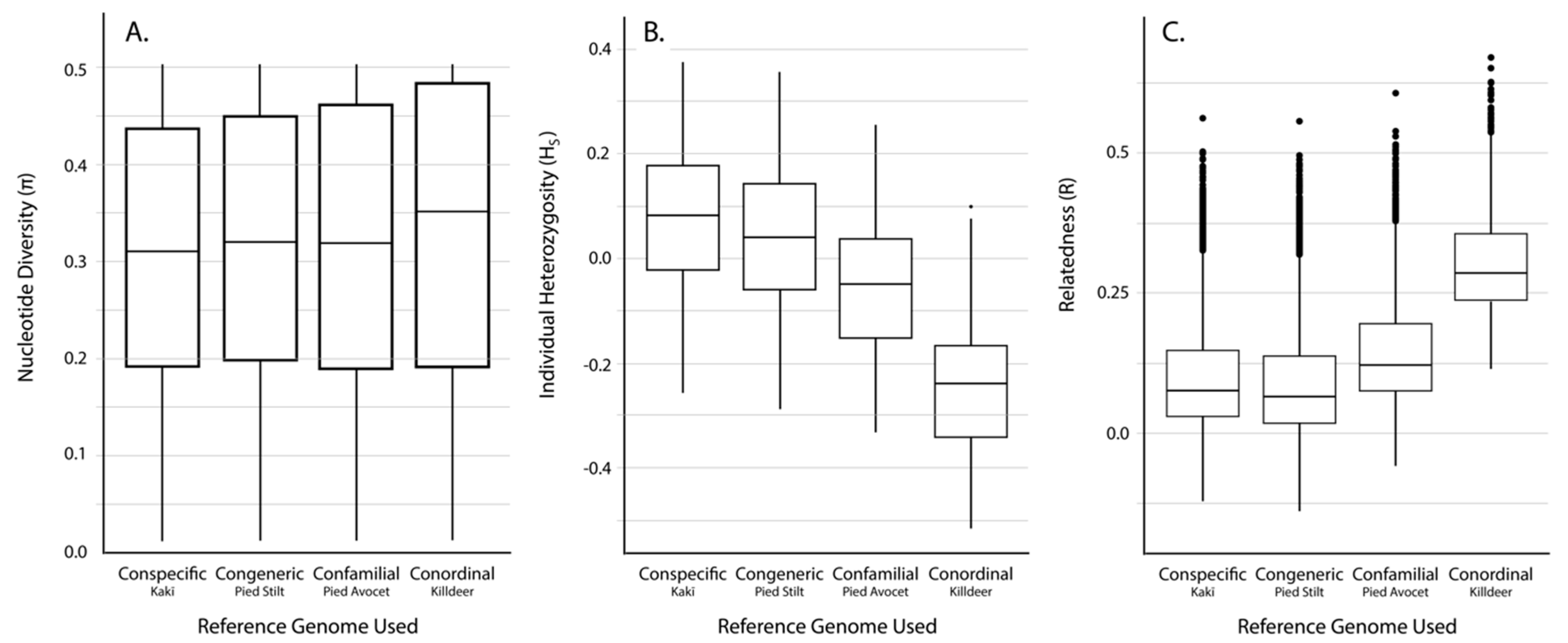
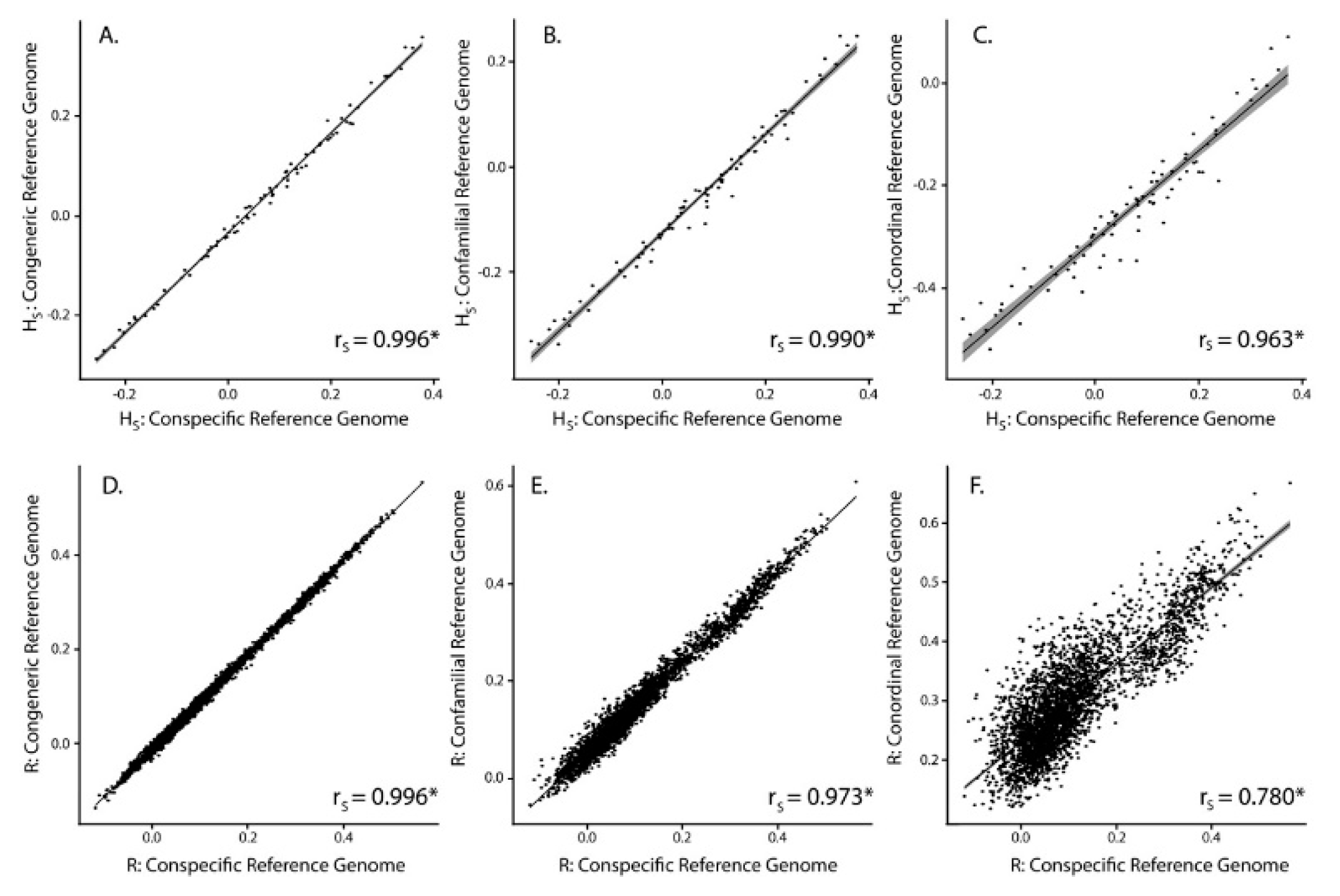
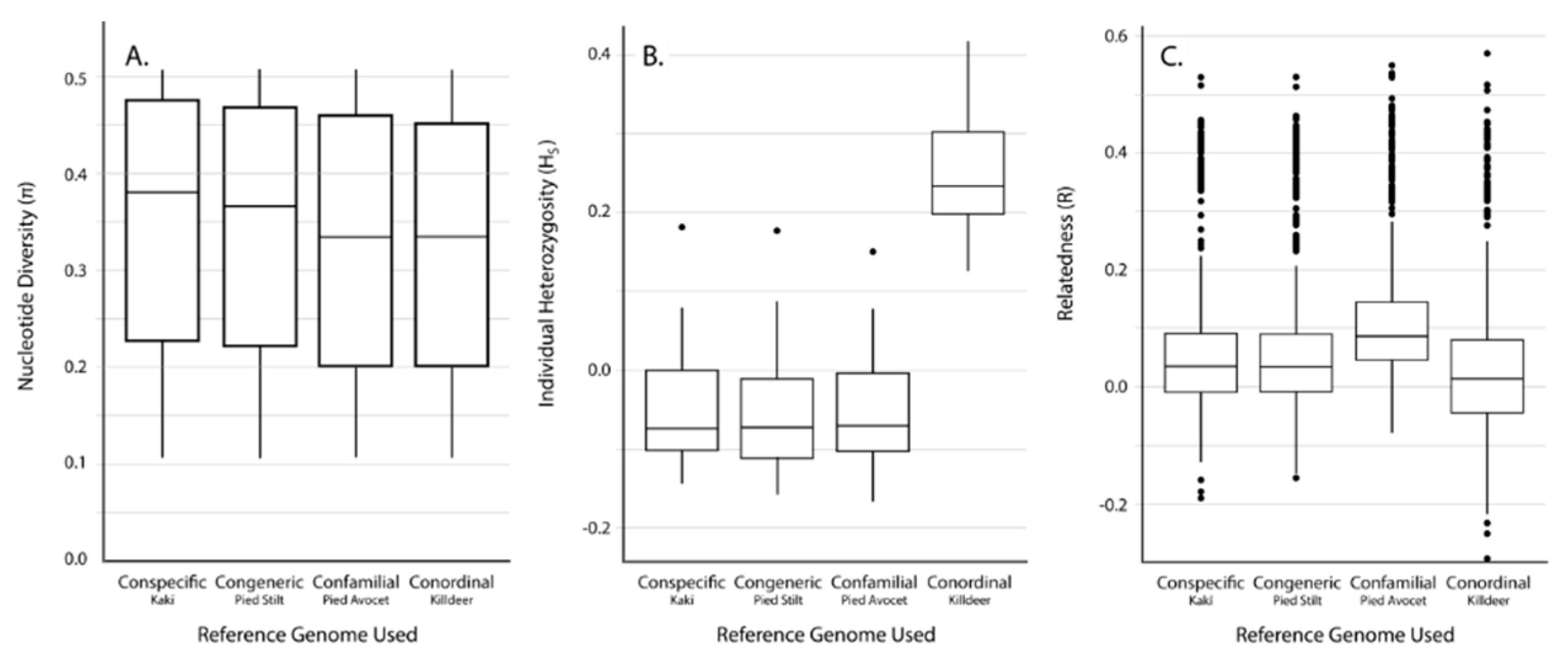
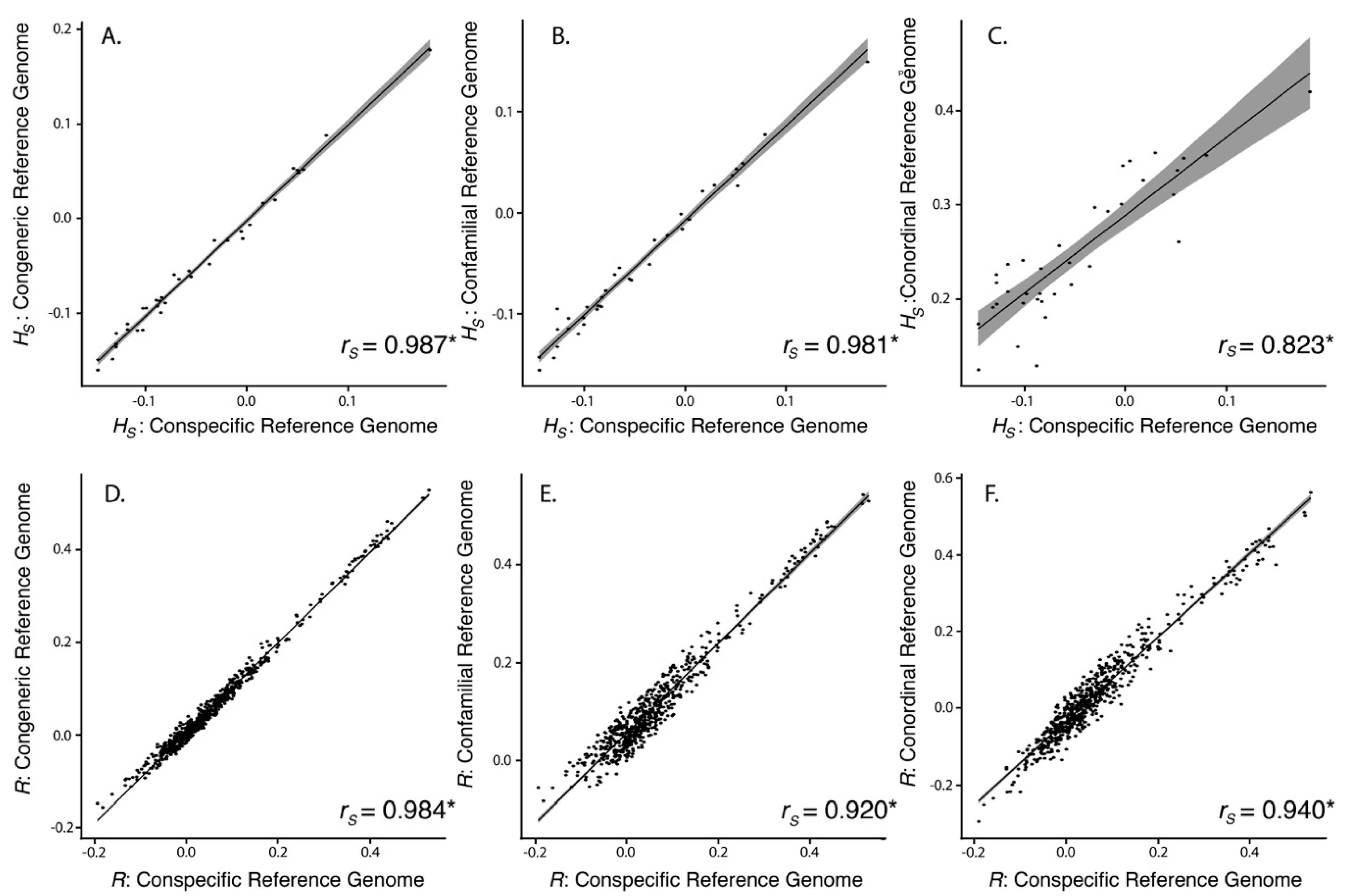
3.2. SNP Discovery and Diversity Estimates—GBS
3.3. SNP Discovery and Diversity Estimates—Resequencing
4. Discussion
5. Conclusions
Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allendorf, F.W.; Hohenlohe, P.A.; Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 2010, 11, 697. [Google Scholar] [CrossRef] [PubMed]
- Harrisson, K.A.; Pavlova, A.; Telonis-Scott, M.; Sunnucks, P. Using genomics to characterize evolutionary potential for conservation of wild populations. Evol. Appl. 2014, 7, 1008–1025. [Google Scholar] [CrossRef] [PubMed]
- Kohn, M.H.; Murphy, W.J.; Ostrander, E.A.; Wayne, R.K. Genomics and conservation genetics. Trends Ecol. Evol. 2006, 21, 629–637. [Google Scholar] [CrossRef]
- Mable, B.K. Conservation of adaptive potential and functional diversity: integrating old and new approaches. Conserv. Genet. 2018, 1–12. [Google Scholar] [CrossRef]
- Luikart, G.; England, P.R.; Tallmon, D.; Jordan, S.; Taberlet, P. The power and promise of population genomics: From genotyping to genome typing. Nat. Rev. Genet. 2003, 4, 981. [Google Scholar] [CrossRef] [PubMed]
- Galla, S.J.; Buckley, T.R.; Elshire, R.; Hale, M.L.; Knapp, M.; McCallum, J.; Moraga, R.; Santure, A.W.; Wilcox, P.; Steeves, T.E. Building strong relationships between conservation genetics and primary industry leads to mutually beneficial genomic advances. Mol. Ecol. 2016, 25, 5267–5281. [Google Scholar] [CrossRef] [PubMed]
- Shafer, A.B.; Wolf, J.B.; Alves, P.C.; Bergström, L.; Bruford, M.W.; Brännström, I.; Colling, G.; Dalén, L.; De Meester, L.; Ekblom, R.; et al. Genomics and the challenging translation into conservation practice. Trends Ecol. Evol. 2015, 30, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.T.; Cowling, R.M.; Rouget, M.; Balmford, A.; Lombard, A.T.; Campbell, B.M. Knowing but not doing: Selecting priority conservation areas and the research-implementation gap. Conserv. Biol. 2008, 22, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.R.; Dussex, N.; van Heezik, Y.J.G.E. Bridging the conservation genetics gap by identifying barriers to implementation for conservation practitioners. Glob. Ecol. Conserv. 2017, 10, 231–242. [Google Scholar] [CrossRef]
- McCormack, J.E.; Hird, S.M.; Zellmer, A.J.; Carstens, B.C.; Brumfield, R.T. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol. Phylogenetics Evol. 2013, 66, 526–538. [Google Scholar] [CrossRef]
- Hayden, E.C. The $1,000 genome. Nature 2014, 507, 294–295. [Google Scholar] [CrossRef] [PubMed]
- Muir, P.; Li, S.; Lou, S.; Wang, D.; Spakowicz, D.J.; Salichos, L.; Zhang, J.; Weinstock, G.M.; Isaacs, F.; Rozowsky, J.; et al. The real cost of sequencing: Scaling computation to keep pace with data generation. Genome Biol. 2016, 17, 53. [Google Scholar] [CrossRef] [PubMed]
- Narum, S.R.; Buerkle, C.A.; Davey, J.W.; Miller, M.R.; Hohenlohe, P.A. Genotyping-by-sequencing in ecological and conservation genomics. Mol. Ecol. 2013, 22, 2841–2847. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499. [Google Scholar] [CrossRef] [PubMed]
- Ilut, D.C.; Nydam, M.L.; Hare, M.P.J.B.R.I. Defining loci in restriction-based reduced representation genomic data from nonmodel species: Sources of bias and diagnostics for optimal clustering. Biomed Res. Int. 2014, 2014, 675158. [Google Scholar] [CrossRef] [PubMed]
- Oyler-McCance, S.J.; Oh, K.P.; Langin, K.M.; Aldridge, C.L. A field ornithologist’s guide to genomics: Practical considerations for ecology and conservation. Auk 2016, 133, 626–648. [Google Scholar] [CrossRef]
- Waldron, A.; Mooers, A.O.; Miller, D.C.; Nibbelink, N.; Redding, D.; Kuhn, T.S.; Roberts, J.T.; Gittleman, J.L. Targeting global conservation funding to limit immediate biodiversity declines. Proc. Natl. Acad. Sci. USA 2013, 110, 12144–12148. [Google Scholar] [CrossRef]
- Ellegren, H. Genome sequencing and population genomics in non-model organisms. Trends Ecol. Evol. 2014, 29, 51–63. [Google Scholar] [CrossRef]
- Genome 10K Community of Scientists. Genome 10K: A proposal to obtain whole-genome sequence for 10 000 vertebrate species. J. Hered. 2009, 100, 659–674. [Google Scholar] [CrossRef]
- Zhang, G.; Li, B.; Li, C.; Gilbert, M.T.P.; Jarvis, E.D.; Wang, J. Comparative genomic data of the Avian Phylogenomics Project. GigaScience 2014, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.E.; Hackett, K.J.; Purcell-Miramontes, M.; Brown, S.J.; Evans, J.D.; Goldsmith, M.R.; Lawson, D.; Okamuro, J.; Robertson, H.M.; Schneider, D.J. Creating a buzz about insect genomes. Science 2011, 331, 1386. [Google Scholar] [CrossRef] [PubMed]
- Matasci, N.; Hung, L.-H.; Yan, Z.; Carpenter, E.J.; Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Ayyampalayam, S.; Barker, M.; et al. Data access for the 1000 Plants (1KP) project. GigaScience 2014, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Duchêne, D.A.; Bragg, J.G.; Duchêne, S.; Neaves, L.E.; Potter, S.; Moritz, C.; Johnson, R.N.; Ho, S.Y.W.; Eldridge, M.D.B. Analysis of phylogenomic tree space resolves relationships among marsupial Families. Syst. Biol. 2017, 67, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Lewin, H.A.; Robinson, G.E.; Kress, W.J.; Baker, W.J.; Coddington, J.; Crandall, K.A.; Durbin, R.; Edwards, S.V.; Forest, F.; Gilbert, M.T.P.; et al. Earth BioGenome Project: Sequencing life for the future of life. Proc. Natl. Acad. Sci. USA 2018, 115, 4325–4333. [Google Scholar] [CrossRef] [PubMed]
- Card, D.C.; Schield, D.R.; Reyes-Velasco, J.; Fujita, M.K.; Andrew, A.L.; Oyler-McCance, S.J.; Fike, J.A.; Tomback, D.F.; Ruggiero, R.P.; Castoe, T.A. Two low coverage bird genomes and a comparison of reference-guided versus de novo genome assemblies. PLoS ONE 2014, 9, e106649. [Google Scholar] [CrossRef]
- Lischer, H.E.; Shimizu, K.K. Reference-guided de novo assembly approach improves genome reconstruction for related species. BMC Bioinform. 2017, 18, 474. [Google Scholar] [CrossRef]
- Der Sarkissian, C.; Ermini, L.; Schubert, M.; Yang, M.A.; Librado, P.; Fumagalli, M.; Jónsson, H.; Bar-Gal, G.K.; Albrechtsen, A.; Vieira, F.G.; et al. Evolutionary genomics and conservation of the endangered Przewalski’s horse. Curr. Biol. 2015, 25, 2577–2583. [Google Scholar] [CrossRef]
- Ng, E.Y.; Garg, K.M.; Low, G.W.; Chattopadhyay, B.; Oh, R.R.; Lee, J.G.; Rheindt, F.E. Conservation genomics identifies impact of trade in a threatened songbird. Biol. Conserv. 2017, 214, 101–108. [Google Scholar] [CrossRef]
- Nuijten, R.J.; Bosse, M.; Crooijmans, R.P.; Madsen, O.; Schaftenaar, W.; Ryder, O.A.; Groenen, M.A.; Megens, H.-J. The use of genomics in conservation management of the endangered visayan warty Pig (Sus cebifrons). Int. J. Genom. 2016, 2016, 5613862. [Google Scholar] [CrossRef]
- Westbury, M.V.; Hartmann, S.; Barlow, A.; Wiesel, I.; Leo, V.; Welch, R.; Parker, D.M.; Sicks, F.; Ludwig, A.; Dalén, L.; et al. Extended and Continuous Decline in Effective Population Size Results in Low Genomic Diversity in the World’s Rarest Hyena Species, the Brown Hyena. Mol. Biol. Evol. 2018, 35, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Organ, C.L.; Shedlock, A.M.; Meade, A.; Pagel, M.; Edwards, S.V. Origin of avian genome size and structure in non-avian dinosaurs. Nature 2007, 446, 180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Consortium, I.C.G.S. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695. [Google Scholar]
- Dalloul, R.A.; Long, J.A.; Zimin, A.V.; Aslam, L.; Beal, K.; Blomberg, L.A.; Bouffard, P.; Burt, D.W.; Crasta, O.; Crooijmans, R.P.; et al. Multi-platform next-generation sequencing of the domestic turkey (Meleagris gallopavo): Genome assembly and analysis. PLoS Biol. 2010, 8, e1000475. [Google Scholar] [CrossRef] [PubMed]
- Warren, W.C.; Clayton, D.F.; Ellegren, H.; Arnold, A.P.; Hillier, L.W.; Künstner, A.; Searle, S.; White, S.; Vilella, A.J.; Fairley, S.; et al. The genome of a songbird. Nature 2010, 464, 757. [Google Scholar] [CrossRef]
- Burga, A.; Wang, W.; Ben-David, E.; Wolf, P.C.; Ramey, A.M.; Verdugo, C.; Lyons, K.; Parker, P.G.; Kruglyak, L. A genetic signature of the evolution of loss of flight in the Galapagos cormorant. Science 2017, 356, eaal3345. [Google Scholar] [CrossRef] [PubMed]
- Callicrate, T.; Dikow, R.; Thomas, J.W.; Mullikin, J.C.; Jarvis, E.D.; Fleischer, R.C. Genomic resources for the endangered Hawaiian honeycreepers. BMC Genom. 2014, 15, 1098. [Google Scholar] [CrossRef]
- Sutton, J.; Helmkampf, M.; Steiner, C.; Bellinger, M.R.; Korlach, J.; Hall, R.; Baybayan, P.; Muehling, J.; Gu, J.; Kingan, S.; et al. A high-quality, long-read de novo genome assembly to aid conservation of Hawaii’s last remaining crow species. bioRxiv 2018, 2018, 349035. [Google Scholar]
- Strigops Habroptilus. 2018. Available online: https://vgp.github.io/genomeark/Strigops_habroptilus. (accessed on 7 November 2018).
- Peona, V.; Weissensteiner, M.H.; Suh, A. How complete are “complete” genome assemblies?—An avian perspective. Mol. Ecol. Resour. 2018, 18, 1188–1195. [Google Scholar] [CrossRef]
- Zhang, G. Genomics: Bird sequencing project takes off. Nature 2015, 522, 34. [Google Scholar] [CrossRef] [PubMed]
- Reed, C.E.M. Management Plan for Captive Black Stilts; Biodiversity Recovery Unit, Department of Conservation: Wellington, New Zealand, 1998.
- Sanders, M.D.; Maloney, R.F. Causes of mortality at nests of ground-nesting birds in the Upper Waitaki Basin, South Island, New Zealand: A 5-year video study. Biol. Conserv. 2002, 106, 225–236. [Google Scholar] [CrossRef]
- Hagen, E.N.; Hale, M.L.; Maloney, R.F.; Steeves, T.E. Conservation genetic management of a critically endangered New Zealand endemic bird: Minimizing inbreeding in the Black Stilt Himantopus novaezelandiae. Ibis 2011, 153, 556–561. [Google Scholar] [CrossRef]
- Ford, M.J.; Parsons, K.; Ward, E.; Hempelmann, J.; Emmons, C.K.; Bradley Hanson, M.; Balcomb, K.C.; Park, L.K. Inbreeding in an endangered killer whale population. Anim. Conserv. 2018. [Google Scholar] [CrossRef]
- Jordan, S.; Giersch, J.J.; Muhlfeld, C.C.; Hotaling, S.; Fanning, L.; Tappenbeck, T.H.; Luikart, G. Loss of genetic diversity and increased subdivision in an endemic alpine stonefly threatened by climate change. PLoS ONE 2016, 11, e0157386. [Google Scholar]
- Pacioni, C.; Hunt, H.; Allentoft, M.E.; Vaughan, T.G.; Wayne, A.F.; Baynes, A.; Haouchar, D.; Dortch, J.; Bunce, M. Genetic diversity loss in a biodiversity hotspot: Ancient DNA quantifies genetic decline and former connectivity in a critically endangered marsupial. Mol. Ecol. 2015, 24, 5813–5828. [Google Scholar] [CrossRef]
- O’Grady, J.J.; Brook, B.W.; Reed, D.H.; Ballou, J.D.; Tonkyn, D.W.; Frankham, R. Realistic levels of inbreeding depression strongly affect extinction risk in wild populations. Biol. Conserv. 2006, 133, 42–51. [Google Scholar] [CrossRef]
- Spielman, D.; Brook, B.W.; Frankham, R. Most species are not driven to extinction before genetic factors impact them. Proc. Natl. Acad. Sci. USA 2004, 101, 15261–15264. [Google Scholar] [CrossRef]
- Baker, A.J.; Pereira, S.L.; Paton, T.A. Phylogenetic relationships and divergence times of Charadriiformes genera: Multigene evidence for the Cretaceous origin of at least 14 clades of shorebirds. Biol. Lett. 2007, 3, 205–210. [Google Scholar] [CrossRef]
- Wallis, G. Genetic Status of New Zealand Black Stilt (Himantopus novaezelandiae) and Impact of Hybridisation; Department of Conservation Wellington: Wellington, New Zealand, 1999.
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 7 November 2018).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Smeds, L.; Künstner, A. ConDeTri-a content dependent read trimmer for Illumina data. PLoS ONE 2011, 6, e26314. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T. Exploring genome characteristics and sequence quality without a reference. Bioinformatics 2014, 30, 1228–1235. [Google Scholar] [CrossRef]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2013, 30, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Bradnam, K.R.; Fass, J.N.; Alexandrov, A.; Baranay, P.; Bechner, M.; Birol, I.; Boisvert, S.; Chapman, J.A.; Chapuis, G.; Chikhi, R.; et al. Assemblathon 2: Evaluating de novo methods of genome assembly in three vertebrate species. GigaScience 2013, 2, 10. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2017, 35, 543–548. [Google Scholar] [CrossRef]
- Zdobnov, E.M.; Tegenfeldt, F.; Kuznetsov, D.; Waterhouse, R.M.; Simao, F.A.; Ioannidis, P.; Seppey, M.; Loetscher, A.; Kriventseva, E.V. OrthoDB v9. 1: Cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 2016, 45, D744–D749. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinf. 2014, 15, 182. [Google Scholar] [CrossRef] [PubMed]
- Crusoe, M.R.; Alameldin, H.F.; Awad, S.; Boucher, E.; Caldwell, A.; Cartwright, R.; Charbonneau, A.; Constantinides, B.; Edvenson, G.; Fay, S.; et al. The khmer software package: Enabling efficient nucleotide sequence analysis. F1000Research 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Goltsman, E.; Ho, I.; Rokhsar, D. Meraculous-2D: Haplotype-sensitive Assembly of Highly Heterozygous genomes. arXiv, 2017; arXiv:1703.09852. [Google Scholar]
- Chapman, J.A.; Ho, I.Y.; Goltsman, E.; Rokhsar, D.S. Meraculous2: Fast accurate short-read assembly of large polymorphic genomes. arXiv, 2016; arXiv:1608.01031. [Google Scholar]
- Kielbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef]
- Tamazian, G.; Dobrynin, P.; Krasheninnikova, K.; Komissarov, A.; Koepfli, K.-P.; O’brien, S.J. Chromosomer: A reference-based genome arrangement tool for producing draft chromosome sequences. GigaScience 2016, 5, 38. [Google Scholar] [CrossRef]
- GigaDB. Genomic Data of the Killdeer (Charadrius Vociferus). 2014. Available online: http://dx.doi.org/10.5524/101007 (accessed on 7 November 2018).
- Gnerre, S.; MacCallum, I.; Przybylski, D.; Ribeiro, F.J.; Burton, J.N.; Walker, B.J.; Sharpe, T.; Hall, G.; Shea, T.P.; Sykes, S.; et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA 2011, 108, 1513–1518. [Google Scholar] [CrossRef]
- Ribeiro, F.; Przybylski, D.; Yin, S.; Sharpe, T.; Gnerre, S.; Abouelleil, A.; Berlin, A.M.; Montmayeur, A.; Shea, T.P.; Walker, B.J.; et al. Finished bacterial genomes from shotgun sequence data. Genome Res. 2012, 22, 2270–2277. [Google Scholar] [CrossRef]
- Moraga, R. SemHelpers [Custom Perl Script]. 2017. Available online: https://github.com/Lanilen/SemHelpers (accessed on 7 November 2018).
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Leigh, D.; Lischer, H.; Grossen, C.; Keller, L. Batch effects in a multiyear sequencing study: False biological trends due to changes in read lengths. Mol. Ecol. Resour. 2018. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.D.; Borevitz, J.O. Axe: Rapid, competitive sequence read demultiplexing using a trie. Bioinformatics 2017. [Google Scholar] [CrossRef] [PubMed]
- Herten, K.; Hestand, M.S.; Vermeesch, J.R.; Van Houdt, J.K. GBSX: A toolkit for experimental design and demultiplexing genotyping by sequencing experiments. BMC Bioinform. 2015, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Moraga, R. Mux Barcodes [Custom Perl Script]. 2017. Available online: https://github.com/sgalla32/mux_barcodes (accessed on 7 November 2018).
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Moraga, R. Pancompare. 2018. Available online: https://github.com/Lanilen/pancompare (accessed on 7 November 2018).
- Moraga, R. Split_bamfile_tasks.pl [Custom Perl Script]. 2018. Available online: https://github.com/Lanilen/pancompare (accessed on 7 November 2018).
- Dodds, K.G.; McEwan, J.C.; Brauning, R.; Anderson, R.M.; Stijn, T.C.; Kristjánsson, T.; Clarke, S.M. Construction of relatedness matrices using genotyping-by-sequencing data. BMC Genom. 2015, 16, 1047. [Google Scholar] [CrossRef]
- Bates, D.; Maechler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Gregory, T.R. Animal Genome Size Database. 2001. Available online: http://www.genomesize.com/ (accessed on 7 November 2018).
- Tigano, A.; Sackton, T.B.; Friesen, V.L. Assembly and RNA-free annotation of highly heterozygous genomes: The case of the thick-billed murre (Uria lomvia). Mol. Ecol. Res. 2018, 18, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Lacy, R.C.; Ballou, J.D.; Pollak, J.P. PMx: Software package for demographic and genetic analysis and management of pedigreed populations. Methods Ecol. Evol. 2012, 3, 433–437. [Google Scholar] [CrossRef]
- Willoughby, J.R.; Fernandez, N.B.; Lamb, M.C.; Ivy, J.A.; Lacy, R.C.; DeWoody, J.A. The impacts of inbreeding, drift and selection on genetic diversity in captive breeding populations. Mol. Ecol. 2015, 24, 98–110. [Google Scholar] [CrossRef]
- Putnam, A.S.; Ivy, J.A. Kinship-based management strategies for captive breeding programs when pedigrees are unknown or uncertain. J. Hered. 2013, 105, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Hammerly, S.C.; de la Cerda, D.A.; Bailey, H.; Johnson, J.A. A pedigree gone bad: Increased offspring survival after using DNA-based relatedness to minimize inbreeding in a captive population. Anim. Conserv. 2016, 19, 296–303. [Google Scholar] [CrossRef]
- Szulkin, M.; Bierne, N.; David, P. Heterozygosity-fitness correlations: A time for reappraisal. Evolution 2010, 64, 1202–1217. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Castillo, J.; Attard, C.R.M.; Marri, S.; Brauer, C.J.; Möller, L.M.; Beheregaray, L.B. swinger: A user-friendly computer program to establish captive breeding groups that minimize relatedness without pedigree information. Mol. Ecol. Res. 2017, 17, 278–287. [Google Scholar] [CrossRef]
- Trapnell, C.; Salzberg, S.L. How to map billions of short reads onto genomes. Nat. Biotechnol. 2009, 27, 455. [Google Scholar] [CrossRef]
- Kajitani, R.; Toshimoto, K.; Noguchi, H.; Toyoda, A.; Ogura, Y.; Okuno, M.; Yabana, M.; Harada, M.; Nagayasu, E.; Maruyama, H.; et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014. [Google Scholar] [CrossRef]
- IUCN. The IUCN Red List of Threatened Species. 2018. Available online: http://www.iucnredlist.org (accessed on 7 November 2018).
- Küpper, C.; Stocks, M.; Risse, J.E.; dos Remedios, N.; Farrell, L.L.; McRae, S.B.; Morgan, T.C.; Karlionova, N.; Pinchuk, P.; Verkuil, Y.I.; et al. A supergene determines highly divergent male reproductive morphs in the ruff. Nat. Genet. 2016, 48, 79. [Google Scholar] [CrossRef]
- Frankham, R. Challenges and opportunities of genetic approaches to biological conservation. Biol. Conserv. 2010, 143, 1919–1927. [Google Scholar] [CrossRef]
- Taylor, H.R. The use and abuse of genetic marker-based estimates of relatedness and inbreeding. Ecol. Evol. 2015, 5, 3140–3150. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Total Assembly Length (Gb) | Total Scaffolds | Scaffold N50 (bp) | Longest Scaffold (bp) | Average Scaffold Length (bp) | Complete Single-Copy BUSCOs (%) |
|---|---|---|---|---|---|---|
| Kakī | 1.18 | 523 | 105,710,992 | 238,324,410 | 2,254,638 | 91.0 |
| Pied Stilt | 1.12 | 1443 | 99,457,149 | 221,521,436 | 773,955 | 85.9 |
| Avocet | 1.02 | 67 | 87,059,367 | 184,945,080 | 15,204,176 | 82.4 |
| Killdeer | 1.22 | 15,167 | 3,657,525 | 21,923,840 | 80,436 | 92.5 |
| Reference Genome | No. of Mapped Tag Pairs | % Tags Shared with Kakī Mapping | No. Unfiltered SNPs | No. Filtered SNPs | Average Missingness | Average Depth | Average π | Average HS | Average R | |
|---|---|---|---|---|---|---|---|---|---|---|
| Kaki | 392,652 | 100 | 634,695 | 19,396 | 0.04 ± 0.04 | 13.73 ± 6.53 | 0.31 ± 0.14 | 0.07 ± 0.15 | 0.11 ± 0.12 | |
| Pied Stilt | 372,906 | 91.04 | 604,573 | 18,625 | 0.04 ± 0.04 | 11.71 ± 5.52 | 0.32 ± 0.14 | 0.03 ± 0.15 | 0.10 ± 0.12 | |
| Avocet | 316,978 | 83.10 | 481,532 | 18,398 | 0.03 ± 0.04 | 13.90 ± 6.58 | 0.31 ± 0.15 | −0.06 ± 0.14 | 0.15 ± 0.11 | |
| Killdeer | 151,546 | 72.42 | 242,493 | 10,440 | 0.02 ± 0.03 | 18.51 ± 8.77 | 0.33 ± 0.15 | −0.25 ± 0.14 | −0.25 ± 0.14 | 0.30 ± 0.09 |
| Reference Genome | Average Alignment Rate (%) | No. Unfiltered SNPs | No. Filtered SNPs | Average Missingness | Average Depth | Average π | Average HS | Average R |
|---|---|---|---|---|---|---|---|---|
| Kaki | 94.6 ± 0.50 | 4,246,100 | 91,854 | 0.002 ± 0.005 | 17.44 ± 6.79 | 0.35 ± 0.13 | −0.05 ± 0.08 | 0.06 ± 0.11 |
| Pied Stilt | 88.1 ± 0.96 | 8,438,866 | 89,419 | 0.002 ± 0.005 | 14.99 ± 6.06 | 0.34 ± 0.13 | −0.05 ± 0.08 | 0.06 ± 0.11 |
| Avocet | 78.5 ± 0.46 | 24,333,620 | 143,343 | 0.002 ± 0.004 | 16.02 ± 6.43 | 0.33 ± 0.14 | −0.05 ± 0.07 | 0.11 ± 0.11 |
| Killdeer | 64.8 ± 4.89 | 62,888,931 | 89,145 | 0.002 ± 0.004 | 13.95 ± 5.54 | 0.32 ± 0.13 | 0.25 ± 0.07 | 0.03 ± 0.13 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galla, S.J.; Forsdick, N.J.; Brown, L.; Hoeppner, M.P.; Knapp, M.; Maloney, R.F.; Moraga, R.; Santure, A.W.; Steeves, T.E. Reference Genomes from Distantly Related Species Can Be Used for Discovery of Single Nucleotide Polymorphisms to Inform Conservation Management. Genes 2019, 10, 9. https://doi.org/10.3390/genes10010009
Galla SJ, Forsdick NJ, Brown L, Hoeppner MP, Knapp M, Maloney RF, Moraga R, Santure AW, Steeves TE. Reference Genomes from Distantly Related Species Can Be Used for Discovery of Single Nucleotide Polymorphisms to Inform Conservation Management. Genes. 2019; 10(1):9. https://doi.org/10.3390/genes10010009
Chicago/Turabian StyleGalla, Stephanie J., Natalie J. Forsdick, Liz Brown, Marc P. Hoeppner, Michael Knapp, Richard F. Maloney, Roger Moraga, Anna W. Santure, and Tammy E. Steeves. 2019. "Reference Genomes from Distantly Related Species Can Be Used for Discovery of Single Nucleotide Polymorphisms to Inform Conservation Management" Genes 10, no. 1: 9. https://doi.org/10.3390/genes10010009
APA StyleGalla, S. J., Forsdick, N. J., Brown, L., Hoeppner, M. P., Knapp, M., Maloney, R. F., Moraga, R., Santure, A. W., & Steeves, T. E. (2019). Reference Genomes from Distantly Related Species Can Be Used for Discovery of Single Nucleotide Polymorphisms to Inform Conservation Management. Genes, 10(1), 9. https://doi.org/10.3390/genes10010009