Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Antibody-Based Technologies (MeRIP-Seq, i/miCLIP)
3. Detection Using Natural or Induced RT-Arrests and Signatures
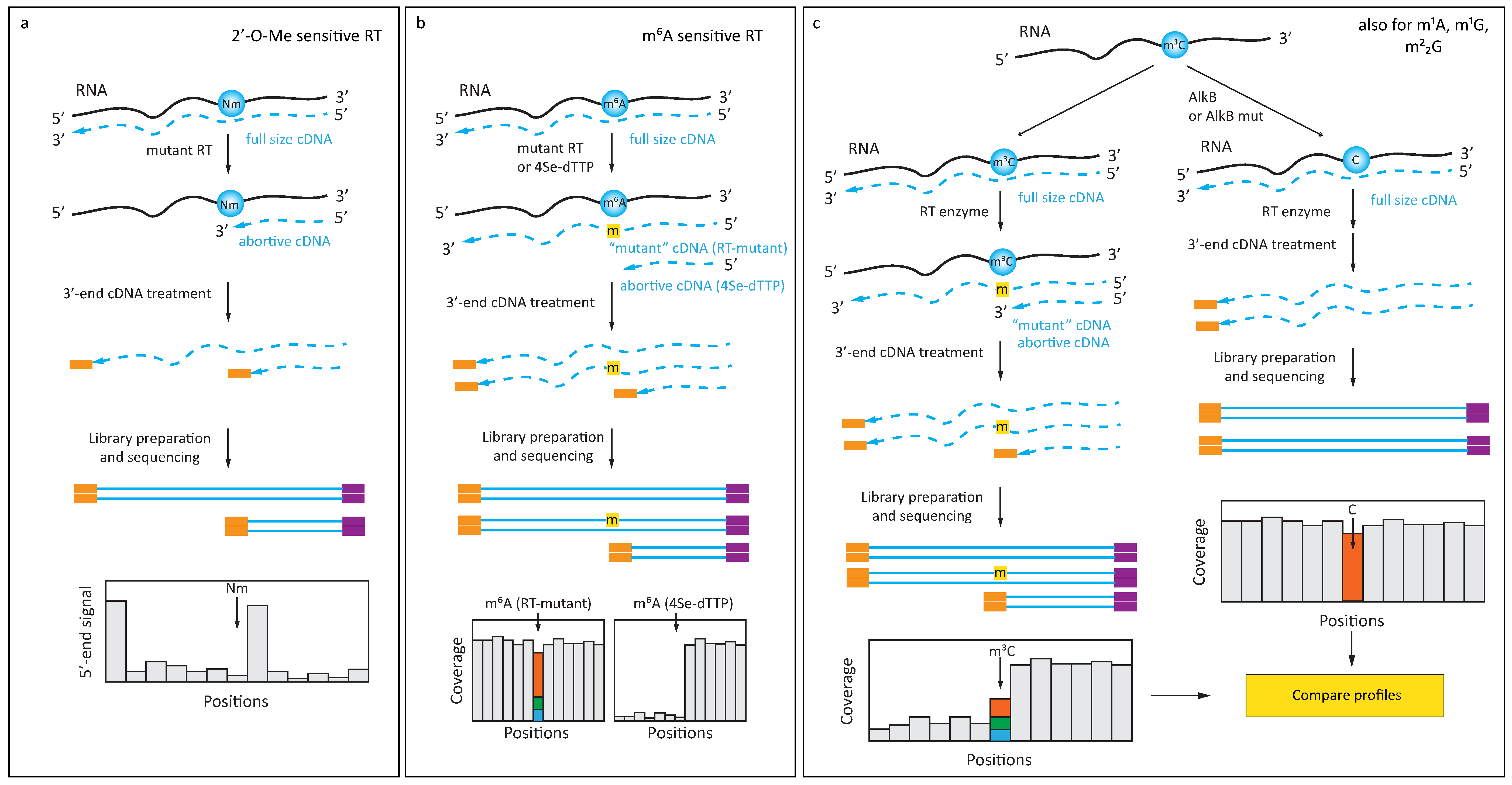
3.1. Naturally Existing RT-Signatures
3.2. Enhancing RT-Signatures with Engineered Enzymes and Substrates
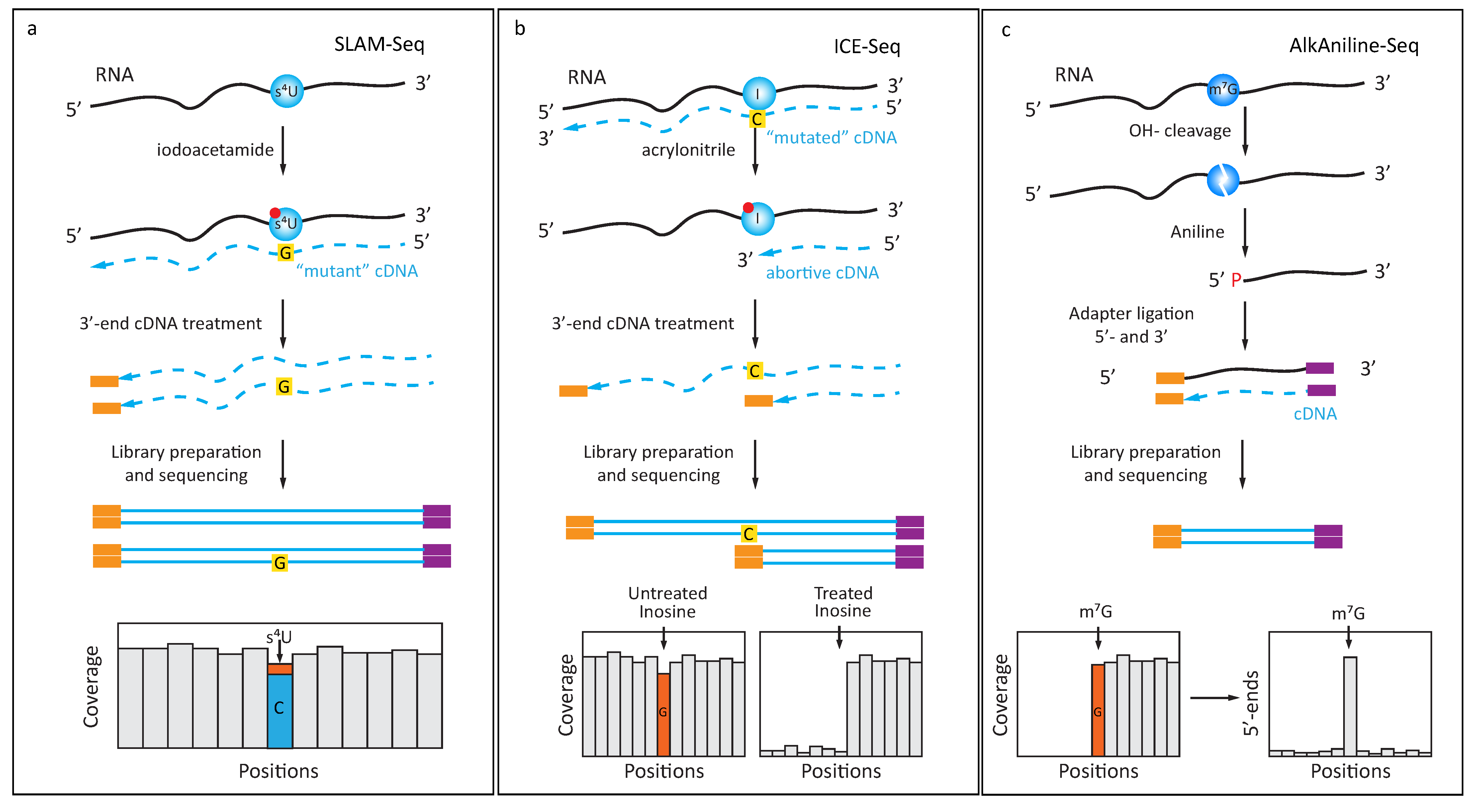
4. Exploiting Specific Chemical Reactivity of Modified Nucleobases
4.1. Chemically Induced Alteration of RT-Profiles
4.2. Protection of RNA from Cleavage
4.3. Specific Enrichment of RNA Fragments by Selective Ligation
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Carlile, T.M.; Rojas-Duran, M.F.; Zinshteyn, B.; Shin, H.; Bartoli, K.M.; Gilbert, W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 2014, 515, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Edelheit, S.; Schwartz, S.; Mumbach, M.R.; Wurtzel, O.; Sorek, R. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet. 2013, 9, e1003602. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Sajini, A.A.; Blanco, S.; Dietmann, S.; Lombard, P.; Sugimoto, Y.; Paramor, M.; Gleeson, J.G.; Odom, D.T.; Ule, J.; et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 2013, 4, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, S.; Horowitz, A.; Nilsen, T.W.; Munns, T.W.; Rottman, F.M. Mapping of N6-methyladenosine residues in bovine prolactin mRNA. Proc. Natl. Acad. Sci. USA 1984, 81, 5667–5671. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Salmon-Divon, M.; Amariglio, N.; Rechavi, G. Transcriptome-wide mapping of N6-methyladenosine by m6A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 2013, 8, 176–189. [Google Scholar] [CrossRef]
- Delatte, B.; Wang, F.; Ngoc, L.V.; Collignon, E.; Bonvin, E.; Deplus, R.; Calonne, E.; Hassabi, B.; Putmans, P.; Awe, S.; et al. RNA biochemistry. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 2016, 351, 282–285. [Google Scholar] [CrossRef]
- Sinclair, W.R.; Arango, D.; Shrimp, J.H.; Zengeya, T.T.; Thomas, J.M.; Montgomery, D.C.; Fox, S.D.; Andresson, T.; Oberdoerffer, S.; Meier, J.L. Profiling cytidine acetylation with specific affinity and reactivity. ACS Chem. Biol. 2017, 12, 2922–2926. [Google Scholar] [CrossRef] [PubMed]
- Arango, D.; Sturgill, D.; Alhusaini, N.; Dillman, A.A.; Sweet, T.J.; Hanson, G.; Hosogane, M.; Sinclair, W.R.; Nanan, K.K.; Mandler, M.D.; et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell 2018, 175, 1872–1886.e24. [Google Scholar] [CrossRef] [PubMed]
- Amort, T.; Rieder, D.; Wille, A.; Khokhlova-Cubberley, D.; Riml, C.; Trixl, L.; Jia, X.-Y.; Micura, R.; Lusser, A. Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol. 2017, 18, 1. [Google Scholar] [CrossRef]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016, 530, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xiong, X.; Zhang, M.; Wang, K.; Chen, Y.; Zhou, J.; Mao, Y.; Lv, J.; Yi, D.; Chen, X.-W.; et al. Base-resolution mapping reveals distinct m1A methylome in nuclear- and mitochondrial-encoded transcripts. Mol. Cell 2017, 68, 993–1005.e9. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Wang, K.; Wang, L.; Shu, X.; Ma, S.; Yi, C. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat. Chem. Biol. 2016, 12, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 2017, 551, 251–255. [Google Scholar] [CrossRef]
- Xiong, X.; Li, X.; Wang, K.; Yi, C. Perspectives on topology of the human m1A methylome at single nucleotide resolution. RNA 2018, 24, 1437–1442. [Google Scholar] [CrossRef]
- Schwartz, S. m1A within cytoplasmic mRNAs at single nucleotide resolution: A reconciled transcriptome-wide map. RNA 2018, 24, 1427–1436. [Google Scholar] [CrossRef]
- Mishima, E.; Jinno, D.; Akiyama, Y.; Itoh, K.; Nankumo, S.; Shima, H.; Kikuchi, K.; Takeuchi, Y.; Elkordy, A.; Suzuki, T.; et al. Immuno-Northern Blotting: Detection of RNA modifications by using antibodies against modified nucleosides. PLoS ONE 2015, 10, e0143756. [Google Scholar] [CrossRef]
- Grozhik, A.V.; Jaffrey, S.R. Distinguishing RNA modifications from noise in epitranscriptome maps. Nat. Chem. Biol. 2018, 14, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Molinie, B.; Wang, J.; Lim, K.S.; Hillebrand, R.; Lu, Z.-X.; Van Wittenberghe, N.; Howard, B.D.; Daneshvar, K.; Mullen, A.C.; Dedon, P.; et al. m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome. Nat. Methods 2016, 13, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Grozhik, A.V.; Linder, B.; Olarerin-George, A.O.; Jaffrey, S.R. Mapping m6A at individual-nucleotide resolution using crosslinking and immunoprecipitation (miCLIP). Methods Mol. Biol. 2017, 1562, 55–78. [Google Scholar] [PubMed]
- Chen, K.; Lu, Z.; Wang, X.; Fu, Y.; Luo, G.-Z.; Liu, N.; Han, D.; Dominissini, D.; Dai, Q.; Pan, T.; et al. High-resolution N(6)-methyladenosine (m6A) map using photo-crosslinking-assisted m6A sequencing. Angew. Chem. Int. Ed. Engl. 2015, 54, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Hauenschild, R.; Tserovski, L.; Schmid, K.; Thüring, K.; Winz, M.-L.; Sharma, S.; Entian, K.-D.; Wacheul, L.; Lafontaine, D.L.J.; Anderson, J.; et al. The reverse transcription signature of N-1-methyladenosine in RNA-Seq is sequence dependent. Nucleic Acids Res. 2015, 43, 9950–9964. [Google Scholar] [CrossRef] [PubMed]
- Ryvkin, P.; Leung, Y.Y.; Silverman, I.M.; Childress, M.; Valladares, O.; Dragomir, I.; Gregory, B.D.; Wang, L.-S. HAMR: High-throughput annotation of modified ribonucleotides. RNA 2013, 19, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Kuksa, P.P.; Leung, Y.Y.; Vandivier, L.E.; Anderson, Z.; Gregory, B.D.; Wang, L.-S. In silico identification of RNA modifications from high-throughput sequencing data using HAMR. Methods Mol. Biol. 2017, 1562, 211–229. [Google Scholar] [PubMed]
- Tserovski, L.; Marchand, V.; Hauenschild, R.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. High-throughput sequencing for 1-methyladenosine (m1A) mapping in RNA. Methods 2016, 107, 110–121. [Google Scholar] [CrossRef]
- Motorin, Y.; Muller, S.; Behm-Ansmant, I.; Branlant, C. Identification of modified residues in RNAs by reverse transcription-based methods. Meth. Enzymol. 2007, 425, 21–53. [Google Scholar]
- Behm-Ansmant, I.; Helm, M.; Motorin, Y. Use of specific chemical reagents for detection of modified nucleotides in RNA. J. Nucleic Acids 2011, 2011, 408053. [Google Scholar] [CrossRef]
- Maden, B.E. Mapping 2′-O-methyl groups in ribosomal RNA. Methods 2001, 25, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Incarnato, D.; Anselmi, F.; Morandi, E.; Neri, F.; Maldotti, M.; Rapelli, S.; Parlato, C.; Basile, G.; Oliviero, S. High-throughput single-base resolution mapping of RNA 2′-O-methylated residues. Nucleic Acids Res. 2017, 45, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.-W.; Shao, P.; Diao, L.-T.; Zhou, H.; Yu, C.-H.; Qu, L.-H. RTL-P: A sensitive approach for detecting sites of 2′-O-methylation in RNA molecules. Nucleic Acids Res. 2012, 40, e157. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, J.; Marx, A. Direct and site-specific quantification of RNA 2′-O-methylation by PCR with an engineered DNA polymerase. Nucleic Acids Res. 2016, 44, 3495–3502. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, J.; Werner, S.; Marchand, V.; Adam, M.; Motorin, Y.; Helm, M.; Marx, A. Engineering of a DNA polymerase for direct m6A sequencing. Angew. Chem. Int. Ed. Engl. 2018, 57, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Yuan, Y.; Chen, Z.; Xi, K.; Wang, T.; Xie, Y.; He, Z.; Su, H.; Zhou, Y.; Tan, Z.-J.; et al. Precise antibody-independent m6A identification via 4SedTTP-involved and FTO-assisted strategy at single-nucleotide resolution. J. Am. Chem. Soc. 2018, 140, 5886–5889. [Google Scholar] [CrossRef]
- Cozen, A.E.; Quartley, E.; Holmes, A.D.; Hrabeta-Robinson, E.; Phizicky, E.M.; Lowe, T.M. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 2015, 12, 879–884. [Google Scholar] [CrossRef]
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambowitz, A.M.; Pan, T. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 2015, 12, 835–837. [Google Scholar] [CrossRef]
- Dai, Q.; Zheng, G.; Schwartz, M.H.; Clark, W.C.; Pan, T. Selective enzymatic demethylation of N2,N2-dimethylguanosine in RNA and its application in high-throughput tRNA sequencing. Angew. Chem. Int. Ed. Engl. 2017, 56, 5017–5020. [Google Scholar] [CrossRef]
- Schaefer, M.; Pollex, T.; Hanna, K.; Lyko, F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2009, 37, e12. [Google Scholar] [CrossRef]
- Müller, M.; Hartmann, M.; Schuster, I.; Bender, S.; Thüring, K.L.; Helm, M.; Katze, J.R.; Nellen, W.; Lyko, F.; Ehrenhofer-Murray, A.E. Dynamic modulation of Dnmt2-dependent tRNA methylation by the micronutrient queuine. Nucleic Acids Res. 2015, 43, 10952–10962. [Google Scholar] [CrossRef] [PubMed]
- Tuorto, F.; Liebers, R.; Musch, T.; Schaefer, M.; Hofmann, S.; Kellner, S.; Frye, M.; Helm, M.; Stoecklin, G.; Lyko, F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 2012, 19, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, G.; Ney, M.; Gaspar, I.; Aigueperse, C.; Schaefer, M.; Kellner, S.; Helm, M.; Motorin, Y. Eukaryotic rRNA modification by yeast 5-methylcytosine-methyltransferases and human proliferation-associated antigen p120. PLoS ONE 2015, 10, e0133321. [Google Scholar] [CrossRef] [PubMed]
- David, R.; Burgess, A.; Parker, B.; Li, J.; Pulsford, K.; Sibbritt, T.; Preiss, T.; Searle, I.R. Transcriptome-wide mapping of RNA 5-Methylcytosine in Arabidopsis mRNAs and noncoding RNAs. Plant Cell 2017, 29, 445–460. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Panneerdoss, S.; Timilsina, S.; Zhu, J.; Mohammad, T.A.; Lu, Z.-L.; de Magalhães, J.P.; Chen, Y.; Rong, R.; Huang, Y.; et al. Topological characterization of human and mouse m5C epitranscriptome revealed by bisulfite sequencing. Int. J. Genom. 2018, 2018, 1351964. [Google Scholar] [CrossRef] [PubMed]
- Legrand, C.; Tuorto, F.; Hartmann, M.; Liebers, R.; Jacob, D.; Helm, M.; Lyko, F. Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs. Genome Res. 2017, 27, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.J.; Marsico, G.; Bachman, M.; Beraldi, D.; Balasubramanian, S. Quantitative sequencing of 5-formylcytosine in DNA at single-base resolution. Nat. Chem. 2014, 6, 435–440. [Google Scholar] [CrossRef]
- Van Haute, L.; Dietmann, S.; Kremer, L.; Hussain, S.; Pearce, S.F.; Powell, C.A.; Rorbach, J.; Lantaff, R.; Blanco, S.; Sauer, S.; et al. Deficient methylation and formylation of mt-tRNAMet wobble cytosine in a patient carrying mutations in NSUN3. Nat. Commun. 2016, 7, 12039. [Google Scholar] [CrossRef]
- Song, C.-X.; Szulwach, K.E.; Dai, Q.; Fu, Y.; Mao, S.-Q.; Lin, L.; Street, C.; Li, Y.; Poidevin, M.; Wu, H.; et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 2013, 153, 678–691. [Google Scholar] [CrossRef]
- Zeng, H.; He, B.; Xia, B.; Bai, D.; Lu, X.; Cai, J.; Chen, L.; Zhou, A.; Zhu, C.; Meng, H.; et al. Bisulfite-Free, Nanoscale analysis of 5-hydroxymethylcytosine at single base resolution. J. Am. Chem. Soc. 2018, 140, 13190–13194. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.-C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Herzog, V.A.; Reichholf, B.; Neumann, T.; Rescheneder, P.; Bhat, P.; Burkard, T.R.; Wlotzka, W.; von Haeseler, A.; Zuber, J.; Ameres, S.L. Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods 2017, 14, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ueda, H.; Okada, S.; Sakurai, M. Transcriptome-wide identification of adenosine-to-inosine editing using the ICE-seq method. Nat. Protoc. 2015, 10, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Carlile, T.M.; Rojas-Duran, M.F.; Gilbert, W.V. Pseudo-Seq: Genome-wide detection of pseudouridine modifications in RNA. Meth. Enzymol. 2015, 560, 219–245. [Google Scholar] [PubMed]
- Carlile, T.M.; Rojas-Duran, M.F.; Gilbert, W.V. Transcriptome-wide identification of pseudouridine modifications using pseudo-seq. Curr. Protoc. Mol. Biol. 2015, 112, 1–24. [Google Scholar]
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; León-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 2014, 159, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, A.F.; Riordan, D.P.; Brown, P.O. Transcriptome-wide mapping of pseudouridines: Pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS ONE 2014, 9, e110799. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, M.A.; Lovejoy, A.F.; Cygan, A.M.; Boothroyd, J.C. mRNA pseudouridylation affects RNA metabolism in the parasite Toxoplasma gondii. RNA 2017, 23, 1834–1849. [Google Scholar] [CrossRef]
- Bakin, A.; Ofengand, J. Four newly located pseudouridylate residues in Escherichia coli 23S ribosomal RNA are all at the peptidyltransferase center: Analysis by the application of a new sequencing technique. Biochemistry 1993, 32, 9754–9762. [Google Scholar] [CrossRef]
- Li, X.; Zhu, P.; Ma, S.; Song, J.; Bai, J.; Sun, F.; Yi, C. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 2015, 11, 592–597. [Google Scholar] [CrossRef]
- Li, X.; Ma, S.; Yi, C. Pseudouridine chemical labeling and profiling. Meth. Enzymol. 2015, 560, 247–272. [Google Scholar] [PubMed]
- Zaringhalam, M.; Papavasiliou, F.N. Pseudouridylation meets next-generation sequencing. Methods 2016, 107, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Safra, M.; Nir, R.; Farouq, D.; Vainberg Slutskin, I.; Schwartz, S. TRUB1 is the predominant pseudouridine synthase acting on mammalian mRNA via a predictable and conserved code. Genome Res. 2017, 27, 393–406. [Google Scholar] [CrossRef]
- Wintermeyer, W.; Zachau, H.G. Tertiary structure interactions of 7-methylguanosine in yeast tRNAPhe as studied by borohydride reduction. FEBS Lett. 1975, 58, 306–309. [Google Scholar] [CrossRef]
- Lin, S.; Liu, Q.; Lelyveld, V.S.; Choe, J.; Szostak, J.W.; Gregory, R.I. Mettl1/Wdr4-mediated m7G tRNA methylome is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Mol. Cell 2018, 71, 244–255.e5. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.; Briney, C.A.; Nance, K.D.; Lopez, J.E.; Thorpe, A.L.; Fox, S.D.; Bortolin-Cavaille, M.-L.; Sas-Chen, A.; Arango, D.; Oberdoerffer, S.; et al. A chemical signature for cytidine acetylation in RNA. J. Am. Chem. Soc. 2018, 140, 12667–12670. [Google Scholar] [CrossRef] [PubMed]
- Birkedal, U.; Christensen-Dalsgaard, M.; Krogh, N.; Sabarinathan, R.; Gorodkin, J.; Nielsen, H. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew. Chem. Int. Ed. Engl. 2015, 54, 451–455. [Google Scholar] [CrossRef]
- Marchand, V.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. Illumina-based RiboMethSeq approach for mapping of 2′-O-Me residues in RNA. Nucleic Acids Res. 2016, 44, e135. [Google Scholar] [CrossRef]
- Marchand, V.; Pichot, F.; Thüring, K.; Ayadi, L.; Freund, I.; Dalpke, A.; Helm, M.; Motorin, Y. Next-generation sequencing-based RiboMethSeq protocol for analysis of tRNA 2′-O-methylation. Biomolecules 2017, 7, 13. [Google Scholar] [CrossRef]
- Gumienny, R.; Jedlinski, D.J.; Schmidt, A.; Gypas, F.; Martin, G.; Vina-Vilaseca, A.; Zavolan, M. High-throughput identification of C/D box snoRNA targets with CLIP and RiboMeth-seq. Nucleic Acids Res. 2017, 45, 2341–2353. [Google Scholar] [CrossRef]
- Dai, Q.; Moshitch-Moshkovitz, S.; Han, D.; Kol, N.; Amariglio, N.; Rechavi, G.; Dominissini, D.; He, C. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat. Methods 2017, 14, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pirnie, S.P.; Carmichael, G.G. High-throughput and site-specific identification of 2′-O-methylation sites using ribose oxidation sequencing (RibOxi-seq). RNA 2017, 23, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Krogh, N.; Kongsbak-Wismann, M.; Geisler, C.; Nielsen, H. Substoichiometric ribose methylations in spliceosomal snRNAs. Org. Biomol. Chem. 2017, 15, 8872–8876. [Google Scholar] [CrossRef] [PubMed]
- Marchand, V.; Ayadi, L.; Ernst, F.G.M.; Hertler, J.; Bourguignon-Igel, V.; Galvanin, A.; Kotter, A.; Helm, M.; Lafontaine, D.L.J.; Motorin, Y. AlkAniline-Seq: Profiling of m7G and m3C RNA modifications at single nucleotide resolution. Angew. Chem. Int. Ed. Engl. 2018, 57, 16785–16790. [Google Scholar] [CrossRef]
- Kubo, K.; Ide, H.; Wallace, S.S.; Kow, Y.W. A novel, sensitive, and specific assay for abasic sites, the most commonly produced DNA lesion. Biochemistry 1992, 31, 3703–3708. [Google Scholar] [CrossRef] [PubMed]
- Ide, H.; Akamatsu, K.; Kimura, Y.; Michiue, K.; Makino, K.; Asaeda, A.; Takamori, Y.; Kubo, K. Synthesis and damage specificity of a novel probe for the detection of abasic sites in DNA. Biochemistry 1993, 32, 8276–8283. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Han, S.; Song, H.; Küpfer, P.A.; Leumann, C.J.; Sonntag, W.E. An assay for RNA oxidation induced abasic sites using the Aldehyde Reactive Probe. Free Radic. Res. 2011, 45, 237–247. [Google Scholar] [CrossRef]
- Tanaka, M.; Han, S.; Küpfer, P.A.; Leumann, C.J.; Sonntag, W.E. Quantification of oxidized levels of specific RNA species using an aldehyde reactive probe. Anal. Biochem. 2011, 417, 142–148. [Google Scholar] [CrossRef]
- Krogh, N.; Jansson, M.D.; Häfner, S.J.; Tehler, D.; Birkedal, U.; Christensen-Dalsgaard, M.; Lund, A.H.; Nielsen, H. Profiling of 2′-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res. 2016, 44, 7884–7895. [Google Scholar] [CrossRef]
- Sharma, S.; Marchand, V.; Motorin, Y.; Lafontaine, D.L.J. Identification of sites of 2′-O-methylation vulnerability in human ribosomal RNAs by systematic mapping. Sci. Rep. 2017, 7, 11490. [Google Scholar] [CrossRef]
- Erales, J.; Marchand, V.; Panthu, B.; Gillot, S.; Belin, S.; Ghayad, S.E.; Garcia, M.; Laforêts, F.; Marcel, V.; Baudin-Baillieu, A.; et al. Evidence for rRNA 2′-O-methylation plasticity: Control of intrinsic translational capabilities of human ribosomes. Proc. Natl. Acad. Sci. USA 2017, 114, 12934–12939. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motorin, Y.; Helm, M. Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies. Genes 2019, 10, 35. https://doi.org/10.3390/genes10010035
Motorin Y, Helm M. Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies. Genes. 2019; 10(1):35. https://doi.org/10.3390/genes10010035
Chicago/Turabian StyleMotorin, Yuri, and Mark Helm. 2019. "Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies" Genes 10, no. 1: 35. https://doi.org/10.3390/genes10010035
APA StyleMotorin, Y., & Helm, M. (2019). Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies. Genes, 10(1), 35. https://doi.org/10.3390/genes10010035