Statistical Issues in the Analysis of ChIP-Seq and RNA-Seq Data


{kind=link}
Abstract
:1. Introduction
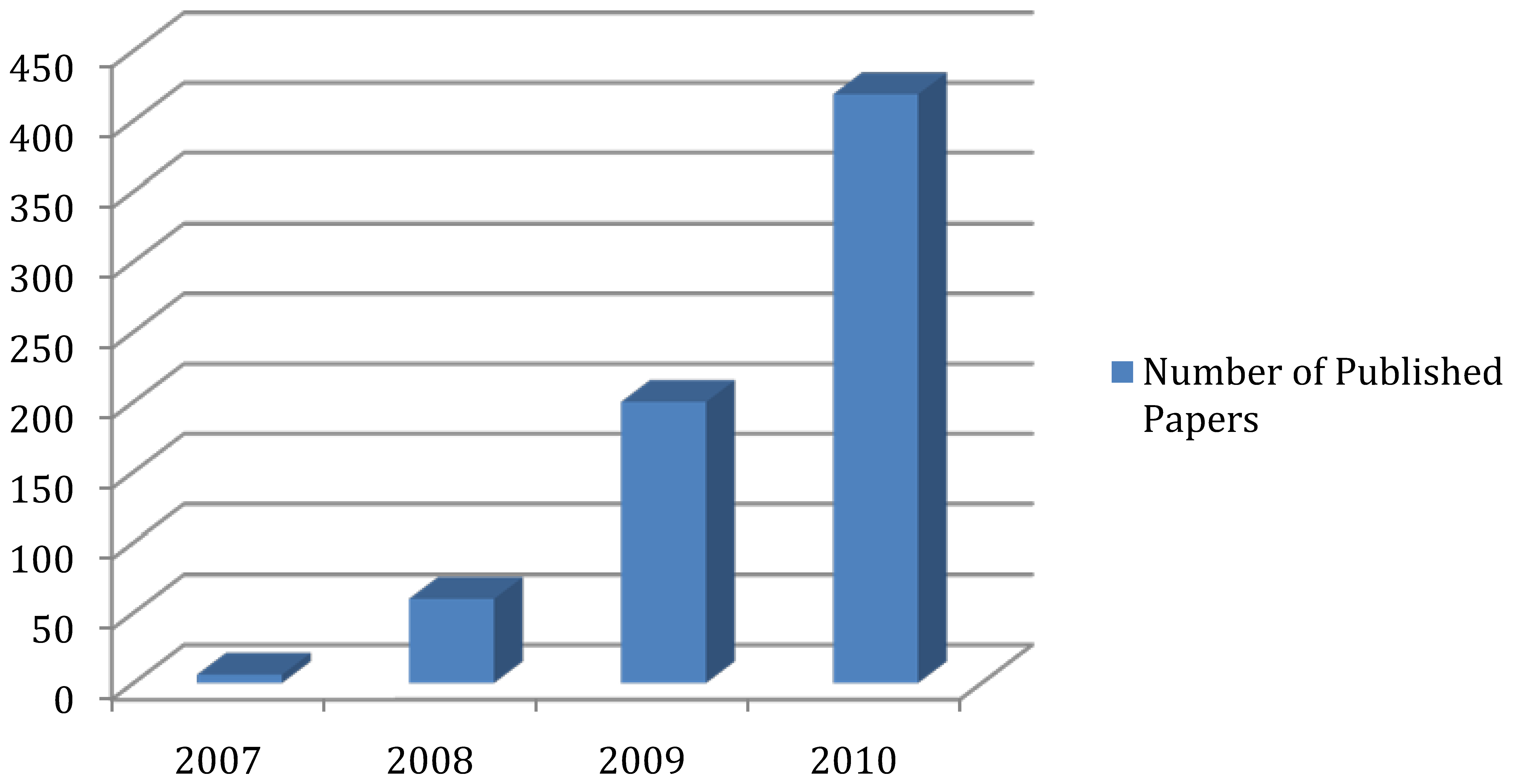
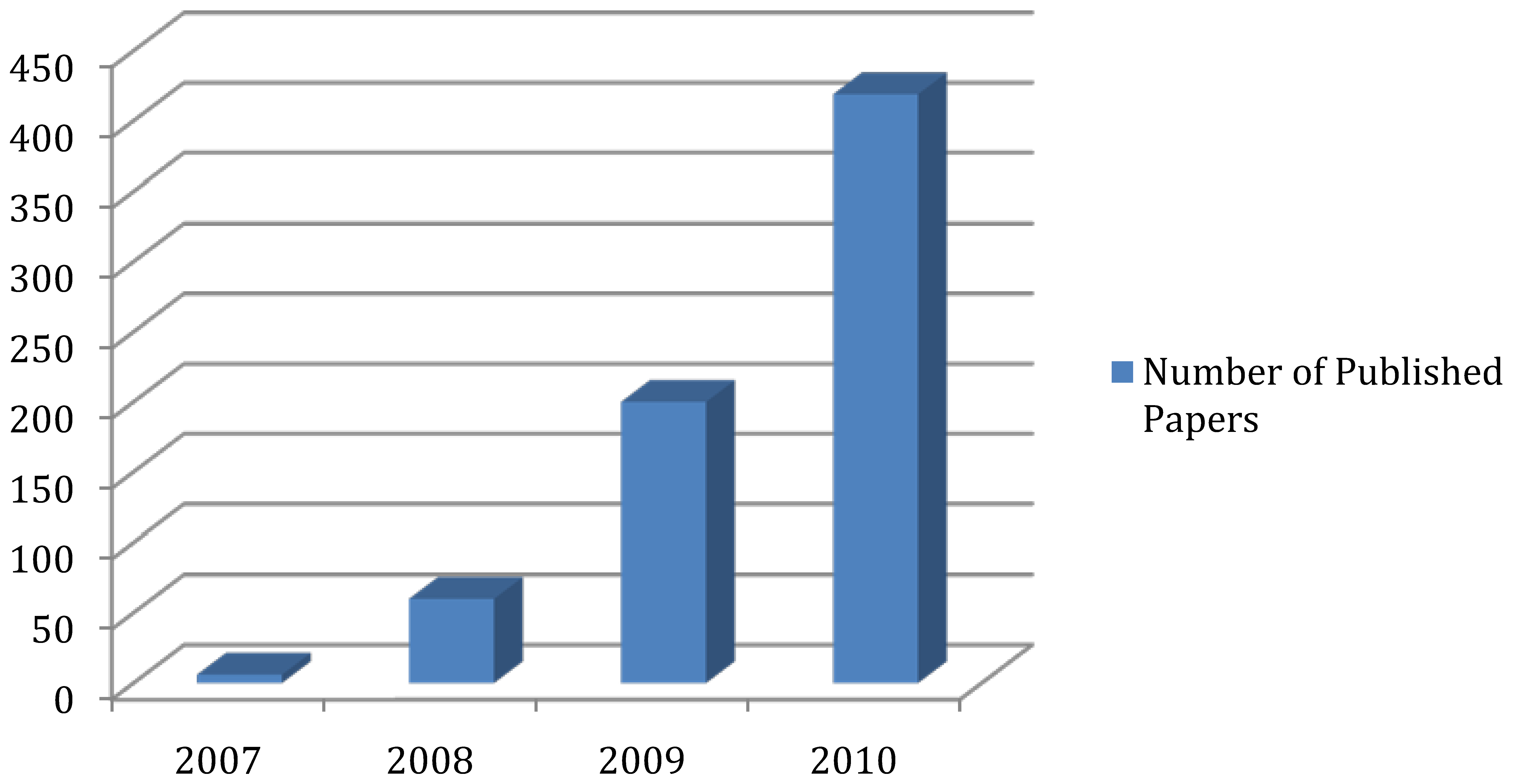
2. Experimental Platform
3. Mapping reads from NGS experiments
4. Statistical methods for ChIP-Seq experiments
4.1. Detecting enriched regions.
- Compute a smooth estimate of the density of the tag counts using a nonparametric kernel density estimator based on a default window size that is guaranteed to be numerically stable;
- Compute an average number of features for window w as nw = nw/L. Here, n is taken to be the number of sequence reads, w is the size of the window, and L is the length of the chromosome.
- Calculate the kernel density at a fixed point, xc, within the window given a random and uniform distribution of the nw features.
- Repeat step 2 k times to obtain a distribution of the kernel density estimates for xc. For large k the kdes become normally distributed.
- The threshold is s SDs above the mean of this normal distribution.
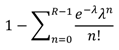
4.2. Follow–up analysis
4.3. Combining ChIP-Seq with ChIP-chip data
5. RNA-Seq experiments: measuring gene expression
6. Experimental design considerations
7. Conclusion and Future Directions
Acknowledgments
References and Notes
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Hirst, M.; Bainbridge, M.; Bilenky, M.; Zhao, Y.; Zeng, T.; Euskirchen, G.; Bernier, B.; Varhol, R.; Delaney, A.; et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 2007, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.L.; Johnson, D.S.; Kim, S.W.; Valouev, A.; Reddy, T.E.; Neff, N.F.; Anton, E.; Medina, C.; Nguyen, L.; Chiao, E.; et al. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009, 19, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Bock, C.; Mikkelsen, T.S.; Jager, N.; Smith, Z.D.; Tomazou, E.; Gnirke, A.; Lander, E.S.; Meissner, A. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat. Methods 2010, 7, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Li, R.; Li, Y.; Tian, G.; Goodman, L.; Fan, W.; Zhang, J.; Li, J.; Zhang, J.; et al. The diploid genome sequence of an Asian individual. Nature 2008, 456, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Ju, Y.S.; Park, H.; Kim, S.; Lee, S.; Yi, J.H.; Mudge, J.; Miller, N.A.; Hong, D.; Bell, C.J.; et al. A highly annotated whole-genome sequence of a Korean individual. Nature 2009, 460, 1011–1015. [Google Scholar] [PubMed]
- Ley, T.J.; Mardis, E.R.; Ding, L.; Fulton, B.; McLellan, M.D.; Chen, K.; Dooling, D.; Dunford-Shore, B.H.; McGrath, S.; Hickenbotham, M.; et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008, 456, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.B.; Sammeth, M.; Gutierrez-Arcelus, M.; Lach, R.P.; Ingle, C.; Nisbett, J.; Guigo, R.; Dermitzakis, E.T. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 2010, 464, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Marioni, J.C.; Pai, A.A.; Degner, J.F.; Engelhardt, B.E.; Nkadori, E.; Veyrieras, J.B.; Stephens, M.; Gilad, Y.; Pritchard, J.K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 2010, 464, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Kaser, M.; Roltgen, K.; Yeboah-Manu, D.; Pluschke, G. Genomic diversity and evolution of Mycobacterium ulcerans revealed by next-generation sequencing. PLoS Pathog. 2009, 5, e1000580. [Google Scholar] [CrossRef]
- Studholme, D.J.; Ibanez, S.G.; MacLean, D.; Dangl, J.L.; Chang, J.H.; Rathjen, J.P. A draft genome sequence and functional screen reveals the repertoire of type III secreted proteins of Pseudomonas syringae pathovar tabaci 11528. BMC Genomics 2009, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, C.; Schuster, S.C.; Sun, Y.; Talbot, S.L.; Qi, J.; Ratan, A.; Tomsho, L.P.; Kasson, L.; Zeyl, E.; Aars, J.; et al. Complete mitochondrial genome of a Pleistocene jawbone unveils the origin of polar bear. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 5053–5057. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.; Drautz, D.I.; Ratan, A.; Pusey, B.; Qi, J.; Lesk, A.M.; Tomsho, L.P.; Packard, M.D.; Zhao, F.; Sher, A.; et al. Sequencing the nuclear genome of the extinct woolly mammoth. Nature 2008, 456, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends. Genet. 2008, 24, 133–141. [Google Scholar] [PubMed]
- Metzker, M.L. Sequencing technologies - the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [PubMed]
- Bennett, S. Solexa Ltd. Pharmacogenomics 2004, 5, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.D.; Buzby, P.R.; Babcock, H.; Beer, E.; Bowers, J.; Braslavsky, I.; Causey, M.; Colonell, J.; Dimeo, J.; Efcavitch, J.W.; et al. Single-molecule DNA sequencing of a viral genome. Science 2008, 320, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Flicek, P.; Birney, E. Sense from sequence reads: methods for alignment and assembly. Nat. Methods 2009, 6, S6–S12. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhang, Z.; Zhang, M.Q.; Ma, B.; Li, M. ZOOM! Zillions of oligos mapped. Bioinformatics 2008, 24, 2431–2437. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009, 10, R25. [Google Scholar] [CrossRef]
- Jiang, H.; Wong, W.H. SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics 2008, 24, 2395–2396. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Watanabe, C.K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Homer, N.; Merriman, B.; Nelson, S.F. BFAST: an alignment tool for large scale genome resequencing. PLoS ONE 2009, 4, e7767. [Google Scholar] [CrossRef]
- Campagna, D.; Albiero, A.; Bilardi, A.; Caniato, E.; Forcato, C.; Manavski, S.; Vitulo, N.; Valle, G. PASS: a program to align short sequences. Bioinformatics 2009, 25, 967–968. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Rumble, S.M.; Lacroute, P.; Dalca, A.V.; Fiume, M.; Sidow, A.; Brudno, M. SHRiMP: accurate mapping of short color-space reads. PLoS Comput. Biol. 2009, 5, e1000386. [Google Scholar] [CrossRef]
- Li, H.; Homer, N. A survey of sequence alignment algorithms for next-generation sequencing. Brief. Bioinform. 2010, 11, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Robert, F.; Wyrick, J.J.; Aparicio, O.; Jennings, E.G.; Simon, I.; Zeitlinger, J.; Schreiber, J.; Hannett, N.; Kanin, E.; et al. Genome-wide location and function of DNA binding proteins. Science 2000, 290, 2306–2309. [Google Scholar] [CrossRef] [PubMed]
- Iyer, V.R.; Horak, C.E.; Scafe, C.S.; Botstein, D.; Snyder, M.; Brown, P.O. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature 2001, 409, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Lieb, J.D.; Liu, X.; Botstein, D.; Brown, P.O. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat. Genet. 2001, 28, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Rinaldi, N.J.; Robert, F.; Odom, D.T.; Bar-Joseph, Z.; Gerber, G.K.; Hannett, N.M.; Harbison, C.T.; Thompson, C.M.; Simon, I.; et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 2002, 298, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.S.; Yu, J.; Shen, J.; Maher, C.A.; Hu, M.; Kalyana-Sundaram, S.; Yu, J.; Chinnaiyan, A.M. HPeak: an HMM-based algorithm for defining read-enriched regions in ChIP-Seq data. BMC Bioinformatics 2010, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Yu, J.; Taylor, J.M.; Chinnaiyan, A.M.; Qin, Z.S. On the detection and refinement of transcription factor binding sites using ChIP-Seq data. Nucleic Acids Res. 2010, 38, 2154–2167. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.P.; Guinney, J.; Crawford, G.E.; Furey, T.S. F-Seq: a feature density estimator for high-throughput sequence tags. Bioinformatics 2008, 24, 2537–2538. [Google Scholar] [CrossRef] [PubMed]
- Nix, D.A.; Courdy, S.J.; Boucher, K.M. Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinformatics 2008, 9, 523. [Google Scholar] [CrossRef] [PubMed]
- Spyrou, C.; Stark, R.; Lynch, A.G.; Tavare, S. BayesPeak: Bayesian analysis of ChIP-seq data. BMC Bioinformatics 2009, 10, 299. [Google Scholar] [CrossRef] [PubMed]
- Rozowsky, J.; Euskirchen, G.; Auerbach, R.K.; Zhang, Z.D.; Gibson, T.; Bjornson, R.; Carriero, N.; Snyder, M.; Gerstein, M.B. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat. Biotechnol. 2009, 27, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Kharchenko, P.V.; Tolstorukov, M.Y.; Park, P.J. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol. 2008, 26, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Jothi, R.; Cuddapah, S.; Barski, A.; Cui, K.; Zhao, K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008, 36, 5221–5231. [Google Scholar] [CrossRef] [PubMed]
- Valouev, A.; Johnson, D.S.; Sundquist, A.; Medina, C.; Anton, E.; Batzoglou, S.; Myers, R.M.; Sidow, A. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat. Methods 2008, 5, 829–834. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nussbaum, C.; Myers, R.M.; Brown, M.; Li, W.; Liu, X.S. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Fejes, A.P.; Robertson, G.; Bilenky, M.; Varhol, R.; Bainbridge, M.; Jones, S.J. FindPeaks 3.1: a tool for identifying areas of enrichment from massively parallel short-read sequencing technology . Bioinformatics 2008, 24, 1729–1730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Robertson, G.; Krzywinski, M.; Ning, K.; Droit, A.; Jones, S.; Gottardo, R. PICS: Probabilistic Inference for ChIP-seq . Biometrics 2010, in press. [Google Scholar]
- Laajala, T.D.; Raghav, S.; Tuomela, S.; Lahesmaa, R.; Aittokallio, T.; Elo, L.L. A practical comparison of methods for detecting transcription factor binding sites in ChIP-seq experiments. BMC Genomics 2009, 10, 618. [Google Scholar] [CrossRef] [PubMed]
- Wilbanks, E.G.; Facciotti, M.T. Evaluation of algorithm performance in ChIP-seq peak detection. PLoS ONE 2010, 5, e11471. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: a pratical and powerful approach to multiple testing. J. Royal Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Jiang, H.; Ma, W.; Johnson, D.S.; Myers, R.M.; Wong, W.H. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat. Biotechnol. 2008, 26, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.E.; Petrie, T. Statistical Inference for Probabilistic Functions of Finite State Markov Chains. Ann. Math. Stat. 1966, 37, 1554–1563. [Google Scholar] [CrossRef]
- Schmidler, S.C.; Liu, J.S.; Brutlag, D.L. Bayesian segmentation of protein secondary structure. J. Comput. Biol. 2000, 7, 233–248. [Google Scholar] [PubMed]
- Li, W.; Meyer, C.A.; Liu, X.S. A hidden Markov model for analyzing ChIP-chip experiments on genome tiling arrays and its application to p53 binding sequences . Bioinformatics 2005, 21 (Suppl. 1), i274–i282. [Google Scholar] [CrossRef] [PubMed]
- Churchill, G.A. Stochastic models for heterogeneous DNA sequences. B. Math. Biol. 1989, 51, 79–94. [Google Scholar] [CrossRef]
- Krogh, A.; Brown, M.; Mian, I.S.; Sjolander, K.; Haussler, D. Hidden Markov-Models in Computational Biology : Applications to Protein Modeling. J. Mol. Biol. 1994, 235, 1501–1531. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Mian, I.S.; Haussler, D. A Hidden Markov Model That Finds Genes in Escherichia-Coli Dna. Nucleic Acids Res. 1994, 22, 4768–4778. [Google Scholar] [CrossRef] [PubMed]
- Baldi, P.; Chauvin, Y.; Hunkapiller, T.; McClure, M.A. Hidden Markov models of biological primary sequence information. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.L.; Eddy, S.R.; Krogh, A.; Mitchison, G. Biological sequence analysis: probabilistic models of proteins and nucleic acids; 1999; Cambridge University Press: Cambridge, UK. [Google Scholar]
- Bergman, N.H.; Passalacqua, K.D.; Hanna, P.C.; Qin, Z.S. Operon prediction for sequenced bacterial genomes without experimental information. Appl. Environ. Microb. 2007, 73, 846–854. [Google Scholar] [CrossRef]
- Eddy, S.R. Multiple alignment using hidden Markov models. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1995, 3, 114–120. [Google Scholar] [PubMed]
- Lander, E.S.; Green, P. Construction of multilocus genetic linkage maps in humans. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Rabiner, L.R. A Tutorial On Hidden Markov-Models and Selected Applications in Speech Recognition. P. IEEE 1989, 77, 257–286. [Google Scholar] [CrossRef]
- Lahiri, S.N. Resampling Methods for Dependent Data; 2003; Springer-Verlag: New York, NY, USA. [Google Scholar]
- Shin, H.; Liu, T.; Manrai, A.K.; Liu, X.S. CEAS: cis-regulatory element annotation system. Bioinformatics 2009, 25, 2605–2606. [Google Scholar] [CrossRef] [PubMed]
- Salmon-Divon, M.; Dvinge, H.; Tammoja, K.; Bertone, P. PeakAnalyzer: Genome-wide annotation of chromatin binding and modification loci. BMC Bioinformatics 2010, 11, 415. [Google Scholar] [CrossRef] [PubMed]
- Blahnik, K.R.; Dou, L.; O'Geen, H.; McPhillips, T.; Xu, X.; Cao, A.R.; Iyengar, S.; Nicolet, C.M.; Ludascher, B.; Korf, I.; et al. Sole-Search: an integrated analysis program for peak detection and functional annotation using ChIP-seq data. Nucleic Acids Res. 2010, 38, e13. [Google Scholar] [CrossRef]
- Kadonaga, J.T. Regulation of RNA polymerase II transcription by sequence-specific DNA binding factors. Cell 2004, 116, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, W.W.; Sandelin, A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev.Genet. 2004, 5, 276–287. [Google Scholar] [CrossRef]
- Lawrence, C.E.; Altschul, S.F.; Boguski, M.S.; Liu, J.S.; Neuwald, A.F.; Wootton, J.C. Detecting Subtle Sequence Signals : a Gibbs Sampling Strategy For Multiple Alignment. Science 1993, 262, 208–214. [Google Scholar] [PubMed]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar] [PubMed]
- Liu, X.; Brutlag, D.L.; Liu, J.S. BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes . Pac. Symp. Biocomput. 2001, 127–138. [Google Scholar]
- Liu, X.S.; Brutlag, D.L.; Liu, J.S. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat. Biotechnol. 2002, 20, 835–839. [Google Scholar] [PubMed]
- Roth, F.P.; Hughes, J.D.; Estep, P.W.; Church, G.M. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat. Biotechnol. 1998, 16, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Bussemaker, H.J.; Li, H.; Siggia, E.D. Building a dictionary for genomes: identification of presumptive regulatory sites by statistical analysis. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 10096–10100. [Google Scholar] [CrossRef] [PubMed]
- Stormo, G.D.; Hartzell III, G.W. Identifying protein-binding sites from unaligned DNA fragments. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Tompa, M.; Li, N.; Bailey, T.L.; Church, G.M.; De Moor, B.; Eskin, E.; Favorov, A.V.; Frith, M.C.; Fu, Y.; Kent, W.J.; et al. Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 2005, 23, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Bussemaker, H.J.; Li, H.; Siggia, E.D. Regulatory element detection using correlation with expression. Nat. Genet. 2001, 27, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Conlon, E.M.; Liu, X.S.; Lieb, J.D.; Liu, J.S. Integrating regulatory motif discovery and genome-wide expression analysis. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3339–3344. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Keles, S. Integrating quantitative information from ChIP-chip experiments into motif finding. Biostatistics 2008, 9, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Nesvizhskii, A.I.; Ghosh, D.; Qin, Z.S. Hierarchical hidden Markov model with application to joint analysis of ChIP-chip and ChIP-seq data. Bioinformatics 2009, 25, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Kahvejian, A.; Quackenbush, J.; Thompson, J.F. What would you do if you could sequence everything? Nat. Biotechnol. 2008, 26, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Dohm, J.C.; Lottaz, C.; Borodina, T.; Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008, 36, e105. [Google Scholar] [CrossRef]
- Gilbert, P.B. A modified false discovery rate multiple-comparisons procedure for discrete data, applied to human immunodeficiency virus genetics. J. Roy. Stat. Soc. C App. 2005, 54, 143–158. [Google Scholar] [CrossRef]
- Ghosh, D. Discrete nonparametric algorithms for outlier detection with genomic data. J. Biopharm. Stat. 2010, 20, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Pounds, S.; Cheng, C. Robust estimation of the false discovery rate. Bioinformatics 2006, 22, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D. Detecting outlier genes from high-dimensional data: a fuzzy approach . Int. J. Syst. Synth. Biol. 2010, in press. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Strimmer, K. A general modular framework for gene set enrichment analysis. BMC Bioinformatics 2009, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Oshlack, A.; Wakefield, M.J. Transcript length bias in RNA-seq data confounds systems biology. Biol. Direct 2009, 4, 14. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.L.; Zhang, W.; Jiang, J.; et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Waterman, M.S. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics 1988, 2, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Wendl, M.C.; Wilson, R.K. Aspects of coverage in medical DNA sequencing. BMC Bioinformatics 2008, 9, 239. [Google Scholar] [CrossRef]
- Shendure, J.; Porreca, G.J.; Reppas, N.B.; Lin, X.; McCutcheon, J.P.; Rosenbaum, A.M.; Wang, M.D.; Zhang, K.; Mitra, R.D.; Church, G.M. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 2005, 309, 1728–1732. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Ghosh, D.; Qin, Z.S. Statistical Issues in the Analysis of ChIP-Seq and RNA-Seq Data. Genes 2010, 1, 317-334. https://doi.org/10.3390/genes1020317
Ghosh D, Qin ZS. Statistical Issues in the Analysis of ChIP-Seq and RNA-Seq Data. Genes. 2010; 1(2):317-334. https://doi.org/10.3390/genes1020317
Chicago/Turabian StyleGhosh, Debashis, and Zhaohui S. Qin. 2010. "Statistical Issues in the Analysis of ChIP-Seq and RNA-Seq Data" Genes 1, no. 2: 317-334. https://doi.org/10.3390/genes1020317
APA StyleGhosh, D., & Qin, Z. S. (2010). Statistical Issues in the Analysis of ChIP-Seq and RNA-Seq Data. Genes, 1(2), 317-334. https://doi.org/10.3390/genes1020317