Breaking Bottlenecks for the TCR Therapy of Cancer


Abstract
1. Immuno-Oncology’s Next Wave
2. The Unique Potential of T-Cell-Receptor (TCR) Therapy
3. Critical Tasks
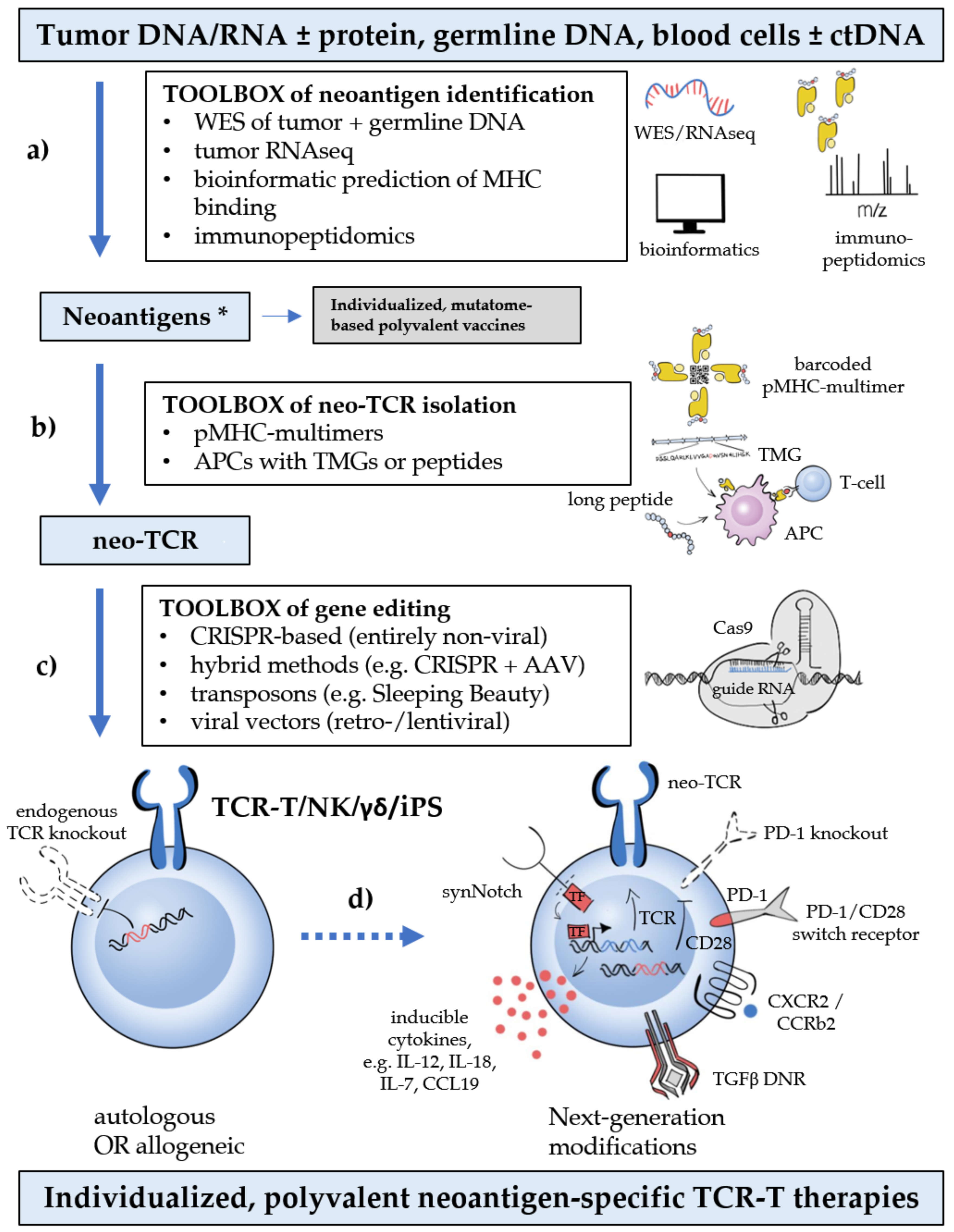
3.1. Neoantigen Multitargeting
3.2. Next-Generation Products
3.2.1. T-Cell Persistence, Memory, and Fitness
3.2.2. Navigating and Prevailing in the TME
3.2.3. Avoiding Toxicity
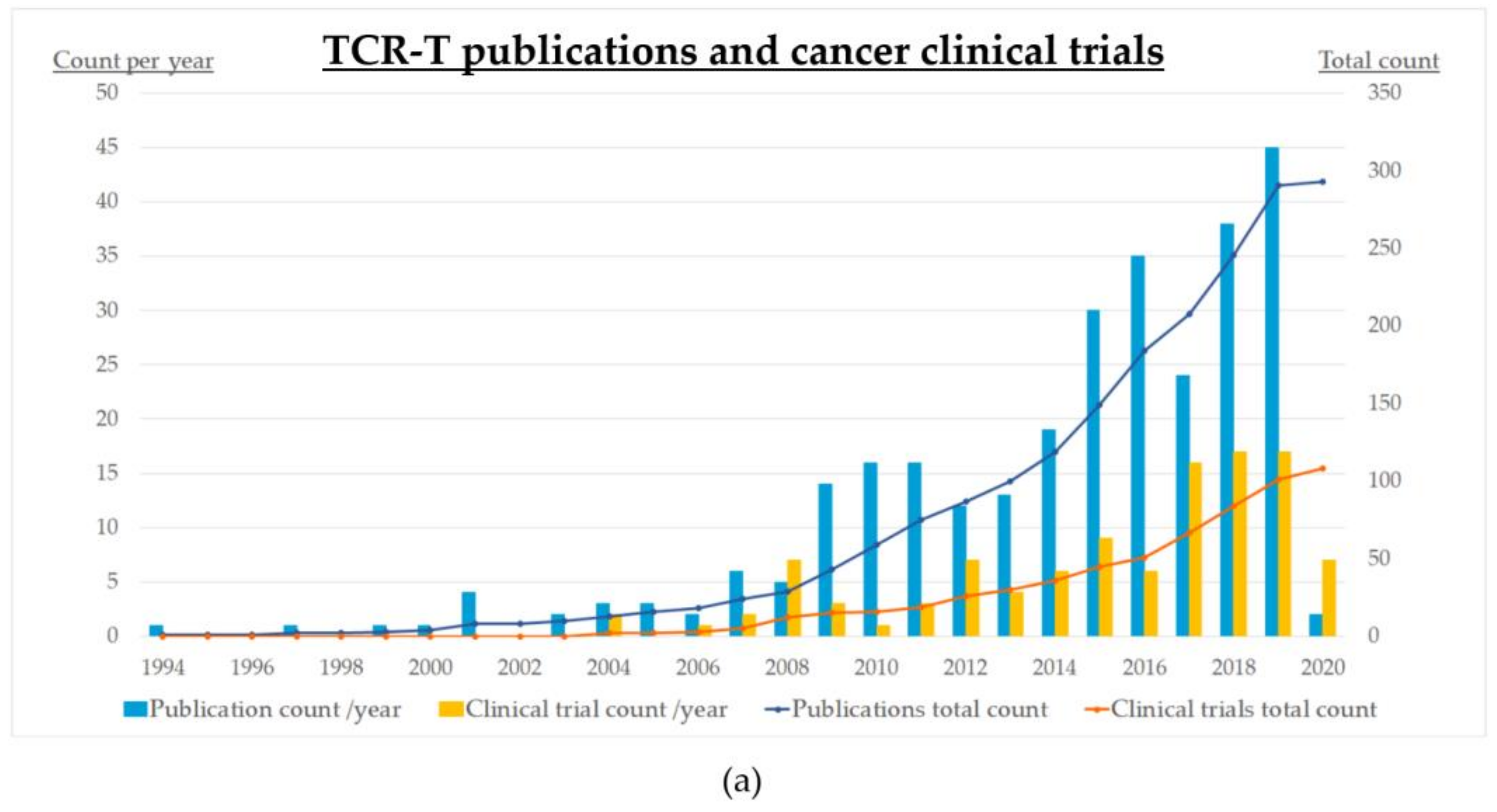
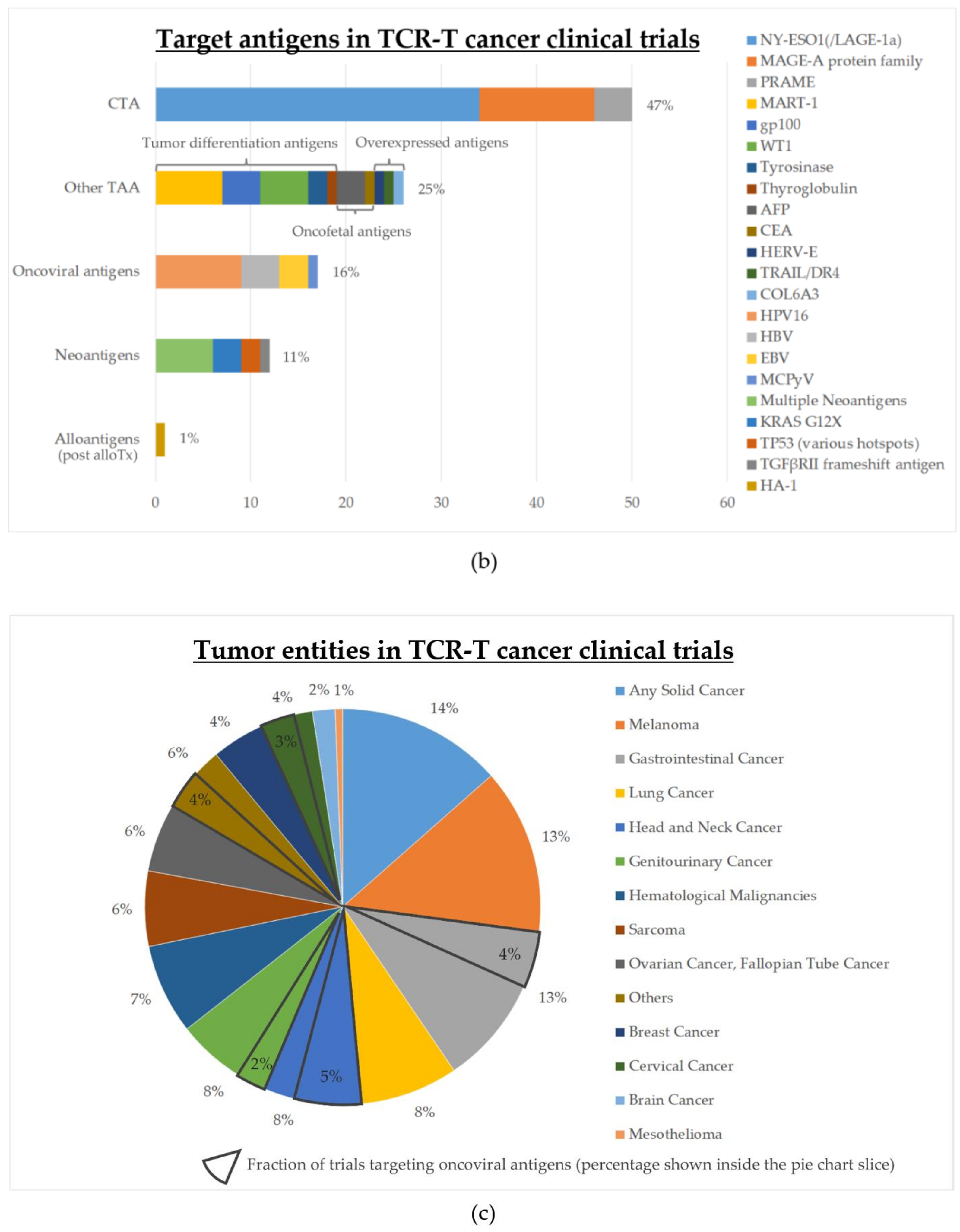
4. Status of Clinical Development
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.-J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-year overall survival for patients with advanced non‒small-cell lung cancer treated with pembrolizumab: Results from the phase I KEYNOTE-001 study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.; Horn, L.; Jackman, D.; Spigel, D.; Antonia, S.; Hellmann, M.; Powderly, J.; Heist, R.; Sequist, L.V.; Smith, D.C.; et al. Five-year follow-up of nivolumab in previously treated advanced non-small-cell lung cancer: Results from the CA209-003 study. J. Clin. Oncol. 2018, 36, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Xin Yu, J.; Hubbard-Lucey, V.M.; Tang, J. Immuno-oncology drug development goes global. Nat. Rev. Drug Discov. 2019, 18, 899–900. [Google Scholar] [CrossRef]
- Spiess, P.J.; Yang, J.C.; Rosenberg, S.A. In vivo antitumor activity of tumor-infiltrating lymphocytes expanded in recombinant interleukin-2. J. Natl. Cancer Inst. 1987, 79, 1067–1075. [Google Scholar]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Ben-Avi, R.; Farhi, R.; Ben-Nun, A.; Gorodner, M.; Greenberg, E.; Markel, G.; Schachter, J.; Itzhaki, O.; Besser, M.J. Establishment of adoptive cell therapy with tumor infiltrating lymphocytes for non-small cell lung cancer patients. Cancer Immunol. Immunother. 2018, 67, 1221–1230. [Google Scholar] [CrossRef]
- Garber, K. Pursuit of tumor-infiltrating lymphocyte immunotherapy speeds up. Nat. Biotechnol. 2019, 37, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Rohaan, M.W.; van den Berg, J.H.; Kvistborg, P.; Haanen, J.B.A.G. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: A viable treatment option. J. Immunother. Cancer 2018, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Usatorre, A.; Carmona, S.J.; Godfroid, C.; Yacoub, M.C.; Labiano, S.; Romero, P. Enhanced phenotype definition for precision isolation of precursor exhausted tumor-infiltrating CD8 T cells. Front. Immunol. 2020, 11, 340. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.N.; Kishton, R.J.; Restifo, N.P. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat. Med. 2019, 25, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, E.; Shahani, S.A.; Shah, N.N. Updates on CAR T-cell therapy in B-cell malignancies. Immunol. Rev. 2019, 290, 39–59. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L. The emerging world of TCR-T cell trials against cancer: A systematic review. Technol. Cancer Res. Treat. 2019, 18, 1533033819831068. [Google Scholar] [CrossRef]
- Garber, K. Driving T-cell immunotherapy to solid tumors. Nat. Biotechnol. 2018, 36, 215–219. [Google Scholar] [CrossRef]
- Puig-Saus, C.; Ribas, A. Gene editing: Towards the third generation of adoptive T-cell transfer therapies. Immuno-Oncol. Technol. 2019, 1, 19–26. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Rosenberg, S.A.; Restifo, N.P. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat. Med. 2016, 22, 26–36. [Google Scholar] [CrossRef]
- Chandran, S.S.; Klebanoff, C.A. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.T.; Kranz, D.M. Adoptive T cell therapies: A comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, Y.J. Engineered T cell therapy for cancer in the clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef]
- Valitutti, S.; Müller, S.; Cella, M.; Padovan, E.; Lanzavecchia, A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature 1995, 375, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Sykulev, Y.; Joo, M.; Vturina, I.; Tsomides, T.J.; Eisen, H.N. Evidence that a single peptide–MHC complex on a target cell can elicit a cytolytic T cell response. Immunity 1996, 4, 565–571. [Google Scholar] [CrossRef]
- Stauss, H.J. Turn to TCRs when CARs fail. Oncotarget 2017, 8, 12538–12539. [Google Scholar] [CrossRef]
- Ellis, J.M.; Henson, V.; Slack, R.; Ng, J.; Hartzman, R.J.; Katovich, H.C. Frequencies of HLA-A2 alleles in five U.S. population groups. Hum. Immunol. 2000, 61, 334–340. [Google Scholar] [CrossRef]
- Rotolo, A.; Karadimitris, A.; Ruella, M. Building upon the success of CART19: Chimeric antigen receptor T cells for hematologic malignancies. Leuk. Lymphoma 2018, 59, 2040–2055. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef]
- Van den Berg, J.H.; Gomez-Eerland, R.; van de Wiel, B.; Hulshoff, L.; van den Broek, D.; Bins, A.; Tan, H.L.; Harper, J.V.; Hassan, N.J.; Jakobsen, B.K.; et al. Case report of a fatal serious adverse event upon administration of T cells transduced with a MART-1-specific T-cell receptor. Mol. Ther. 2015, 23, 1541–1550. [Google Scholar] [CrossRef]
- Gotter, J.; Brors, B.; Hergenhahn, M.; Kyewski, B. Medullary epithelial cells of the human thymus express a highly diverse selection of tissue-specific genes colocalized in chromosomal clusters. J. Exp. Med. 2004, 199, 155–166. [Google Scholar] [CrossRef]
- Zhong, S.; Malecek, K.; Johnson, L.A.; Yu, Z.; de Vega-Saenz, M.E.; Darvishian, F.; McGary, K.; Huang, K.; Boyer, J.; Corse, E.; et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc. Natl. Acad. Sci. USA 2013, 110, 6973–6978. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, T.M.; Aggen, D.H.; Stromnes, I.M.; Dossett, M.L.; Richman, S.A.; Kranz, D.M.; Greenberg, P.D. Enhanced-affinity murine T-cell receptors for tumor/self-antigens can be safe in gene therapy despite surpassing the threshold for thymic selection. Blood 2013, 122, 348–356. [Google Scholar] [CrossRef]
- Obenaus, M.; Leitão, C.; Leisegang, M.; Chen, X.; Gavvovidis, I.; van der Bruggen, P.; Uckert, W.; Schendel, D.J.; Blankenstein, T. Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nat. Biotechnol. 2015, 33, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Moysey, R.; Molloy, P.E.; Vuidepot, A.-L.; Mahon, T.; Baston, E.; Dunn, S.; Liddy, N.; Jacob, J.; Jakobsen, B.K.; et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005, 23, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Li, Y.F.; El-Gamil, M.; Zhao, Y.; Wargo, J.A.; Zheng, Z.; Xu, H.; Morgan, R.A.; Feldman, S.A.; Johnson, L.A.; et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J. Immunol. 2008, 180, 6116–6131. [Google Scholar] [CrossRef] [PubMed]
- Chervin, A.S.; Aggen, D.H.; Raseman, J.M.; Kranz, D.M. Engineering higher affinity T cell receptors using a T cell display system. J. Immunol. Methods 2008, 339, 175–184. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.-A.N.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 c259T cells in synovial sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Border, E.C.; Sanderson, J.P.; Weissensteiner, T.; Gerry, A.B.; Pumphrey, N.J. Affinity-enhanced T-cell receptors for adoptive T-cell therapy targeting MAGE-A10: Strategy for selection of an optimal candidate. Oncoimmunology 2019, 8, e1532759. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef]
- Kaluza, K.M.; Kottke, T.; Diaz, R.M.; Rommelfanger, D.; Thompson, J.; Vile, R. Adoptive transfer of cytotoxic T lymphocytes targeting two different antigens limits antigen loss and tumor escape. Hum. Gene Ther. 2012, 23, 1054–1064. [Google Scholar] [CrossRef]
- Blankenstein, T.; Leisegang, M.; Uckert, W.; Schreiber, H. Targeting cancer-specific mutations by T cell receptor gene therapy. Curr. Opin. Immunol. 2015, 33, 112–119. [Google Scholar] [CrossRef]
- Schiller, J.T.; Lowy, D.R. Vaccines to prevent infections by oncoviruses. Annu. Rev. Microbiol. 2010, 64, 23–41. [Google Scholar] [CrossRef]
- Immatics US, Inc.; M.D. Anderson Cancer Center. Phase I Adoptive Cellular Therapy Trial With Endogenous CD8+ T Cells (ACTolog® IMA101) Alone or in Combination with Atezolizumab in Patients With Relapsed and/or Refractory Solid Cancers: NCT02876510, IMA101-101. Available online: https://clinicaltrials.gov/ct2/show/NCT02876510 (accessed on 7 June 2020).
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-cell responses to TP53 “hotspot” mutations and unique neoantigens expressed by human ovarian cancers. Clin. Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef]
- Anonymous. The problem with neoantigen prediction. Nat. Biotechnol. 2017, 35, 97. [Google Scholar] [CrossRef]
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for neoantigen identification. Front. Immunol. 2019, 10, 1392. [Google Scholar] [CrossRef] [PubMed]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Purcell, A.W.; Ramarathinam, S.H.; Ternette, N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019, 14, 1687–1707. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Burris, H.A.; Patel, M.R.; Cho, D.C.; Clarke, J.M.; Gutierrez, M.; Zaks, T.Z.; Frederick, J.; Hopson, K.; Mody, K.; Binanti-Berube, A.; et al. A phase I multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in patients with resected solid tumors and in combination with pembrolizumab in patients with unresectable solid tumors. J. Clin. Oncol. 2019, 37, 2523. [Google Scholar] [CrossRef]
- Bethune, M.T.; Joglekar, A.V. Personalized T cell-mediated cancer immunotherapy: Progress and challenges. Curr. Opin. Biotechnol. 2017, 48, 142–152. [Google Scholar] [CrossRef]
- Arnaud, M.; Duchamp, M.; Bobisse, S.; Renaud, P.; Coukos, G.; Harari, A. Biotechnologies to tackle the challenge of neoantigen identification. Curr. Opin. Biotechnol. 2020, 65, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef] [PubMed]
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; de Miranda, N.F.; Goodall, C.P.; Blair, T.C.; Fox, B.A.; McDermott, J.E.; et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018, 9, 2724. [Google Scholar] [CrossRef] [PubMed]
- Wolfl, M.; Kuball, J.; Ho, W.Y.; Nguyen, H.; Manley, T.J.; Bleakley, M.; Greenberg, P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood 2007, 110, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Puig-Saus, C.; Sennino, B.; Purandare, B.; An, D.; Quach, B.; Peng, S.; Xia, H.; Zhao, S.; Pan, Z.; Ma, Y.; et al. Landscape analysis of neoepitope-specific T-cell responses to immunotherapy. In The 111th Annual Meeting of the American Association for Cancer Research, Proceedings of the AACR Annual Meeting 2020, Philadelphia, PA, USA, 22–24 June 2020; AACR: Philadelphia, PA, USA, 2020. [Google Scholar]
- Parkhurst, M.; Gros, A.; Pasetto, A.; Prickett, T.; Crystal, J.S.; Robbins, P.; Rosenberg, S.A. Isolation of T-cell receptors specifically reactive with mutated tumor-associated antigens from tumor-infiltrating lymphocytes based on CD137 expression. Clin. Cancer Res. 2017, 23, 2491–2505. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Seliktar-Ofir, S.; Merhavi-Shoham, E.; Itzhaki, O.; Yunger, S.; Markel, G.; Schachter, J.; Besser, M.J. Selection of shared and neoantigen-reactive T cells for adoptive cell therapy based on CD137 separation. Front. Immunol. 2017, 8, 1211. [Google Scholar] [CrossRef]
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018, 3, e112467. [Google Scholar] [CrossRef]
- Liu, S.; Matsuzaki, J.; Wei, L.; Tsuji, T.; Battaglia, S.; Hu, Q.; Cortes, E.; Wong, L.; Yan, L.; Long, M.; et al. Efficient identification of neoantigen-specific T-cell responses in advanced human ovarian cancer. J. Immunother. Cancer 2019, 7, 156. [Google Scholar] [CrossRef]
- Ali, M.; Foldvari, Z.; Giannakopoulou, E.; Böschen, M.-L.; Strønen, E.; Yang, W.; Toebes, M.; Schubert, B.; Kohlbacher, O.; Schumacher, T.N.; et al. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 2019, 14, 1926–1943. [Google Scholar] [CrossRef]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; de Rooij, M.A.J.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K.; et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2019, 25, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Strønen, E.; Toebes, M.; Kelderman, S.; van Buuren, M.M.; Yang, W.; van Rooij, N.; Donia, M.; Böschen, M.-L.; Lund-Johansen, F.; Olweus, J.; et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016, 352, 1337–1341. [Google Scholar] [CrossRef] [PubMed]
- Dwarshuis, N.J.; Parratt, K.; Santiago-Miranda, A.; Roy, K. Cells as advanced therapeutics: State-of-the-art, challenges, and opportunities in large scale biomanufacturing of high-quality cells for adoptive immunotherapies. Adv. Drug Deliv. Rev. 2017, 114, 222–239. [Google Scholar] [CrossRef] [PubMed]
- Anczurowski, M.; Hirano, N. Two weeks’ notice from allogeneic sources. Clin. Cancer Res. 2018, 24, 5195–5197. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Leisegang, M.; Park, J.-H.; Ren, L.; Kato, T.; Ikeda, Y.; Harada, M.; Kiyotani, K.; Lengyel, E.; Fleming, G.F.; et al. Induction of neoantigen-specific cytotoxic T cells and construction of T-cell receptor-engineered T cells for ovarian cancer. Clin. Cancer Res. 2018, 24, 5357–5367. [Google Scholar] [CrossRef]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- Albers, J.J.; Ammon, T.; Gosmann, D.; Audehm, S.; Thoene, S.; Winter, C.; Secci, R.; Wolf, A.; Stelzl, A.; Steiger, K.; et al. Gene editing enables T-cell engineering to redirect antigen specificity for potent tumor rejection. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.-Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Berdien, B.; Mock, U.; Atanackovic, D.; Fehse, B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. 2014, 21, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.D.; Cohen, C.J.; Yang, S.; Hsu, C.; Jones, S.; Zhao, Y.; Zheng, Z.; Rosenberg, S.A.; Morgan, R.A. Efficient nonviral sleeping beauty transposon-based TCR gene transfer to peripheral blood lymphocytes confers antigen-specific antitumor reactivity. Gene Ther. 2009, 16, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.R.; Maus, M.V. Gene editing for immune cell therapies. Nat. Biotechnol. 2019, 37, 1425–1434. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef]
- Schuldt, N.J.; Binstadt, B.A. Dual TCR T cells: Identity crisis or multitaskers? J. Immunol. 2019, 202, 637–644. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Morris, G.P. The highly alloreactive nature of dual TCR T cells. Curr. Opin. Organ. Transpl. 2016, 21, 22–28. [Google Scholar] [CrossRef]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front. Immunol. 2018, 9, 14. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4+ T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2020. [Google Scholar] [CrossRef]
- Sillito, F.; Holler, A.; Stauss, H.J. Engineering CD4+ T cells to enhance cancer immunity. Cells 2020, 9, 1721. [Google Scholar] [CrossRef]
- Anderson, V.E.; Weber, A.M.; Wiedermann, G.E.; Pachnio, A.; Dauleh, S.; Ahmed, T.; Docta, R.Y.; Quattrini, A.; Pope, G.; Quinn, L.; et al. Abstract 2313: Enhanced activity of second-generation MAGE-A4 SPEAR T-cells through co-expression of a CD8α homodimer. In Immunology, Proceedings of the AACR Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019; AACR: Philadelphia, PA, USA, 2019; p. 2313. [Google Scholar]
- Ghorashian, S.; Veliça, P.; Chua, I.; McNicol, A.-M.; Carpenter, B.; Holler, A.; Nicholson, E.; Ahmadi, M.; Zech, M.; Xue, S.-A.; et al. CD8 T cell tolerance to a tumor-associated self-antigen is reversed by CD4 T cells engineered to express the same T cell receptor. J. Immunol. 2015, 194, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Holler, P.D.; Kranz, D.M. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity 2003, 18, 255–264. [Google Scholar] [CrossRef]
- Mensali, N.; Dillard, P.; Hebeisen, M.; Lorenz, S.; Theodossiou, T.; Myhre, M.R.; Fåne, A.; Gaudernack, G.; Kvalheim, G.; Myklebust, J.H.; et al. NK cells specifically TCR-dressed to kill cancer cells. EBioMedicine 2019, 40, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Parlar, A.; Sayitoglu, E.C.; Ozkazanc, D.; Georgoudaki, A.-M.; Pamukcu, C.; Aras, M.; Josey, B.J.; Chrobok, M.; Branecki, S.; Zahedimaram, P.; et al. Engineering antigen-specific NK cell lines against the melanoma-associated antigen tyrosinase via TCR gene transfer. Eur. J. Immunol. 2019, 49, 1278–1290. [Google Scholar] [CrossRef]
- Anonymous. HLA loss facilitates immune escape. Cancer Discov. 2018, 8, 8. [Google Scholar] [CrossRef]
- Minagawa, A.; Yoshikawa, T.; Yasukawa, M.; Hotta, A.; Kunitomo, M.; Iriguchi, S.; Takiguchi, M.; Kassai, Y.; Imai, E.; Yasui, Y.; et al. Enhancing T cell receptor stability in rejuvenated iPSC-derived T cells improves their use in cancer immunotherapy. Cell Stem Cell 2018, 23, 850–858.e4. [Google Scholar] [CrossRef]
- Van der Veken, L.T.; Hagedoorn, R.S.; van Loenen, M.M.; Willemze, R.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Alphabeta T-cell receptor engineered gammadelta T cells mediate effective antileukemic reactivity. Cancer Res. 2006, 66, 3331–3337. [Google Scholar] [CrossRef]
- Iriguchi, S.; Kaneko, S. Toward the development of true “off-the-shelf” synthetic T-cell immunotherapy. Cancer Sci. 2019, 110, 16–22. [Google Scholar] [CrossRef]
- Sadelain, M.; Rivière, I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef]
- Krause, A.; Guo, H.-F.; Latouche, J.-B.; Tan, C.; Cheung, N.-K.V.; Sadelain, M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Kim, C.H. Evolution of chimeric antigen receptor (CAR) T cell therapy: Current status and future perspectives. Arch. Pharmacal Res. 2019, 42, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, L.; Masoumi, E.; Fallah-Mehrjardi, K.; Mirzaei, H.R.; Hadjati, J. Prolonged persistence of Chimeric Antigen Receptor (CAR) T cell in adoptive cancer immunotherapy: Challenges and ways forward. Front. Immunol. 2020, 11, 702. [Google Scholar] [CrossRef]
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Investig. 2008, 118, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, C.S.; Borman, Z.A.; Cassard, L.; Gattinoni, L.; Spolski, R.; Yu, Z.; Sanchez-Perez, L.; Muranski, P.; Kern, S.J.; Logun, C.; et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 17469–17474. [Google Scholar] [CrossRef]
- Busch, D.H.; Fräßle, S.P.; Sommermeyer, D.; Buchholz, V.R.; Riddell, S.R. Role of memory T cell subsets for adoptive immunotherapy. Semin. Immunol. 2016, 28, 28–34. [Google Scholar] [CrossRef]
- Stemberger, C.; Dreher, S.; Tschulik, C.; Piossek, C.; Bet, J.; Yamamoto, T.N.; Schiemann, M.; Neuenhahn, M.; Martin, K.; Schlapschy, M.; et al. Novel serial positive enrichment technology enables clinical multiparameter cell sorting. PLoS ONE 2012, 7, e35798. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Zhou, J.; Jin, L.; Wang, F.; Zhang, Y.; Liu, B.; Zhao, T. Chimeric Antigen Receptor T (CAR-T) cells expanded with IL-7/IL-15 mediate superior antitumor effects. Protein Cell 2019, 10, 764–769. [Google Scholar] [CrossRef]
- McLellan, A.D.; Ali, H.R.S.M. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol. 2019, 97, 664–674. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Catakovic, K.; Klieser, E.; Neureiter, D.; Geisberger, R. T cell exhaustion: From pathophysiological basics to tumor immunotherapy. Cell Commun. Signal. 2017, 15, 1. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Menger, L.; Sledzinska, A.; Bergerhoff, K.; Vargas, F.A.; Smith, J.; Poirot, L.; Pule, M.; Herrero, J.; Peggs, K.S.; Quezada, S.A. TALEN-mediated inactivation of PD-1 in tumor-reactive lymphocytes promotes intratumoral T-cell persistence and rejection of established tumors. Cancer Res. 2016, 76, 2087–2093. [Google Scholar] [CrossRef]
- Prosser, M.E.; Brown, C.E.; Shami, A.F.; Forman, S.J.; Jensen, M.C. Tumor PD-L1 co-stimulates primary human CD8+ cytotoxic T cells modified to express a PD1: CD28 chimeric receptor. Mol. Immunol. 2012, 51, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Ankri, C.; Shamalov, K.; Horovitz-Fried, M.; Mauer, S.; Cohen, C.J. Human T cells engineered to express a programmed death 1/28 costimulatory retargeting molecule display enhanced antitumor activity. J. Immunol. 2013, 191, 4121–4129. [Google Scholar] [CrossRef]
- Hoogi, S.; Eisenberg, V.; Mayer, S.; Shamul, A.; Barliya, T.; Cohen, C.J. A TIGIT-based chimeric co-stimulatory switch receptor improves T-cell anti-tumor function. J. Immunother. Cancer 2019, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Morello, A.; Tano, Z.; Adusumilli, P.S. CAR T-cell intrinsic PD-1 checkpoint blockade: A two-in-one approach for solid tumor immunotherapy. Oncoimmunology 2017, 6, e1273302. [Google Scholar] [CrossRef]
- Oda, S.K.; Daman, A.W.; Garcia, N.M.; Wagener, F.; Schmitt, T.M.; Tan, X.; Chapuis, A.G.; Greenberg, P.D. A CD200R-CD28 fusion protein appropriates an inhibitory signal to enhance T-cell function and therapy of murine leukemia. Blood 2017, 130, 2410–2419. [Google Scholar] [CrossRef]
- Shin, J.H.; Park, H.B.; Oh, Y.M.; Lim, D.P.; Lee, J.E.; Seo, H.H.; Lee, S.J.; Eom, H.S.; Kim, I.-H.; Lee, S.H.; et al. Positive conversion of negative signaling of CTLA4 potentiates antitumor efficacy of adoptive T-cell therapy in murine tumor models. Blood 2012, 119, 5678–5687. [Google Scholar] [CrossRef]
- Massagué, J. TGFbeta in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, J.; Wang, Z.; Zhang, J.; Xiao, M.; Wang, C.; Lu, Y.; Qin, Z. Endogenous interleukin-4 promotes tumor development by increasing tumor cell resistance to apoptosis. Cancer Res. 2008, 68, 8687–8694. [Google Scholar] [CrossRef] [PubMed]
- Leen, A.M.; Sukumaran, S.; Watanabe, N.; Mohammed, S.; Keirnan, J.; Yanagisawa, R.; Anurathapan, U.; Rendon, D.; Heslop, H.E.; Rooney, C.M.; et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol. Ther. 2014, 22, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, Z.; Muranski, P.; Palmer, D.C.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. 2013, 20, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Roswell Park Cancer Institute; National Cancer Institute. A Phase I/II a Study of TGFß Blockade in TCR-Engineered T-Cell Cancer Immunotherapy in Patients with Advanced Malignancies: NCT02650986, I 258514. Available online: https://clinicaltrials.gov/ct2/show/NCT02650986 (accessed on 25 June 2020).
- Daniel-Meshulam, I.; Horovitz-Fried, M.; Cohen, C.J. Enhanced antitumor activity mediated by human 4-1BB-engineered T cells. Int. J. Cancer 2013, 133, 2903–2913. [Google Scholar] [CrossRef] [PubMed]
- Miyao, K.; Terakura, S.; Okuno, S.; Julamanee, J.; Watanabe, K.; Hamana, H.; Kishi, H.; Sakemura, R.; Koyama, D.; Goto, T.; et al. Introduction of genetically modified cd3zeta improves proliferation and persistence of antigen-specific CTLs. Cancer Immunol Res. 2018, 6, 733–744. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef]
- Zebley, C.C.; Gottschalk, S.; Youngblood, B. Rewriting history: Epigenetic reprogramming of CD8+ T cell differentiation to enhance immunotherapy. Trends Immunol. 2020, 41, 665–675. [Google Scholar] [CrossRef]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T cells into tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef]
- Idorn, M.; Skadborg, S.K.; Kellermann, L.; Halldórsdóttir, H.R.; Holmen, O.G.; Met, Ö.; Thor, S.P. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. Oncoimmunology 2018, 7, e1450715. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.S.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Alsaieedi, A.; Holler, A.; Velica, P.; Bendle, G.; Stauss, H.J. Safety and efficacy of Tet-regulated IL-12 expression in cancer-specific T cells. Oncoimmunology 2019, 8, 1542917. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-residing batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 2017, 31, 711–723.e4. [Google Scholar] [CrossRef]
- Galon, J.; Rossi, J.; Turcan, S.; Danan, C.; Locke, F.L.; Neelapu, S.S.; Miklos, D.B.; Bartlett, N.L.; Jacobson, C.A.; Braunschweig, I.; et al. Characterization of anti-CD19 chimeric antigen receptor (CAR) T cell-mediated tumor microenvironment immune gene profile in a multicenter trial (ZUMA-1) with axicabtagene ciloleucel (axi-cel, KTE-C19). J. Clin. Oncol. 2017, 35, 3025. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold tumors: A therapeutic challenge for immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Gaissmaier, L.; Christopoulos, P. Immune modulation in lung cancer: Current concepts and future strategies. Respiration 2020, in press. [Google Scholar] [CrossRef]
- Lai, J.; Mardiana, S.; House, I.G.; Sek, K.; Henderson, M.A.; Giuffrida, L.; Chen, A.X.Y.; Todd, K.L.; Petley, E.V.; Chan, J.D.; et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat. Immunol. 2020, 21, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.T.; Moffett, H.F.; Stephan, S.B.; Opel, C.F.; Dumigan, A.G.; Jiang, X.; Pillarisetty, V.G.; Pillai, S.P.S.; Wittrup, K.D.; Stephan, M.T. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Investig. 2017, 127, 2176–2191. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [PubMed]
- Chinnasamy, D.; Yu, Z.; Theoret, M.R.; Zhao, Y.; Shrimali, R.K.; Morgan, R.A.; Feldman, S.A.; Restifo, N.P.; Rosenberg, S.A. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J. Clin. Investig. 2010, 120, 3953–3968. [Google Scholar] [CrossRef] [PubMed]
- Foley, K.C.; Nishimura, M.I.; Moore, T.V. Combination immunotherapies implementing adoptive T-cell transfer for advanced-stage melanoma. Melanoma Res. 2018, 28, 171–184. [Google Scholar] [CrossRef]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 125, 3905–3916. [Google Scholar] [CrossRef] [PubMed]
- Minn, I.; Rowe, S.P.; Pomper, M.G. Enhancing CAR T-cell therapy through cellular imaging and radiotherapy. Lancet Oncol. 2019, 20, e443–e451. [Google Scholar] [CrossRef]
- Rosewell, S.A.; Suzuki, M. Oncolytic viruses partner with T-cell therapy for solid tumor treatment. Front. Immunol. 2018, 9, 2103. [Google Scholar] [CrossRef]
- Christopoulos, P.; Pfeifer, D.; Bartholomé, K.; Follo, M.; Timmer, J.; Fisch, P.; Veelken, H. Definition and characterization of the systemic T-cell dysregulation in untreated indolent B-cell lymphoma and very early CLL. Blood 2011, 117, 3836–3846. [Google Scholar] [CrossRef]
- Christopoulos, P.; Follo, M.; Fisch, P.; Veelken, H. The peripheral helper T-cell repertoire in untreated indolent B-cell lymphomas: Evidence for antigen-driven lymphomagenesis. Leukemia 2008, 22, 1952–1954. [Google Scholar] [CrossRef]
- Christopoulos, P.; Dopfer, E.P.; Malkovsky, M.; Esser, P.R.; Schaefer, H.-E.; Marx, A.; Kock, S.; Rupp, N.; Lorenz, M.R.; Schwarz, K.; et al. A novel thymoma-associated immunodeficiency with increased naive T cells and reduced CD247 expression. J. Immunol. 2015, 194, 3045–3053. [Google Scholar] [CrossRef]
- Christopoulos, P.; Chung, I.; Bozorgmehr, F.; Muley, T.; Meister, M.; Kobinger, S.; Marx, A.; Thomas, M.; Winter, H.; Herpel, E.; et al. Deficient CD247 expression is a typical histopathological characteristic of thymomas with cortical features. Histopathology 2018, 73, 1040–1043. [Google Scholar] [CrossRef]
- Christopoulos, P.; Fisch, P. Acquired T-cell immunodeficiency in thymoma patients. Crit. Rev. Immunol. 2016, 36, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Chen, L. A paradigm shift in cancer immunotherapy: From enhancement to normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, P.; Schneider, M.A.; Bozorgmehr, F.; Kuon, J.; Engel-Riedel, W.; Kollmeier, J.; Baum, V.; Muley, T.; Schnabel, P.A.; Bischoff, H.; et al. Large cell neuroendocrine lung carcinoma induces peripheral T-cell repertoire alterations with predictive and prognostic significance. Lung Cancer 2018, 119, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Van Loenen, M.M.; de Boer, R.; Amir, A.L.; Hagedoorn, R.S.; Volbeda, G.L.; Willemze, R.; van Rood, J.J.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc. Natl. Acad. Sci. USA 2010, 107, 10972–10977. [Google Scholar] [CrossRef]
- Bunse, M.; Bendle, G.M.; Linnemann, C.; Bies, L.; Schulz, S.; Schumacher, T.N.; Uckert, W. RNAi-mediated TCR knockdown prevents autoimmunity in mice caused by mixed TCR dimers following TCR gene transfer. Mol. Ther. 2014, 22, 1983–1991. [Google Scholar] [CrossRef]
- Cohen, C.J.; Zhao, Y.; Zheng, Z.; Rosenberg, S.A.; Morgan, R.A. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006, 66, 8878–8886. [Google Scholar] [CrossRef]
- Reuß, S.; Sebestyén, Z.; Heinz, N.; Loew, R.; Baum, C.; Debets, R.; Uckert, W. TCR-engineered T cells: A model of inducible TCR expression to dissect the interrelationship between two TCRs. Eur. J. Immunol. 2014, 44, 265–274. [Google Scholar] [CrossRef]
- Ochi, T.; Fujiwara, H.; Okamoto, S.; An, J.; Nagai, K.; Shirakata, T.; Mineno, J.; Kuzushima, K.; Shiku, H.; Yasukawa, M. Novel adoptive T-cell immunotherapy using a WT1-specific TCR vector encoding silencers for endogenous TCRs shows marked antileukemia reactivity and safety. Blood 2011, 118, 1495–1503. [Google Scholar] [CrossRef]
- Okamoto, S.; Amaishi, Y.; Goto, Y.; Ikeda, H.; Fujiwara, H.; Kuzushima, K.; Yasukawa, M.; Shiku, H.; Mineno, J. A Promising Vector for TCR Gene Therapy: Differential Effect of siRNA, 2A Peptide, and Disulfide Bond on the Introduced TCR Expression. Mol. Ther. Nucleic Acids 2012, 1, e63. [Google Scholar] [CrossRef] [PubMed]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Schober, K.; Müller, T.R.; Busch, D.H. Orthotopic T-Cell Receptor Replacement-An “Enabler” for TCR-Based Therapies. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Legut, M.; Cole, D.K.; Sewell, A.K. The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell. Mol. Immunol. 2015, 12, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [PubMed]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.S.; Lamb, L.S.; Goldman, F.; Di Stasi, A. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Bonini, C.; Stanghellini, M.T.L.; Bondanza, A.; Traversari, C.; Salomoni, M.; Turchetto, L.; Colombi, S.; Bernardi, M.; Peccatori, J.; et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): A non-randomised phase I–II study. Lancet Oncol. 2009, 10, 489–500. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| CAR | TCR | |
|---|---|---|
| Target Ag | Surface proteins, glycoproteins, glycolipids, carbohydrates | Peptides from surface and intracellular proteins |
| Ag recognition | MHC-independent | MHC-dependent |
| Receptor structure | Single-chain, scFv 3 ITAMs | αβ heterodimer 10 ITAMs |
| Affinity for target | Nanomolar range | Micromolar range |
| Required target density for response | >103/cell | ∼1–50/cell |
| Type of Antigen | Number of Trials | % of Active Trials 1 | % of Completed Trials | Start Year of the First Trial |
|---|---|---|---|---|
| Cancer-testis antigens (CTA) | 50 | 56% (28/50) | 12% (6/50) | 2008 |
| Other tumor-associated antigens (TAA) | 25 | 44% (11/25) | 28% (7/25) | 2004 |
| Oncoviral antigens | 17 | 88% (15/17) | 12% (2/17) | 2014 |
| Neoantigens (public or private) | 12 | 75% (9/12) | 8% (1/12) | 2006 (public) 2018 (private) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaissmaier, L.; Elshiaty, M.; Christopoulos, P. Breaking Bottlenecks for the TCR Therapy of Cancer. Cells 2020, 9, 2095. https://doi.org/10.3390/cells9092095
Gaissmaier L, Elshiaty M, Christopoulos P. Breaking Bottlenecks for the TCR Therapy of Cancer. Cells. 2020; 9(9):2095. https://doi.org/10.3390/cells9092095
Chicago/Turabian StyleGaissmaier, Lena, Mariam Elshiaty, and Petros Christopoulos. 2020. "Breaking Bottlenecks for the TCR Therapy of Cancer" Cells 9, no. 9: 2095. https://doi.org/10.3390/cells9092095
APA StyleGaissmaier, L., Elshiaty, M., & Christopoulos, P. (2020). Breaking Bottlenecks for the TCR Therapy of Cancer. Cells, 9(9), 2095. https://doi.org/10.3390/cells9092095