Autologous, Non-Invasively Available Mesenchymal Stem Cells from the Outer Root Sheath of Hair Follicle Are Obtainable by Migration from Plucked Hair Follicles and Expandable in Scalable Amounts

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
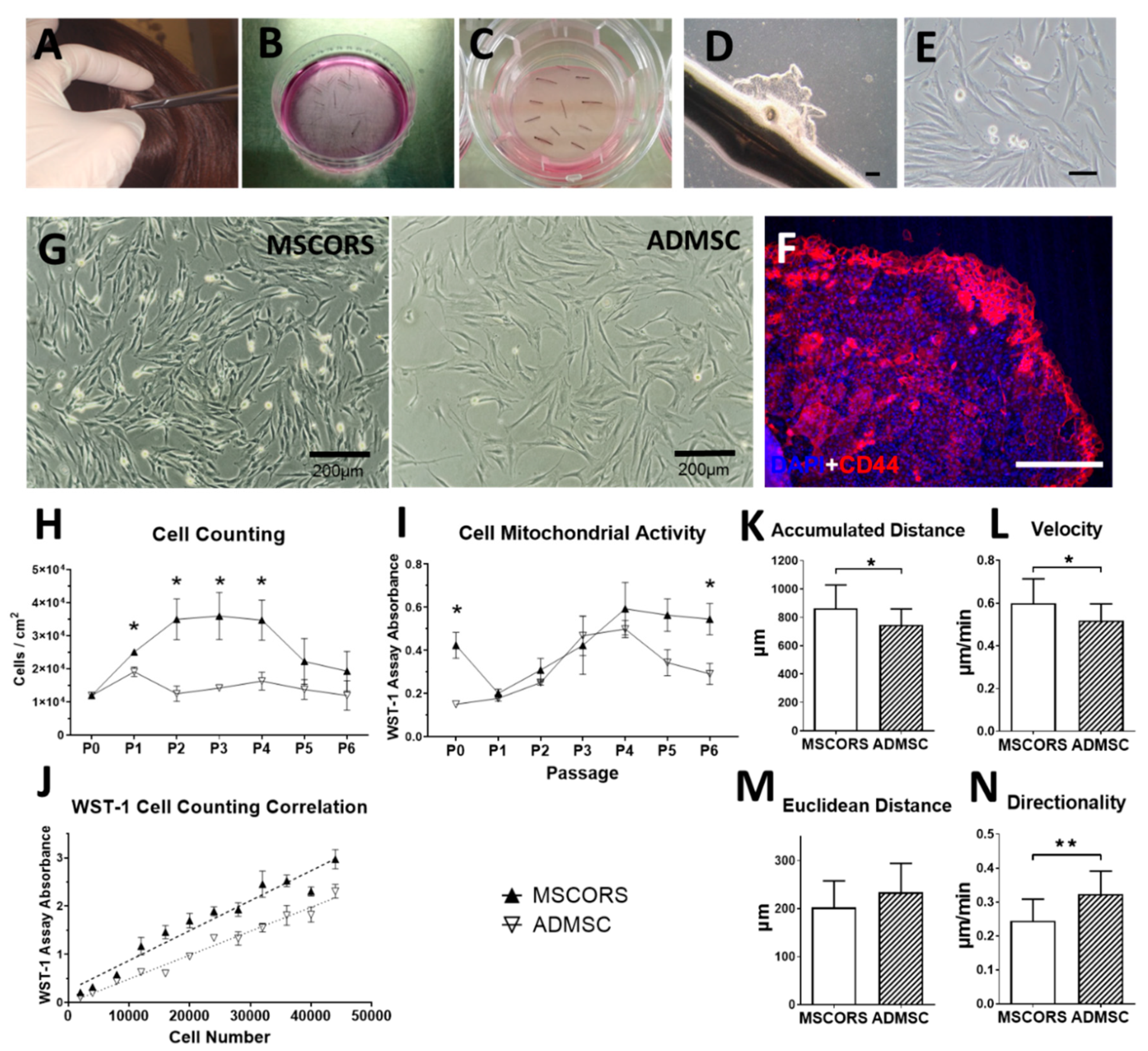
2.1. Isolation and Culture of MSCORS
2.2. Isolation and Culture of Adipose Derived MSCs (ADMSCs)
2.3. Determination of Cell Count and Cell Mitochondrial Activity
2.4. Determination of Cell Movement: Live Cell Imaging Time Lapse
2.5. Determination of Gene Expression: Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.6. Determination of Cell Surface Marker: Fluorescence-Activated Cell Sorting (FACS)
2.7. Determination of Cell Surface Marker and Differentiation Markers: Immunofluorescence Staining
2.8. Osteogenic Differentiation
2.9. Chondrogenic Differentiation
2.10. Adipogenic Differentiation
2.11. Endothelial Differentiation
2.12. Smooth Muscle Differentiation
2.13. Determination of Cell Immunomodulatory Effects
2.14. Statistical Analysis
3. Results
3.1. MSCORS Isolation by Migration and Adherent Culture
3.2. Cell Proliferation and Mitochondrial Activity
3.3. Cell Motility and Migration Parameters
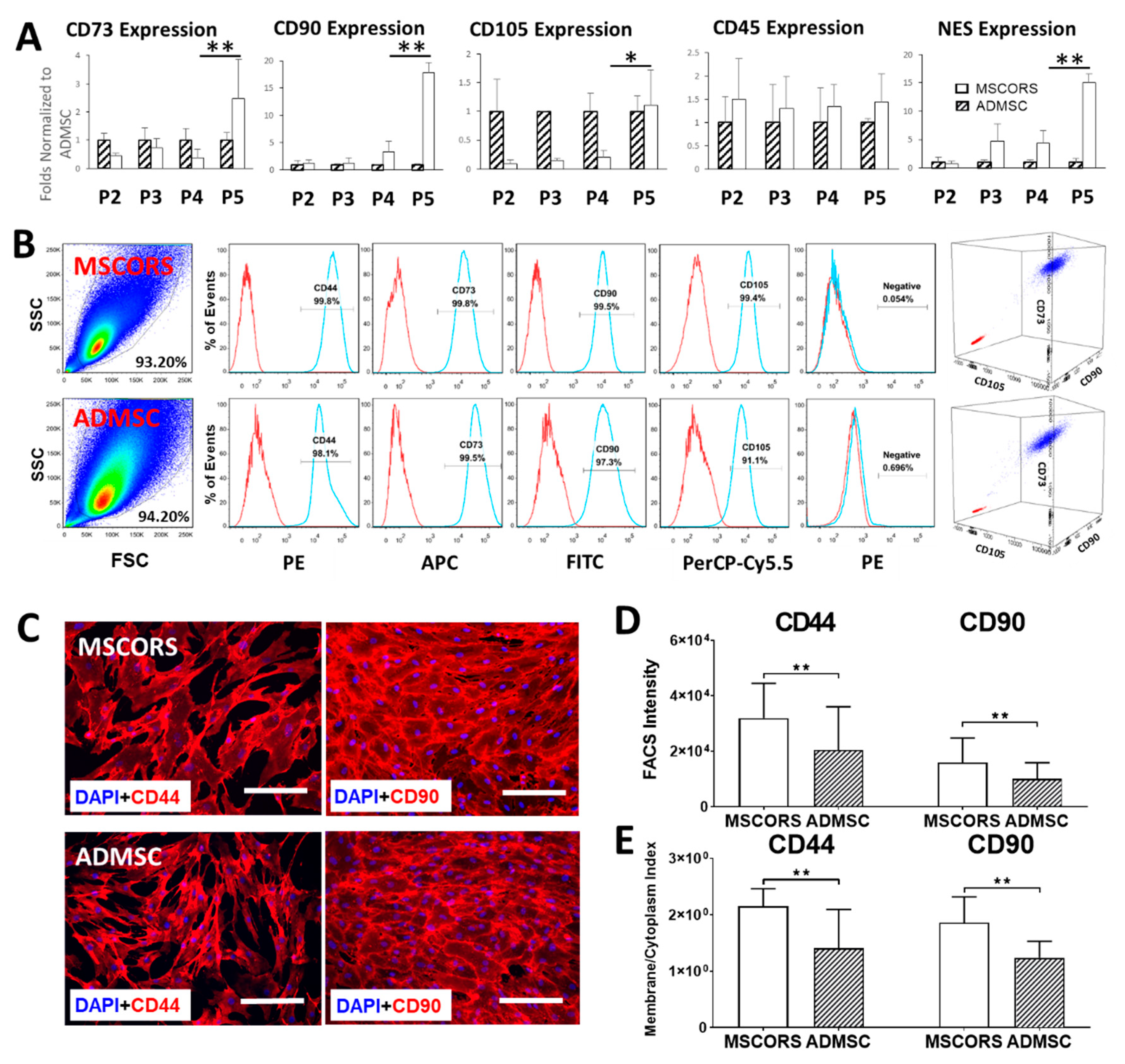
3.4. Gene Expression
3.5. MSC Surface Marker Expression
3.6. Protein Expression and Subcellular Distribution
3.7. Differentiation Potentials
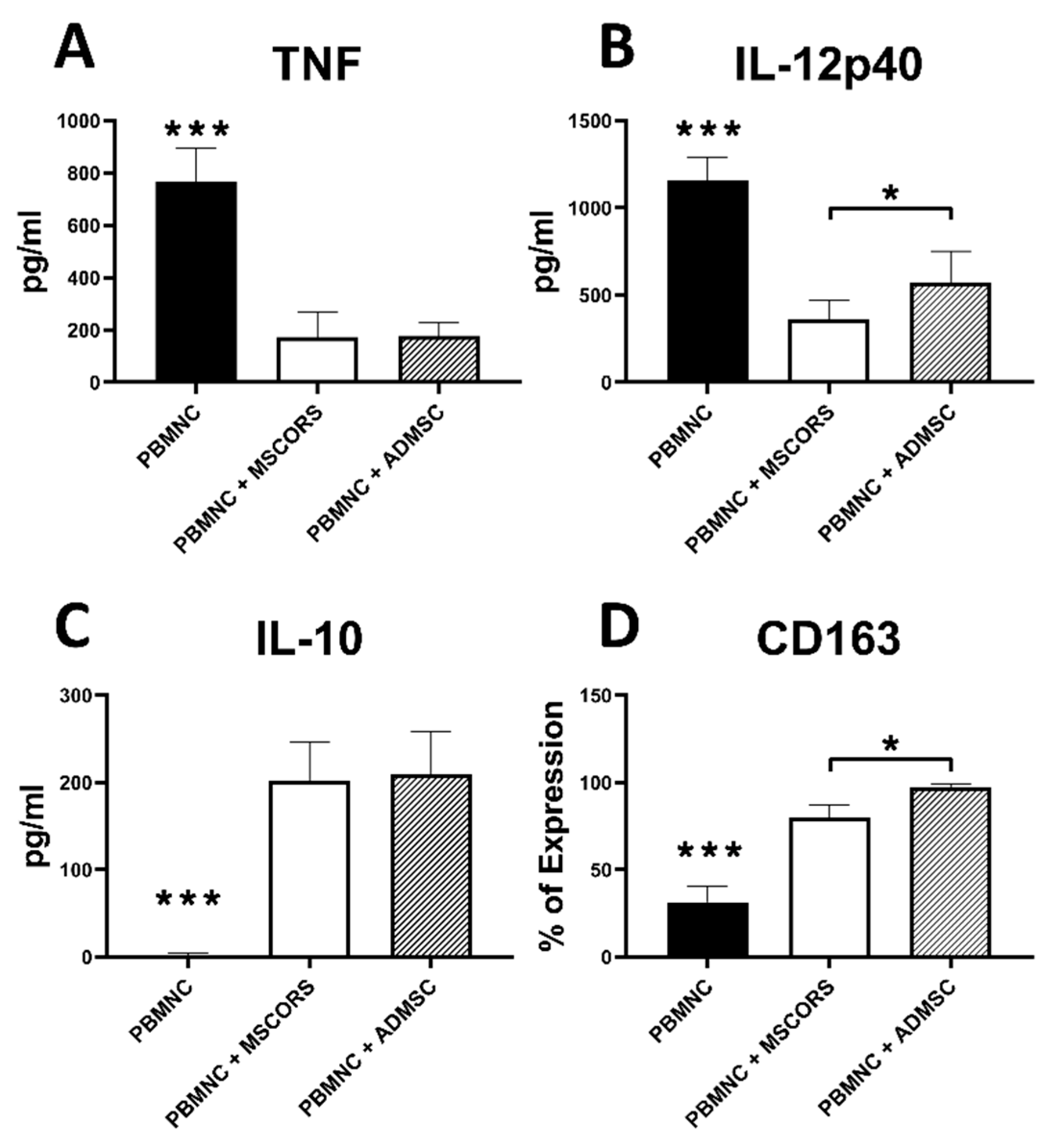
3.8. Immunomodulatory Effects of MSCORS vs. ADMSC
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Savkovic, V.; Li, H.; Seon, J.K.; Hacker, M.; Franz, S.; Simon, J.C. Mesenchymal stem cells in cartilage regeneration. Curr. Stem Cell Res. 2014, 9, 469–488. [Google Scholar] [CrossRef] [PubMed]
- Bartel, R.L. Chapter 8—Stem Cells and Cell Therapy: Autologous Cell Manufacturing. In Translational Regenerative Medicine; Atala, A., Allickson, J.G., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 107–112. [Google Scholar]
- EUCouncil. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating To Medicinal Products for Human Use. Off. J. Eur. Communities L 2001, 311, 62. [Google Scholar]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Park, C.W.; Kim, K.-S.; Bae, S.; Son, H.K.; Myung, P.-K.; Hong, H.J.; Kim, H. Cytokine secretion profiling of human mesenchymal stem cells by antibody array. Int. J. Stem Cells 2009, 2, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusuma, G.D.; Carthew, J.; Lim, R.; Frith, J.E. Effect of the Microenvironment on Mesenchymal Stem Cell Paracrine Signaling: Opportunities to Engineer the Therapeutic Effect. Stem Cells Dev. 2017, 26, 617–631. [Google Scholar] [CrossRef]
- Dominici, M.; Blanc, K.L.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. J. Cytotherapy 2006, 8, 315–317. [Google Scholar]
- Viswanathan, S.; Shi, Y.; Galipeau, J.; Krampera, M.; Leblanc, K.; Martin, I.; Nolta, J.; Phinney, D.G.; Sensebe, L. Mesenchymal stem versus stromal cells: International Society for Cell & Gene Therapy (ISCT) Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy 2019, 21, 1019–1024. [Google Scholar]
- Frith, J.E.; Cameron, A.R.; Menzies, D.J.; Ghosh, P.; Whitehead, D.L.; Gronthos, S.; Zannettino, A.C.; Cooper-White, J.J. An injectable hydrogel incorporating mesenchymal precursor cells and pentosan polysulphate for intervertebral disc regeneration. Biomaterials 2013, 34, 9430–9440. [Google Scholar] [CrossRef]
- Haynesworth, S.E.; Goshima, J.; Goldberg, V.M.; Caplan, A.I. Characterization of cells with osteogenic potential from human marrow. Bone 1992, 13, 81–88. [Google Scholar] [CrossRef]
- Cao, C.; Dong, Y. Study on culture and in vitro osteogenesis of blood-derived human mesenchymal stem cells. Chin. J. Reparative Reconstr. Surg. 2005, 19, 642–647. [Google Scholar]
- Zuk, P.A.; Zhu, M.; Ashjian, P.; De Ugarte, D.A.; Huang, J.I.; Mizuno, H.; Alfonso, Z.C.; Fraser, J.K.; Benhaim, P.; Hedrick, M.H. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 2002, 13, 4279–4295. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.K.; Wulur, I.; Alfonso, Z.; Hedrick, M.H. Fat tissue: An underappreciated source of stem cells for biotechnology. Trends Biotechnol. 2006, 24, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, M.J.D.; Bonnet, D.; Janes, S.M. Stem cells of the alveolar epithelium. Lancet 2005, 366, 249–260. [Google Scholar] [CrossRef]
- Morsczeck, C.; Gotz, W.; Schierholz, J.; Zeilhofer, F.; Kuhn, U.; Mohl, C.; Sippel, C.; Hoffmann, K.H. Isolation of precursor cells (PCs) from human dental follicle of wisdom teeth. Matrix Biol. 2005, 24, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.I.; Hong, H.-H.; Lin, W.-R.; Fu, J.-F.; Chang, C.-C.; Wang, I.K.; Huang, W.-H.; Weng, C.-H.; Hsu, C.-W.; Yen, T.-H. Isolation of Mesenchymal Stem Cells from Human Deciduous Teeth Pulp. BioMed Res. Int. 2017, 2017, 2851906. [Google Scholar] [CrossRef]
- Jones, E.A.; English, A.; Henshaw, K.; Kinsey, S.E.; Markham, A.F.; Emery, P.; McGonagle, D. Enumeration and phenotypic characterization of synovial fluid multipotential mesenchymal progenitor cells in inflammatory and degenerative arthritis. Arthritis Rheum. 2004, 50, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhao, Y.; Wu, X.; Yin, S.; Chuai, Y.; Wang, A. The utility of human fallopian tube mucosa as a novel source of multipotent stem cells for the treatment of autologous reproductive tract injury. Stem Cell Res. 2015, 6, 98. [Google Scholar] [CrossRef] [Green Version]
- Barlow, S.; Brooke, G.; Chatterjee, K.; Price, G.; Pelekanos, R.; Rossetti, T.; Doody, M.; Venter, D.; Pain, S.; Gilshenan, K. Comparison of human placenta-and bone marrow–derived multipotent mesenchymal stem cells. Stem Cells Dev. 2008, 17, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Anker, P.S.; Scherjon, S.A.; van der Keur, C.K.; de Groot-Swings, G.M.J.S.; Claas, F.H.J.; Fibbe, W.E.; Kanhai, H.H.H. Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta. Stem Cells 2004, 22, 1338–1345. [Google Scholar] [CrossRef]
- Savkovic, V.; Li, H.; Simon, J.-C.; Etz, C.; Schneider, M. Method of Culturing Mesenchymal Stem Cells. U.S. Patent No. PCT/EP2020/070027, 13 July 2019. [Google Scholar]
- Ohyama, M.; Terunuma, A.; Tock, C.L.; Radonovich, M.F.; Pise-Masison, C.A.; Hopping, S.B.; Brady, J.N.; Udey, M.C.; Vogel, J.C. Characterization and isolation of stem cell–enriched human hair follicle bulge cells. J. Clin. Investig. 2006, 116, 249–260. [Google Scholar] [CrossRef]
- Amoh, Y.; Li, L.; Katsuoka, K.; Penman, S.; Hoffman, R. Multipotent nestin-positive, keratin-negative hair-follicle bulge stem cells can form neurons. Proc. Natl. Acad. Sci. USA 2005, 102, 5530–5534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoh, Y.; Li, L.; Campillo, R.; Kawahara, K.; Katsuoka, K.; Penman, S.; Hoffman, R.M. Implanted hair follicle stem cells form Schwann cells that support repair of severed peripheral nerves. Proc. Natl. Acad. Sci. USA 2005, 102, 17734–17738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Kumar, S.M.; Kossenkov, A.V.; Showe, L.; Xu, X. Stem cells with neural crest characteristics derived from the bulge region of cultured human hair follicles. J. Investig. Dermatol. 2010, 130, 1227–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, V.; Lindon, C.; Harfe, B.D.; Morgan, B.A. Distinct stem cell populations regenerate the follicle and interfollicular epidermis. Dev. Cell 2005, 9, 855–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snippert, H.J.; Haegebarth, A.; Kasper, M.; Jaks, V.; van Es, J.H.; Barker, N.; van de Wetering, M.; van den Born, M.; Begthel, H.; Vries, R.G.; et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science 2010, 327, 1385–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaks, V.; Kasper, M.; Toftgård, R. The hair follicle—a stem cell zoo. Exp. Cell Res. 2010, 316, 1422–1428. [Google Scholar] [CrossRef]
- Barker, N.; Shawna Tan, S.; Clevers, H. Lgr proteins in epithelial stem cell biology. Development 2013, 140, 2484–2494. [Google Scholar] [CrossRef] [Green Version]
- Blanpain, C. Skin regeneration and repair. Nature 2010, 464, 686. [Google Scholar] [CrossRef]
- Shih, D.T.; Lee, D.C.; Chen, S.C.; Tsai, R.Y.; Huang, C.T.; Tsai, C.C.; Shen, T.E.Y.; Chiu, W.T. Isolation and characterization of neurogenic mesenchymal stem cells in human scalp tissue. Stem Cells 2005, 23, 1012–1020. [Google Scholar] [CrossRef]
- Ma, D.; Kua, J.E.; Lim, W.K.; Lee, S.T.; Chua, A.W. In vitro characterization of human hair follicle dermal sheath mesenchymal stromal cells and their potential in enhancing diabetic wound healing. Cytotherapy 2015, 17, 1036–1051. [Google Scholar] [CrossRef]
- Li, P.; Liu, F.; Wu, C.; Jiang, W.; Zhao, G.; Liu, L.; Bai, T.; Wang, L.; Jiang, Y.; Guo, L.; et al. Feasibility of human hair follicle-derived mesenchymal stem cells/CultiSpher®-G constructs in regenerative medicine. Cell Tissue Res. 2015, 362, 69–86. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Gao, Y.; Liu, X.; Bai, T.; Li, M.; Li, L.; Chi, G.; Xu, H.; Liu, F.; et al. Maintenance of high proliferation and multipotent potential of human hair follicle-derived mesenchymal stem cells by growth factors. Int. J. Mol. Med. 2013, 31, 913–921. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, J.; Tan, X.; Li, G.; Gao, Y.; Liu, X.; Zhang, L.; Li, Y. Induced pluripotent stem cells from human hair follicle mesenchymal stem cells. Stem Cell Rev. 2013, 9, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplan, A.I. Mesenchymal Stem Cells: Time to Change the Name! Stem Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas-Camarena, L. Lipoaspiration and its complications: A safe operation. Plast Reconstr. Surg. 2003, 112, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Reig, G.; Pulgar, E.; Concha, M.L. Cell migration: From tissue culture to embryos. Development 2014, 141, 1999–2013. [Google Scholar] [CrossRef] [Green Version]
- Perez, R.L.; Münz, F.; Vidoni, D.; Rühle, A.; Trinh, T.; Sisombath, S.; Zou, B.; Wuchter, P.; Debus, J.; Grosu, A.-L.; et al. Mesenchymal stem cells preserve their stem cell traits after exposure to antimetabolite chemotherapy. Stem Cell Res. 2019, 40, 101536. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [Green Version]
- Marquez-Curtis, L.A.; Janowska-Wieczorek, A. Enhancing the Migration Ability of Mesenchymal Stromal Cells by Targeting the SDF-1/CXCR4 Axis. BioMed Res. Int. 2013, 2013, 561098. [Google Scholar] [CrossRef] [Green Version]
- Rolf, H.; Kevin, J.M. Compositions and Methods for Treating and Repairing Tendons. U.S. Patent No. EP2956543, 12 February 2014. [Google Scholar]
- Inoue, K.; Yoshimura, K. Skin Stem Cells. In Methods in Molecular Biology (Methods and Protocols); Humana Press: Totowa, NJ, USA, 2013. [Google Scholar]
- Tsuji, K.; Ojima, M.; Otabe, K.; Horie, M.; Koga, H.; Ekiya, I.; Muneta, T. Effects of Different Cell-Detaching Methods on the Viability and Cell Surface Antigen Expression of Synovial Mesenchymal Stem Cells. Cell Transpl. 2017, 26, 1089–1102. [Google Scholar] [CrossRef] [Green Version]
- Savkovic, V.; Dieckmann, C.; Milkova, L.; Simon, J.C. Improved method of differentiation, selection and amplification of human melanocytes from the hair follicle cell pool. Exp. Derm. 2012, 21, 948–950. [Google Scholar] [CrossRef]
- Schneider, M.; Dieckmann, C.; Rabe, K.; Simon, J.-C.; Vuk Savkovic, V. Differentiating the Stem Cell Pool of Human Hair Follicle Outer Root Sheath into Functional Melanocytes. Stem Cells Tissue Repair 2014, 1020, 203–227. [Google Scholar]
- Sülflow, K.; Schneider, M.; Loth, T.; Kascholke, C.; Schulz-Siegmund, M.; Hacker, M.C.; Simon, J.C.; Savkovic, V. Melanocytes from the outer root sheath of human hair and epidermal melanocytes display improved melanotic features in the niche provided by cGEL, oligomer-cross-linked gelatin-based hydrogel. J. Biomed. Mater. Res. A 2016, 104, 3115–3126. [Google Scholar] [CrossRef]
- Savkovic, V.; Dieckmann, C.; Simon, J.-C.; Schulz-Siegmund, M.; Hacker, M. Method for Deriving Melanocytes from the Hair Follicle Outer Root Sheath and Preparation for Grafting. U.S. Patent No. EP11186944, 20 June 2013. [Google Scholar]
- Grayson, W.L.; Zhao, F.; Izadpanah, R.; Bunnell, B.; Ma, T. Effects of hypoxia on human mesenchymal stem cell expansion and plasticity in 3D constructs. J. Cell Physiol. 2006, 207, 331–339. [Google Scholar] [CrossRef]
- Ito, M.; Liu, Y.; Yang, Z.; Nguyen, J.; Liang, F.; Morris, R.J.; Cotsarelis, G. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med. 2005, 11, 1351–1354. [Google Scholar] [CrossRef] [PubMed]
- Al-Rekabi, Z.; Fura, A.M.; Juhlin, I.; Yassin, A.; Popowics, T.E.; Sniadecki, N.J. Hyaluronan-CD44 interactions mediate contractility and migration in periodontal ligament cells. Cell Adhes. Migr. 2019, 13, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Rege, T.A.; Hagood, J.S. Thy-1 as a regulator of cell-cell and cell-matrix interactions in axon regeneration, apoptosis, adhesion, migration, cancer, and fibrosis. FASEB J. 2006, 20, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Bertolo, A.; Gemperli, A.; Gruber, M.; Gantenbein, B.; Baur, M.; Pötzel, T.; Stoyanov, J. In vitro cell motility as a potential mesenchymal stem cell marker for multipotency. Stem Cells Transl. Med. 2015, 4, 84–90. [Google Scholar] [CrossRef]
- de Witte, S.F.H.; Luk, F.; Parraga, J.M.S.; Gargesha, M.; Merino, A.; Korevaar, S.S.; Shankar, A.S.; O’Flynn, L.; Elliman, S.J.; Roy, D. Immunomodulation by therapeutic mesenchymal stromal cells (MSC) is triggered through phagocytosis of MSC by monocytic cells. Stem Cells 2018, 36, 602–615. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Garza, D.M.; Cantú-Rodríguez, O.G.; Jaime-Pérez, J.C.; Gutiérrez-Aguirre, C.H.; Góngora-Rivera, J.F.; Gómez-Almaguer, D. Current state and perspectives of stem cell therapy for stroke. Med. Univ. 2016, 18, 169–180. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Hair Follicles | Hair Weight (mg) | Cell Yield in P0 (Million Cells) | Cell Yield in P1 (Million Cells) | Cell Yield in P2 (Million Cells) |
|---|---|---|---|---|
| 60 | 21.45 ± 12.17 | 5.00 ± 2.68 | 21.90 ± 9.46 | 50.31 ± 27.79 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Masieri, F.F.; Schneider, M.; Kottek, T.; Hahnel, S.; Yamauchi, K.; Obradović, D.; Seon, J.-K.; Yun, S.J.; Ferrer, R.A.; et al. Autologous, Non-Invasively Available Mesenchymal Stem Cells from the Outer Root Sheath of Hair Follicle Are Obtainable by Migration from Plucked Hair Follicles and Expandable in Scalable Amounts. Cells 2020, 9, 2069. https://doi.org/10.3390/cells9092069
Li H, Masieri FF, Schneider M, Kottek T, Hahnel S, Yamauchi K, Obradović D, Seon J-K, Yun SJ, Ferrer RA, et al. Autologous, Non-Invasively Available Mesenchymal Stem Cells from the Outer Root Sheath of Hair Follicle Are Obtainable by Migration from Plucked Hair Follicles and Expandable in Scalable Amounts. Cells. 2020; 9(9):2069. https://doi.org/10.3390/cells9092069
Chicago/Turabian StyleLi, Hanluo, Federica Francesca Masieri, Marie Schneider, Tina Kottek, Sebastian Hahnel, Kensuke Yamauchi, Danilo Obradović, Jong-Keun Seon, Sook Jung Yun, Rubén A. Ferrer, and et al. 2020. "Autologous, Non-Invasively Available Mesenchymal Stem Cells from the Outer Root Sheath of Hair Follicle Are Obtainable by Migration from Plucked Hair Follicles and Expandable in Scalable Amounts" Cells 9, no. 9: 2069. https://doi.org/10.3390/cells9092069
APA StyleLi, H., Masieri, F. F., Schneider, M., Kottek, T., Hahnel, S., Yamauchi, K., Obradović, D., Seon, J.-K., Yun, S. J., Ferrer, R. A., Franz, S., Simon, J.-C., Lethaus, B., & Savković, V. (2020). Autologous, Non-Invasively Available Mesenchymal Stem Cells from the Outer Root Sheath of Hair Follicle Are Obtainable by Migration from Plucked Hair Follicles and Expandable in Scalable Amounts. Cells, 9(9), 2069. https://doi.org/10.3390/cells9092069