ER Stress-Induced Secretion of Proteins and Their Extracellular Functions in the Heart



1
Department of Internal Medicine III (Cardiology, Angiology, and Pneumology), Heidelberg University Hospital, Im Neuenheimer Feld 669, 69120 Heidelberg, Germany
2
DZHK (German Centre for Cardiovascular Research), Partner Site Heidelberg/Mannheim, 69120 Heidelberg, Germany
*
Author to whom correspondence should be addressed.
Cells 2020, 9(9), 2066; https://doi.org/10.3390/cells9092066
Submission received: 5 August 2020
/
Revised: 2 September 2020
/
Accepted: 3 September 2020
/
Published: 10 September 2020
(This article belongs to the Special Issue The Role of Proteostasis Derailment in Cardiac Diseases)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Endoplasmic reticulum (ER) stress is a result of conditions that imbalance protein homeostasis or proteostasis at the ER, for example ischemia, and is a common event in various human pathologies, including the diseased heart. Cardiac integrity and function depend on the active secretion of mature proteins from a variety of cell types in the heart, a process that requires an intact ER environment for efficient protein folding and trafficking to the secretory pathway. As a consequence of ER stress, most protein secretion by the ER secretory pathway is decreased. Strikingly, there is a select group of proteins that are secreted in greater quantities during ER stress. ER stress resulting from the dysregulation of ER Ca2+ levels, for instance, stimulates the secretion of Ca2+-binding ER chaperones, especially GRP78, GRP94, calreticulin, and mesencephalic astrocyte-derived neurotrophic factor (MANF), which play a multitude of roles outside the cell, strongly depending on the cell type and tissue. Here we review current insights in ER stress-induced secretion of proteins, particularly from the heart, and highlight the extracellular functions of these proteins, ranging from the augmentation of cardiac cell viability to the modulation of pro- and anti-apoptotic, oncogenic, and immune-stimulatory cell signaling, cell invasion, extracellular proteostasis, and more. Many of the roles of ER stress-induced protein secretion remain to be explored in the heart. This article is part of a special issue entitled “The Role of Proteostasis Derailment in Cardiac Diseases.”
1. Protein Secretion in the Heart
The heart is made of highly differentiated cells that must communicate with each other and with cells outside the heart to facilitate normal organ development and function [1]. The four most abundant cardiac cell types are fibroblasts, endothelial cells, cardiac myocytes, and smooth muscle cells [2]. Active protein secretion is a crucial element of intercellular communication and mediates autocrine, paracrine, or endocrine signaling, depending on whether secreted proteins have an effect on the secreting cell, neighboring cells, or distant tissues, respectively. Passive secretion, by contrast, refers to the release of cellular contents from dying cells, following necrotic tissue damage. Proteins actively secreted from cardiac cells are called cardiokines [3,4] and are released via the classical [5] or non-classical [6] secretory pathways (Figure 1). In the classical pathway, proteins are secreted after they are synthesized and translocated into the endoplasmic reticulum (ER). They are then transported to the Golgi and subsequently secreted after secretory vesicles fuse with the plasma membrane. Non-classical protein secretion does not involve the ER-Golgi pathway.
In classical protein secretion, the mRNAs encoding many secreted and membrane proteins are specifically localized to the cytosolic face of the ER membrane by several mechanisms, the most studied of which involves signal recognition particles (SRP) [7]. In eukaryotes, SRP binds to the signal sequence of a newly synthesized peptide as it emerges from the ribosome and through interactions with the SRP receptor, targets the ribosome-nascent chain complex to dock with the protein-conducting channel, also known as the translocon, in the ER membrane. Upon ribosome docking to the ER, nascent polypeptides are co-translationally inserted into the ER lumen the translocon [8]. SRP-independent pathways for the delivery of mRNAs to the ER have been proposed [9,10], with new evidence suggesting that in addition to SRP-targeted signal sequences in the peptide nascent chain, alternative signal motifs in the mRNA may also be responsible for directing protein synthesis to the ER [11]. Subsequent to protein folding and modification, proteins are routed in vesicle-mediated transport alongside the ER-Golgi intermediate compartment (ERGIC) and the Golgi apparatus towards their final destination [12]. The last step of classical protein secretion can be constitutive or regulated, depending on whether the secretory vesicles fuse with the plasma membrane directly after packaging, or whether they are stored until the appropriate stimulus activates their secretion (Figure 1; Classical secretion). One prominent example of a classically secreted cardiokine is the atrial natriuretic peptide (ANP), or atrial natriuretic factor (ANF), a small peptide hormone that reduces blood pressure and volume in an endocrine manner; ANF is released from atrial myocytes by regulated secretion in response to atrial stretch [13,14,15].
Non-classical secretion, on the other hand, describes ER-/Golgi-independent protein trafficking through various kinds of either vesicle- or nonvesicle-mediated pathways [16]. At least five non-classical protein secretion pathways have been described [17]. They include: (1) lysosomal secretion, (2) cleavage and release of a membrane-bound protein, (3) release via exosomes derived from multivesicular bodies, (4) membrane blebbing, and (5) direct translocation of proteins across the plasma membrane through membrane transporters (Figure 1; Non-classical secretion 1–5). One important example, S100A1, a cardiac myocyte-specific member of the EF-hand Ca2+-binding S100 protein family, is a regulator of cardiac myocyte function and is secreted via the non-classical secretory pathway during myocardial infarction. Extracellular S100A1 is taken up by cells via Ca2+-dependent, clathrin-mediated endocytosis and protects cardiac myocytes from apoptosis by signaling through phospholipase C (PLC) and protein kinase C (PKC) associated with endosomes [18].
Taken together, actively secreted proteins play many roles in the healthy heart, as well as in the damaged heart, including cardiac tissue remodeling and repair [3]. Considering the role of the ER in classical protein secretion, the status of the ER is an important determinant of cardiac function.
2. ER Stress in the Heart
2.1. Cardiac Proteostasis at the ER
The quality and quantity of cellular protein is managed by an extensive network of molecular mechanisms that collectively constitute the dynamic balance of protein homeostasis, or proteostasis [19]. Proteostasis involves the synthesis, folding, and trafficking of proteins, as well as the degradation of excess protein, terminally unfolded, or misfolded proteins, and responses to stresses that risk protein quality by dysregulating proteostasis. Proteostasis is essential for cellular function in and outside the heart [20], leading to longer life span when maintained properly [21]. Accordingly, cardiac ageing is associated with gradual derailment of cardiac proteostasis, which underlies the development of numerous cardiac diseases [22]. An optimal ER environment is required for efficient protein synthesis, folding, and modification. Many proteins need to be folded into the appropriate three-dimensional configuration to be functional [23]. Efficient protein folding in the ER is determined by the availability of ER Ca2+ [24], N-linked glycosylation [25], redox status of the ER with regards to disulfide bond formation [26], and the abundance of ER-resident molecular chaperones that assist in protein folding [27]. Terminally unfolded or misfolded proteins, which have failed dynamic protein folding and quality control, are removed from the ER via the ubiquitin/proteasome-dependent process of ER-associated protein degradation (ERAD) [28]. Since there are no proteasomes in the lumen of the ER, misfolded proteins in the ER must be translocated out of the ER and into the cytosol, where they are ubiquitylated by ER-transmembrane E3 ubiquitin-protein ligases and subsequently degraded by proteasomes on the cytosolic face of the ER [29]. In addition to the ER, specialized cells types, such as cardiac myocytes, comprise further intracellular membrane networks, i.e., the sarcoplasmic reticulum (SR). The attachment of ribosomes to the SR suggests that protein synthesis may also take place at the SR [30]. The SR is best known for its function in Ca2+ storage, release, and reuptake for excitation-contraction coupling in muscle cells [31]. Nonetheless, proper definition and distinction of the ER and SR with regards to essential components of cellular proteostasis, for example localization of protein synthesis, remain to be determined.
2.2. ER Stress and Heart Disease
Age- and disease-linked alterations of the ER environment, or genetically encoded mutations that misbalance proteostasis at the ER, lead to a mismatch of ER protein folding capacity and load, resulting in ER stress and the accumulation of misfolded proteins in the ER [32]. Misfolded proteins not only lack biological function, but may gain potentially toxic activity by exposing hydrophobic regions to nearby proteins, which can result in inappropriate binding and aggregation of nonfunctional structures [33]. To avoid proteotoxic effects in stressed cells, the ER senses protein folding status within the ER by monitoring levels of folding-deficient proteins, which leads to the activation of a conserved signaling network, called the unfolded protein response (UPR) [34] (Figure 2).
As a consequence, ER protein folding capacity is enhanced to match its demand by focusing cellular resources on synthesizing proteins that are involved in the folding process, such as protein disulfide isomerases and ER-resident chaperones [36]. Meanwhile, ER protein folding load is reduced by suppression of overall cellular protein synthesis [36], along with the augmentation of ER-associated degradation (ERAD) of misfolded proteins [37]. Together, these aspects of the UPR ultimately contribute to relieve ER stress [38]. Three ER-resident integral membrane proteins, PERK (protein kinase RNA (PKR)-like ER kinase) [39,40], IRE1α (inositol requiring enzyme 1α) [41,42,43,44], and ATF6 (activating transcription factor 6) [45,46,47], function as ER stress sensors (Figure 2). Downstream UPR signaling can be activated by the dissociation of the ER-luminal chaperone, glucose-regulated protein 78 kDa (GRP78), from the ER-transmembrane sensors, to assist the refolding of misfolded proteins or direct them towards ERAD [48]. Additional evidence suggests that misfolded proteins can also directly associate with the ER stress sensors, particularly IRE1α [49]; however, the exact mechanisms by which ER stress is sensed are still not clearly understood. Upon activation, PERK, IRE1α, and ATF6 transmit the information about detected ER stress into the cytosol through regulation of translational control (PERK), mRNA splicing (IRE1α), and regulated proteolysis (ATF6). From there, the corresponding transcription factors ATF4, XBP1, and ATF6 enter the nucleus, where they bind to specific elements in ER stress response genes and regulate their transcription [34]. Overall, the UPR is oriented toward the reestablishment of proteostasis in order to promote cell survival, which is adaptive. However, if the adaptive UPR is insufficient to reestablish proteostasis, depending on the strength and duration of ER stress, the UPR becomes maladaptive, guiding the cell towards apoptosis [50,51,52]. In particular, prolonged activation of the PERK branch of the UPR determines the apoptotic cell fate [53]. Programmed cell death upon unresolved ER stress is an especially important problem in the heart, given the limited regenerative potential of cardiac muscle [54].
UPR signaling is activated in cardiac myocytes under various pathologic conditions, such as ischemia, hypertrophy, or heart failure [55,56,57]. A prominent pathologic setting that causes ER stress in the heart is ischemia, the deprivation of oxygen and nutrient delivery to cardiac cells due to inadequate blood flow, often as a result of atherosclerosis or myocardial infarction. While ischemic cardiac myocytes in an infarct zone undergo cell death within 15–30 min of loss of blood supply, the surviving myocardium in the periphery of an infarct is exposed to milder ischemia [58]. Several studies have aimed to determine the extent to which each UPR pathway and subcellular signaling of its components are adaptive and can be used for therapeutic approaches. For example, under conditions when energy sources are not abundant, such as ischemia, activation of the ATF6 branch of the UPR reprograms cardiac myocytes in ways that limit growth, thus, conserving energy [56,59]. In this case, the induction of anti-growth-oriented genes during ER stress is considered protective, since it contributes to protect cardiac tissue against pathological hypertrophy, which is a hallmark of the diseased heart [60]. However, the UPR is multifaceted and under other conditions, when energy sources are abundant, ATF6 serves as regulator of the induction of major growth-promoting pathways in cardiac myocytes [61].
3. Protein Secretion during ER Stress
Cardiac proteostasis is crucial for the secretion of mature, properly folded proteins from cardiac cells. ER stress impairs most of protein processing and transport through the classical secretory pathway, ultimately limiting active protein secretion in stressed cells [62]. In addition to PERK-dependent translational attenuation, the UPR contributes to the reduction of ER protein trafficking by regulated IRE1α-dependent decay of mRNA (RIDD) [63], a mechanism of selective degradation of ER-bound mRNAs, and selective release of secreted and membrane protein-encoding mRNAs from the ER to the cytosol [64]. Secretion of selective proteins, however, is induced during ER stress, even when secretion of most other proteins that are secreted via the ER-Golgi secretory pathway has been attenuated. Proteins selectively secreted during ER stress fall into at least one of two categories: (1) functional proteins that are secreted upon the pathologic alteration of the ER environment by a distinct stress stimulus, i.e., decreased ER Ca2+ levels during ischemia [65], and (2) functional proteins whose secretion is directly regulated by the UPR [66]. Both kinds of proteins can exert protective effects after secretion, from outside cells, which will be discussed in the following sections in more detail.
4. Secretion of Ca2+-Binding ER Chaperones
4.1. Secretion during Ca2+-Mediated ER Stress
Ca2+ handling in cardiac cells is essential for cardiac contractibility [67]. Dysregulation of Ca2+ handling in cardiac myocytes contributes substantially to the initiation and progression of cardiac arrhythmia and heart failure [68]. ER Ca2+ depletion by chemical stressors, for example thapsigargin, a strong inhibitor of ER Ca2+-ATPases, is a common method for examining the effects of ER stress, in particular ER stress mediated by ER Ca2+ depletion [69]. Thapsigargin mimics the pathological decrease of ER Ca2+ levels during ischemic conditions, under which, for example, ATP production and thereby the activity of ATPases is limited. Moreover, the majority of ER-resident molecular chaperones and folding enzymes (protein disulfide isomerases and peptidyl prolyl isomerases) are low-affinity, high-capacity Ca2+-binding proteins and the activities of many of these are Ca2+-regulated [70,71]. Therefore, chaperone and folding functions are disturbed upon perturbation of ER-luminal Ca2+ levels [72,73,74,75].
Studies in which ER stress is induced either by ER Ca2+ depletion, for example by thapsigargin treatment or, alternatively, by ER protein glycosylation inhibition, by compounds such as tunicamycin, have shown that only Ca2+-mediated ER stress induces the secretion of a select group of ER proteins [62,65]. These findings suggest that different ER stressors have different effects on protein secretion and highlight the particular importance of altered ER Ca2+ levels for ER stress-induced protein secretion. Several studies in cardiac myocytes, fibroblasts, and other cell types, have reported on the secretion of ER-luminal chaperones with Ca2+-binding properties during Ca2+-mediated ER stress. Among these proteins are GRP78, GRP94, calreticulin [65], and mesencephalic astrocyte-derived neurotrophic factor (MANF) [76], all of which require Ca2+ for essential protein-protein interactions. In the ER, these proteins are typically involved in protein, as well as, Ca2+ homeostasis. Beyond the ER, they can be found, in part, in various other subcellular compartments, including the cytoplasm, nucleus, mitochondria, and the cell surface, with distinct functions depending on their localization [77,78,79] and novel functions continuously being discovered [80].
ER-luminal protein mobility was first observed in the 1990s in immunogold electron microscopy studies that showed, for example, the export of GRP78 and GRP94 from the ER and routing through the classical secretory pathway in rat exocrine pancreatic cells [81]. Although these Ca2+-binding ER chaperones possess a C-terminal retention motif that facilitates binding to the KDEL-receptor [82], which fosters the retrieval of proteins from the cis-Golgi back to the ER [83], they manage to overcome ER retention under certain conditions, such as ER Ca2+ depletion. Current explanations include an overwhelmed capacity of the KDEL retrieval system, alteration of KDEL system components, masking of the KDEL motif, for example by glycosylation of the protein sequence adjacent to KDEL [84], and decreased ER retention caused by drastically reduced ER Ca2+ levels during certain ER stress [85]. Notably, GRP78, GRP94, calreticulin, and MANF are also ER stress-inducible transcriptional targets of the UPR, particularly of the transcription factors ATF6 and XBP1. Nevertheless, their secretion happens within minutes of ER Ca2+ depletion, suggesting that it is independent from their transcriptional induction [76]. Since their transcripts efficiently escape translational inhibition, UPR-induced upregulation of these proteins can increase their extracellular levels further.
4.2. Extracellular Functions of ER Chaperones in the Cardiovascular System
Accumulating evidence suggests that secreted Ca2+-binding ER chaperones in the heart contribute to enhanced cardiac cell viability and cardioprotection. Our recent study on the cardiac myocyte secretome during ER stress reported the selective secretion of ER-resident proteins, i.e., GRP78, calreticulin, and GRP94, from cardiac myocytes in vitro upon ER Ca2+ depletion by thapsigargin or stimulated ischemia [62]. These proteins were cytoprotective in a culture media volume-dependent, auto- and paracrine manner, supporting their potential role in the regulation of cell viability in the onset of Ca2+-mediated ER stress. Mechanistically, GRP78 was shown to interact with the protein, Cripto, on the cell surface of cardiac myocytes to activate pro-survival AKT signaling and to inhibit death-promoting SMAD2 signaling (Figure 3A–C). This study demonstrated for the first time that GRP78 secreted as a result of ER stress can protect cardiac myocytes in a Cripto-dependent manner. The small, glycosylphosphatidylinositol (GPI)-anchored signaling protein, Cripto, plays important roles in vertebrate embryogenesis as well as tumorigenesis (e.g., inhibition of TGFβ signaling) [86,87]. Previously, GRP78 was identified as a necessary mediator of oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways at the cell surface of non-cardiac cell types, such as cancer cells [88,89]. Importantly, in addition to its secretion in response to ER stress, a subfraction of GRP78 can relocate to the cell surface during ER stress [84], where it resides mostly as a peripheral protein via interaction with other proteins [90], and acts as a multifunctional receptor in various signaling pathways [91] (see more in Section 4.3). Moreover, Bi et al. showed that overexpression of GRP78 in neonatal rat ventricular myocytes protected myocytes from ischemia/reperfusion-induced cell death, and cardiac myocyte-specific overexpression of GRP78 decreased ischemia/reperfusion damage to the heart [92]. GRP78-mediated cytoprotection involved plasma membrane translocation of GRP78 and interaction with PI3 kinase, resulting in AKT activation.
Another study identified GRP78 on the surface of endothelium and monocyte/macrophage-like cells in atherosclerotic lesions, where it was shown to negatively regulate tissue factor (TF) procoagulant activity via direct binding to and functional inhibition of TF [93]. Meanwhile, calreticulin protects against mild ischemia/reperfusion injury via association with the cardiokine C1q/TNF-related protein 9 (CTRP9) on the cell surface of cardiac myocytes through activation of the PKA/CREB pathway, ultimately inhibiting cardiac myocyte apoptosis [94]. Moreover, MANF is also known to be selectively released during Ca2+-mediated ER stress, including hypoxia, ischemia, and treatment with thapsigargin, and after its release, MANF protects cardiac myocytes from ischemia/reperfusion damage in vitro and in vivo [76,85]. MANF family proteins are highly evolutionarily conserved and both human and Drosophila orthologues promote dopamine neuron survival [95,96,97,98]. For over a decade since the cytoprotective effects of extracellular MANF were first reported [95,99], no receptor for MANF was known. However, recently, lipid sulfatide, also called 3-O-sulfogalactosylceramide, a sulfoglycolipid synthesized in ER and Golgi, which distributes to the extracellular leaflet of the plasma membrane of many eukaryotic cells, was reported to act as a direct mediator of MANF’s cytoprotective function, as it is located in the outer cell membrane of target cells and promotes cellular uptake of MANF by endocytosis [100]. Overall, our knowledge of the adaptive roles for ER stress-induced Ca2+-binding chaperone secretion from cardiac cells is finite, yet very promising. Therefore, identifying and understanding the mechanisms of action of these kinds of secreted proteins remains a key challenge of the field.
4.3. Extracellular Functions of ER Chaperones in Non-Cardiovascular Cells and Tissues
Secreted and cell surface-associated GRP78, GRP94, calreticulin, and MANF have been investigated in many non-cardiac cell types in different contexts, including ER stress. To complement the available knowledge of extracellular functions of these proteins in the heart, we have decided to include in this review findings pertaining to the extracellular functions of these proteins from other fields. This, we hope, can help guide future studies of these proteins in the heart. Therefore, this section aims to provide a short overview of extracellular functions identified for these proteins outside the heart.
4.3.1. 78 kDa Glucose-Regulated Protein (GRP78; Binding Immunoglobulin Protein)
In recent years, the critical role of cell surface and secreted GRP78 in the tumor microenvironment, and its potential as a target in anti-cancer therapies, have become well-recognized [101]. For example, GRP78 is preferentially present on the surface of cancer cells, where it collaborates with other surface proteins to mediate anti-apoptotic and proliferative signaling [102]. Furthermore, GRP78 secreted from tumor cells was recently shown to act as a chemokine that recruits macrophages into tumor tissues through cytoskeletal remodeling [103]. Another study reported the translocation of GRP78 to the cell surface and a novel role of GRP78 in neuroprotection, as it contributes to a decrease in maladaptive PERK activation at the onset of ER stress by direct binding to serine protease tissue-type plasminogen activator (tPA) at the cell surface [104]. In pancreatic beta cells, by contrast, inflammatory cytokines induce surface translocation of GRP78, where it initiates pro-apoptotic signaling cascades, including increased caspase 3/7 activity, as well as Chop and Bax mRNA expression. This study found that GRP78 is shuttled through the Golgi apparatus and secretory granules, and identified the DNAJ homolog subfamily C member 3 (DNAJC3) as a GRP78-interacting protein that facilitates its membrane translocation. The pro-apoptotic signaling was found to be mediated by extracellular, soluble GRP78 binding to cell surface GRP78 [105]. In different cell types, extracellular GRP78 displays anti-inflammatory and immunomodulatory properties [106]; for example, GRP78 can mediate endocytosis of toll-like receptor (TLR) 4 with CD14 to resolve inflammation [107]. At the cell surface, GRP78 can also act as a co-receptor to modulate cell invasion, i.e., virus entry in host cells, as shown, for example, for Coxsackie virus A9 [108], dengue virus serotype 2 [109], Borna disease virus [110], and Japanese encephalitis virus [111]. Previously, GRP78 was reported to augment virus attachment of certain coronavirus lineages (e.g., MERS-CoV) onto host cells [112], to which a recent study added the prediction of GRP78 binding sites to the spike proteins of the novel SARS-CoV-2, the strain of coronavirus that causes coronavirus disease 2019 (COVID-19) [113]. This prediction was made based on molecular modeling by docking and structural bioinformatics, and is highly relevant in consideration of the current worldwide COVID-19 pandemic.
4.3.2. Other Ca2+-Binding ER Chaperones: Calreticulin, 94 kDa Glucose-Regulated Protein (GRP94; Endoplasmin; Gp96), MANF
Since the identification of calreticulin in the extracellular matrix [114], its secretion, and localization to the cell surface, particularly upon reduced ER Ca2+ [65,115], studies have reported extracellular functions of calreticulin in multiple physiological and pathological processes. These include cutaneous wound healing [116], immunological responses, i.e., induction of immunogenic cancer cell death [117] and promotion of phagocytic uptake of cancer [118], viable and apoptotic cells [119] as an “eat me” signal, fibrosis [120], cancer cell survival [121], and more. Previous reviews have highlighted the multifunctionality of calreticulin outside the ER [24,78], which is underscored by the continuous characterization of its extracellular protein interactions and functions [122]. By contrast, extracellular GRP94 has been acknowledged, thus far, mostly for its contribution to immunological responses, e.g., by GRP94 surface binding, uptake, and peptide cross-presentation [123], and delivery of co-secreted client proteins [124]. Additionally, proinflammatory properties of GRP94 were elucidated in GRP94 overexpressing transgenic mice, where overexpression of an engineered form of GRP94 that remains on the cell surface induced dendritic cell activation and spontaneous autoimmune diseases [125]. Originally identified as a secreted trophic factor in neurons, protective roles of secreted MANF continue to be discovered in various contexts, including neuroprotection [97], cardioprotection (see, Section 4.2.), and diabetes (reviewed in [126]).
5. Secretion of Other Proteins during ER Stress
5.1. Other Cardiokines
Ischemia in vivo and simulated ischemia in vitro, the latter of which is defined as 0.1% O2 and lack of glucose, cause ER stress in cardiac cells [3,56]. A recent study of the secretome of primary cardiac myocytes provided evidence that protein secretion can be regulated by hypoxia [127]. The secreted proteins were found to contribute to the cellular response to hypoxia, the immune system, cell death, and cellular wounding. Moreover, the study showed increased secretion of the calcium-dependent serine endoprotease proprotein convertase subtilisin/kexin type 6 (PCSK6), an unexplored protein in cardiac ischemia, from cardiac myocytes by hypoxia. Significant increases in PCSK6 mRNA and protein levels were detected in hypoxic cardiac myocytes, infarct and infarct border zone, as well as in blood samples from patients suffering from acute ST-elevation myocardial infarction. Secreted PCSK6 was found to regulate TGFβ signaling and fibrosis in cardiac fibroblasts. Regulation of expression and secretion of PCSK6 during ER stress conditions, including ischemia in glucose-free conditions and lower oxygen levels, remain to be elucidated.
While some cardioprotective secreted proteins, such as follistatin-like 1 (Fstl1) and tumor necrosis factor (TNF)-α, can still be secreted during ischemic stress, secretion of other cardiokines, for example, encephalin and calcitonin gene-related peptide, is even enhanced by mild ischemia, which contributes to preserving cardiac function [3]. For an overview of ischemic stress-induced protein secretion in the heart the readers are referred to [3].
5.2. ERdj3 (Endoplasmic Reticulum DNA J Domain-Containing Protein 3; DnaJ Homolog Subfamily B Member 11)
ERdj3, a chaperone of the heat shock protein (HSP)-40 family, is an exemplar of how intracellular and extracellular proteostasis are linked, particularly during ER stress. In the ER, ERdj3 normally directs misfolded proteins to GRP78 [128]. In contrast to GRP78, ERdj3 lacks a KDEL ER retention motif. Therefore, at least half of newly synthesized ERdj3 is secreted via the classical secretory pathway. In 2015, Genereux et al. demonstrated that ERdj3 is an UPR-induced secreted chaperone, whose extracellular levels increase in response to ER stress and stress-independent activation of the UPR-associated transcription factor ATF6 [66]. Increased secretion of ERdj3 acts in extracellular proteostasis in two ways: (a) Free ERdj3 binds and attenuates extracellular misfolded protein aggregates that escaped ER protein quality control. (b) ERdj3 is co-secreted with destabilized, presumably misfolded protein clients when the chaperone capacity of intracellular GRP78 has been overwhelmed. Both mechanisms protect tissues from the potentially toxic extracellular aggregation of proteins, a maladaptive issue that contributes to the pathological phenotypes of many protein misfolding diseases. It also suggests a shift of proteins involved in extracellular proteostasis during ER stress. Secretion of the prominent extracellular chaperone, clusterin, for example, is reduced under similar ER stress conditions, in a process involving stress-induced retrotanslocation out of the ER and into the cytosol [129].
5.3. Angiogenin
Angiogenin, a cytoplasmic ribonuclease, is a secreted RNase of the pancreatic RNase A superfamily, showing tRNA substrate cleavage, and consequently inhibiting protein synthesis [130,131]. Mutations in angiogenin are linked to the development of amyotrophic lateral sclerosis (ALS) [132]. Recombinant angiogenin extends the lifespan of transgenic ALS mice [133] and protects primary motoneurons against hypoxic injury [134]. Angiogenin is secreted from neurons, and then endocytosed by astroglia; in this way, angiogenin mediates neuroprotection in a paracrine manner by eliciting astrocytes to induce RNA cleavage [135]. During pathologic conditions, for instance in acute kidney injury, cytoplasmic angiogenin interferes with translational initiation through cleavage, and thereby production of tRNA fragments, which contributes to decreased protein synthesis and alleviation of ER stress [136]. In a recent study on UPR signaling in the kidney, novel extracellular functions of angiogenin have been identified [137]. This study found that during ER stress, angiogenin expression and secretion are increased. Angiogenin secretion is under the selective control of ER stress sensor and UPR regulator, IRE-1α. The transcription factors, XBP1s and p65, which are activated by IRE-1α upon ER stress, bind to the angiogenin promoter, thereby upregulating angiogenin expression, leading to increased secretion. Similar to secretion of the ER stress-induced proinflammatory cytokine IL-6, secretion of angiogenin requires the ER-Golgi pathway. Upon secretion, extracellular angiogenin promotes macrophage reprogramming towards an activated and proinflammatory phenotype through receptor-mediated PI3K/AKT signaling and/or activation of intracellular pattern recognition receptors by tRNA fragments produced by angiogenin after endocytosis. Therefore, angiogenin has been characterized as a mediator of an ER stress-dependent inflammatory response of acute kidney ischemia that promotes kidney tissue adaption [137]. It remains to be determined whether ER stress-induced secretion of ribonucleases plays a role in tissues other than the kidney.
6. Conclusions and Future Directions
Characterization of proteins whose secretion is activated, maintained, or enhanced during ER stress holds promising therapeutic potential for a variety of human diseases, including heart diseases, as well as protein misfolding and autoimmune diseases, and cancer. Our view of ER chaperones has evolved considerably over the past two decades and affirmed their immense multifunctionality through ever-expanding findings of their roles inside and outside cells. Moreover, the UPR has been redefined as a critical regulator of cellular proteostasis that has the potential to link intra- and extracellular mechanisms of stress management. Considering the importance of maintaining precise control over intracellular Ca2+ levels that regulate cardiac myocyte contraction, further studies of Ca2+-mediated ER stress-inducible protein secretion could contribute substantially to our understanding of how secreted proteins can mediated cardioprotection.
In summary, most ER chaperones secreted during ER stress are secreted due to decreased ER Ca2+. Moreover, ERdj3 is the first metazoan chaperone whose secretion is directly regulated by the UPR. Considering that ER proteostasis involves a number of chaperones whose novel functions are continuously being discovered, the importance and innovation capacity of this field is highlighted here. We expect further characterization of the underlying mechanisms of secretion and extracellular actions of these kinds of proteins in the future, by examining their roles in pro-survival, oncogenic, and immune-stimulatory cell signaling, extracellular proteostasis, and more, both inside and outside the heart.
Author Contributions
Conceptualization, B.A.M. and S.D.; writing—original draft preparation, B.A.M.; writing—review and editing, B.A.M. and S.D.; visualization, S.D.; supervision, S.D.; funding acquisition, S.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the German Centre for Cardiovascular Research (DZHK).
Acknowledgments
The authors would like to thank Tobias Jakobi for his valuable comments on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kakkar, R.; Lee, R.T. Intramyocardial fibroblast myocyte communication. Circ. Res. 2010, 106, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Pu, W.T. Recounting Cardiac Cellular Composition. Circ. Res. 2016, 118, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Doroudgar, S.; Glembotski, C.C. The cardiokine story unfolds: Ischemic stress-induced protein secretion in the heart. Trends Mol. Med. 2011, 17, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimano, M.; Ouchi, N.; Walsh, K. Cardiokines: Recent progress in elucidating the cardiac secretome. Circulation 2012, 126, e327–e332. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.S.; Miller, E.A.; Goldberg, J.; Orci, L.; Schekman, R. Bi-Directional Protein Transport Between the Er and Golgi. Annu. Rev. Cell Dev. Biol. 2004, 20, 87–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, W.; Rabouille, C. Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 2009, 10, 148–155. [Google Scholar] [CrossRef]
- Elvekrog, M.M.; Walter, P. Dynamics of co-translational protein targeting. Curr. Opin. Chem. Biol. 2015, 29, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Cabrita, L.D.; Dobson, C.M.; Christodoulou, J. Protein folding on the ribosome. Curr. Opin. Struct. Biol. 2010, 20, 33–45. [Google Scholar] [CrossRef]
- Kraut-Cohen, J.; Gerst, J.E. Addressing mRNAs to the ER: Cis sequences act up! Trends Biochem. Sci. 2010, 35, 459–469. [Google Scholar] [CrossRef]
- Weis, B.L.; Schleiff, E.; Zerges, W. Protein targeting to subcellular organelles via mRNA localization. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 260–273. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Zontag, O.; Baez, C.; Lim, L.Q.J.; Olender, T.; Schirman, D.; Dahary, D.; Pilpel, Y.; Gerst, J.E. A secretion-enhancing cis regulatory targeting element (SECReTE) involved in mRNA localization and protein synthesis. PLoS Genet. 2019, 15, e1008248. [Google Scholar] [CrossRef] [PubMed]
- Palade, G. Intracellular Aspects of the Process of Protein Synthesis. Science 1975, 189, 867. [Google Scholar] [CrossRef] [PubMed]
- Bloch, K.D.; Seidman, J.G.; Naftilan, J.D.; Fallon, J.T.; Seidman, C.E. Neonatal atria and ventricles secrete atrial natriuretic factor via tissue-specific secretory pathways. Cell 1986, 47, 695–702. [Google Scholar] [CrossRef]
- Sei, C.A.; Hand, G.L.; Murray, S.F.; Glembotski, C.C. The cosecretional maturation of atrial natriuretic factor by primary atrial myocytes. Mol. Endocrinol. 1992, 6, 309–319. [Google Scholar] [CrossRef]
- Maack, T. Role of atrial natriuretic factor in volume control. Kidney Int. 1996, 49, 1732–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Gee, H.Y.; Lee, M.G. Unconventional protein secretion— New insights into the pathogenesis and therapeutic targets of human diseases. J. Cell Sci. 2018, 131, jcs213686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, W.; Seedorf, M. Unconventional Mechanisms of Protein Transport to the Cell Surface of Eukaryotic Cells. Annu. Rev. Cell Dev. Biol. 2008, 24, 287–308. [Google Scholar] [CrossRef]
- Most, P.; Boerries, M.; Eicher, C.; Schweda, C.; Völkers, M.; Wedel, T.; Söllner, S.; Katus, H.A.; Remppis, A.; Aebi, U.; et al. Distinct subcellular location of the Ca2+-binding protein S100A1 differentially modulates Ca2+-cycling in ventricular rat cardiomyocytes. J. Cell Sci. 2005, 118, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Treaster, S.B.; Ridgway, I.D.; Richardson, C.A.; Gaspar, M.B.; Chaudhuri, A.R.; Austad, S.N. Superior proteome stability in the longest lived animal. Age (Omaha) 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henning, R.H.; Brundel, B.J.J.M. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637. [Google Scholar] [CrossRef] [PubMed]
- Christis, C.; Lubsen, N.H.; Braakman, I. Protein folding includes oligomerization—Examples from the endoplasmic reticulum and cytosol. FEBS J. 2008, 275, 4700–4727. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef]
- Mohorko, E.; Glockshuber, R.; Aebi, M. Oligosaccharyltransferase: The central enzyme of N-linked protein glycosylation. J. Inherit. Metab. Dis. 2011, 34, 869–878. [Google Scholar] [CrossRef]
- Hudson, D.A.; Gannon, S.A.; Thorpe, C. Oxidative protein folding: From thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic. Biol. Med. 2015, 80, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Gidalevitz, T.; Stevens, F.; Argon, Y. Orchestration of secretory protein folding by ER chaperones. Biochim. Biophys. Acta 2013, 1833, 2410–2424. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, J.L. Cleaning Up: ER-associated degradation to the rescue. Cell 2012, 151, 1163–1167. [Google Scholar] [CrossRef] [Green Version]
- Claessen, J.H.L.; Kundrat, L.; Ploegh, H.L. Protein quality control in the ER: Balancing the ubiquitin checkbook. Trends Cell Biol. 2012, 22, 22–32. [Google Scholar] [CrossRef]
- Chyn, T.L.; Martonosi, A.N.; Morimoto, T.; Sabatini, D.D. In vitro synthesis of the Ca2+ transport ATPase by ribosomes bound to sarcoplasmic reticulum membranes. Proc. Natl. Acad. Sci. USA 1979, 76, 1241–1245. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.E.; Dirksen, R.T. Sarcoplasmic reticulum: The dynamic calcium governor of muscle. Muscle Nerve 2006, 33, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLendon, P.M.; Robbins, J. Proteotoxicity and Cardiac Dysfunction. Circ. Res. 2015, 116, 1863–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Paxman, R.J.; Plate, L.; Kelly, J.W.; Wiseman, R.L.; Glembotski, C.C. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat. Commun. 2019, 10, 187. [Google Scholar] [CrossRef] [Green Version]
- Kilberg, M.S.; Shan, J.; Su, N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009, 20, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Plemper, R.K.; Wolf, D.H. Endoplasmic reticulum degradation. Reverse protein transport and its end in the proteasome. Mol. Biol. Rep. 1999, 26, 125–130. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic- reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and Characterization of Pancreatic Eukaryotic Initiation Factor 2 α-Subunit Kinase, PEK, Involved in Translational Control. Mol. Cell. Biol. 1998, 18, 7499–7509. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.Z.; Harding, H.P.; Zhang, Y.; Jolicoeur, E.M.; Kuroda, M.; Ron, D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998, 17, 5708–5717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, M.; Sidrauski, C.; Kaufman, R.J.; Walter, P. A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell 1999, 99, 691–702. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Johansen, F.E.; Prywes, R. Interaction of ATF6 and serum response factor. Mol. Cell. Biol. 1997, 17, 4957–4966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose- regulated proteins: Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef] [Green Version]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxidants Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front. Mol. Biosci. 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.K.; Lawrence, D.A.; Lu, M.; Tan, J.; Harnoss, J.M.; Marsters, S.A.; Liu, P.; Sandoval, W.; Martin, S.E.; Ashkenazi, A. Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Mol. Cell 2018, 71, 629.e5–636.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, C.; Katus, H.A.; Doroudgar, S. Protein Misfolding in Cardiac Disease. Circulation 2019, 139, 2085–2088. [Google Scholar] [CrossRef]
- Thuerauf, D.J.; Marcinko, M.; Gude, N.; Rubio, M.; Sussman, M.A.; Glembotski, C.C. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ. Res. 2006, 99, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Doroudgar, S.; Thuerauf, D.J.; Marcinko, M.C.; Belmont, P.J.; Glembotski, C.C. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J. Biol. Chem. 2009, 284, 29735–29745. [Google Scholar] [CrossRef] [Green Version]
- Groenendyk, J.; Sreenivasaiah, P.K.; Kim, D.H.; Agellon, L.B.; Michalak, M. Biology of endoplasmic reticulum stress in the heart. Circ. Res. 2010, 107, 1185–1197. [Google Scholar] [CrossRef] [Green Version]
- Buja, L.M. Myocardial ischemia and reperfusion injury. Cardiovasc. Pathol. 2005, 14, 170–175. [Google Scholar] [CrossRef]
- Doroudgar, S.; Glembotski, C.C. New concepts of endoplasmic reticulum function in the heart: Programmed to conserve. J. Mol. Cell. Cardiol. 2013, 55, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Engl. J. Med. 2008, 358, 1370. [Google Scholar] [CrossRef]
- Blackwood, E.A.; Hofmann, C.; Santo Domingo, M.; Bilal, A.S.; Sarakki, A.; Stauffer, W.; Arrieta, A.; Thuerauf, D.J.; Kolkhorst, F.W.; Müller, O.J.; et al. ATF6 Regulates Cardiac Hypertrophy by Transcriptional Induction of the mTORC1 Activator, Rheb. Circ. Res. 2019, 124, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.A.; Thuerauf, D.J.; Stastna, M.; Stephens, H.; Sand, Z.; Pentoney, A.; Azizi, K.; Jakobi, T.; Van Eyk, J.E.; Katus, H.A.; et al. Proteomic analysis of the cardiac myocyte secretome reveals extracellular protective functions for the ER stress response. J. Mol. Cell. Cardiol. 2020, 143, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.W.; Chen, Q.; Tay, A.S.L.; Shenolikar, S.; Nicchitta, C.V. The unfolded protein response triggers selective mRNA release from the endoplasmic reticulum. Cell 2014, 158, 1362–1374. [Google Scholar] [CrossRef] [Green Version]
- Peters, L.R.; Raghavan, M. Endoplasmic Reticulum Calcium Depletion Impacts Chaperone Secretion, Innate Immunity, and Phagocytic Uptake of Cells. J. Immunol. 2011, 187, 919–931. [Google Scholar] [CrossRef] [Green Version]
- Genereux, J.C.; Qu, S.; Zhou, M.; Ryno, L.M.; Wang, S.; Shoulders, M.D.; Kaufman, R.J.; Lasmézas, C.I.; Kelly, J.W.; Wiseman, R.L. Unfolded protein response-induced ER dj3 secretion links ER stress to extracellular proteostasis. EMBO J. 2015, 34, 4–19. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium Cycling and Signaling in Cardiac Myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [Green Version]
- Davlouros, P.A.; Gkizas, V.; Vogiatzi, C.; Giannopoulos, G.; Alexopoulos, D.; Deftereos, S. Calcium Homeostasis and Kinetics in Heart Failure. Med. Chem. 2016, 12, 151–161. [Google Scholar] [CrossRef]
- da Silva, D.C.; Valentão, P.; Andrade, P.B.; Pereira, D.M. Endoplasmic reticulum stress signaling in cancer and neurodegenerative disorders: Tools and strategies to understand its complexity. Pharmacol. Res. 2020, 155. [Google Scholar] [CrossRef]
- Wetmore, D.R.; Hardman, K.D. Roles of the propeptide and metal ions in the folding and stability of the catalytic domain of stromelysin (matrix metalloproteinase 3). Biochemistry 1996, 35, 6549–6558. [Google Scholar] [CrossRef]
- Halperin, L.; Jung, J.; Michalak, M. The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 2014, 66, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Corbett, E.F.; Oikawa, K.; Francois, P.; Tessier, D.C.; Kay, C.; Bergeron, J.J.M.; Thomas, D.Y.; Krause, K.-H.; Michalak, M. Ca2+ Regulation of Interactions between Endoplasmic Reticulum Chaperones. J. Biol. Chem. 1999, 274, 6203–6211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, W.-J.; Bergeron, J.J.M.; Li, Y.; Kang, C.Y.; Thomas, D.Y. Conformational Changes Induced in the Endoplasmic Reticulum Luminal Domain of Calnexin by Mg-ATP and Ca. J. Biol. Chem. 1995, 270, 18051–18059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, C.K.; Bonifacino, J.S.; Lin, A.Y.; Davis, M.M.; Klausner, R.D. Regulating the retention of T-cell receptor alpha chain variants within the endoplasmic reticulum: Ca(2+)-dependent association with BiP. J. Cell Biol. 1991, 114, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Klappa, P.; Kietzmann, D.T. The Endoplasmic Reticulum: Folding, Calcium Homeostasis, Signaling, and Redox Control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef]
- Glembotski, C.C.; Thuerauf, D.J.; Huang, C.; Vekich, J.A.; Gottlieb, R.A.; Doroudgar, S. Mesencephalic astrocyte-derived neurotrophic factor protects the heart from ischemic damage and is selectively secreted upon sarco/endoplasmic reticulum calcium depletion. J. Biol. Chem. 2012, 287, 25893–25904. [Google Scholar] [CrossRef] [Green Version]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef]
- Gold, L.I.; Eggleton, P.; Sweetwyne, M.T.; Van Duyn, L.B.; Greives, M.R.; Naylor, S.; Michalak, M.; Murphy-Ullrich, J.E. Calreticulin: Non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010, 24, 665–683. [Google Scholar] [CrossRef] [Green Version]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 774–787. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ma, F.; Liu, Z.; Su, Q.; Liu, Y.; Liu, Z.; Li, Y. The ER-localized Ca2+-binding protein calreticulin couples ER stress to autophagy by associating with microtubule-associated protein 1A/1B light chain 3. J. Biol. Chem. 2019, 294, 772–782. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, H.; Yoshimori, T.; Yamamoto, A.; Miyata, Y.; Yahara, I.; Inoue, K.; Tashiro, Y. Heavy chain binding protein (BiP/GRP78) and endoplasmin are exported from the endoplasmic reticulum in rat exocrine pancreatic cells, similar to protein disulfide-isomerase. Arch. Biochem. Biophys. 1992, 296, 129–136. [Google Scholar] [CrossRef]
- Capitani, M.; Sallese, M. The KDEL receptor: New functions for an old protein. FEBS Lett. 2009, 583, 3863–3871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, S.R. Unsolved Mysteries in Membrane Traffic. Annu. Rev. Biochem. 2007, 76, 629–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, R.; Ni, M.; Gill, P.; Lee, A.S.S. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J. Biol. Chem. 2010, 285, 15065–15075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glembotski, C.C. Functions for the cardiomyokine, MANF, in cardioprotection, hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 512–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Castro, N.P.; Rangel, M.C.; Nagaoka, T.; Salomon, D.S.; Bianco, C. Cripto-1: An embryonic gene that promotes tumorigenesis. Future Oncol. 2010, 6, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.C.; Vale, W. Cripto/GRP78 modulation of the TGF-β pathway in development and oncogenesis. FEBS Lett. 2012, 586, 1836–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shani, G.; Fischer, W.H.; Justice, N.J.; Kelber, J.A.; Vale, W.; Gray, P.C. GRP78 and Cripto Form a Complex at the Cell Surface and Collaborate To Inhibit Transforming Growth Factor β Signaling and Enhance Cell Growth. Mol. Cell. Biol. 2008, 28, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelber, J.A.; Panopoulos, A.D.; Shani, G.; Booker, E.C.; Belmonte, J.C.; Vale, W.W.; Gray, P.C. Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways. Oncogene 2009, 28, 2324–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.L.; Zhang, Y.; Tseng, C.C.; Stanciauskas, R.; Pinaud, F.; Lee, A.S. Characterization and mechanism of stress-induced translocation of 78-kilodalton glucose-regulated protein (GRP78) to the cell surface. J. Biol. Chem. 2015, 290, 8049–8064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Gronow, M.; Selim, M.A.; Papalas, J.; Pizzo, S.V. GRP78: A multifunctional receptor on the cell surface. Antioxidants Redox Signal. 2009, 11, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Zhang, G.; Wang, X.; Nguyen, C.; May, H.I.; Li, X.; Al-Hashimi, A.A.; Austin, R.C.; Gillette, T.G.; Fu, G.; et al. Endoplasmic Reticulum Chaperone GRP78 Protects Heart From Ischemia/Reperfusion Injury Through Akt Activation. Circ. Res. 2018, 122, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, G.; Ahamed, J.; Pedersen, B.; El-Sheikh, A.; Mackman, N.; Ruf, W.; Liu, C.; Edgington, T.S. Regulation of tissue factor-mediated initiation of the coagulatiion cascade by cell surface Grp78. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1737–1743. [Google Scholar] [CrossRef]
- Zhao, D.; Feng, P.; Sun, Y.; Qin, Z.; Zhang, Z.; Tan, Y.; Gao, E.; Lau, W.B.; Ma, X.; Yang, J.; et al. Cardiac-derived CTRP9 protects against myocardial ischemia/reperfusion injury via calreticulin-dependent inhibition of apoptosis article. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Petrova, P.S.; Raibekas, A.; Pevsner, J.; Vigo, N.; Anafi, M.; Moore, M.K.; Peaire, A.E.; Shridhar, V.; Smith, D.I.; Kelly, J.; et al. MANF. J. Mol. Neurosci. 2003, 20, 173–187. [Google Scholar] [CrossRef]
- Palgi, M.; Lindström, R.; Peränen, J.; Piepponen, T.P.; Saarma, M.; Heino, T.I. Evidence that DmMANF is an invertebrate neurotrophic factor supporting dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2009, 106, 2429–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neves, J.; Zhu, J.; Sousa-Victor, P.; Konjikusic, M.; Riley, R.; Chew, S.; Qi, Y.; Jasper, H.; Lamba, D.A. Immune modulation by MANF promotes tissue repair and regenerative success in the retina. Science 2016, 353. [Google Scholar] [CrossRef] [Green Version]
- Lindström, R.; Lindholm, P.; Kallijärvi, J.; Palgi, M.; Saarma, M.; Heino, T.I. Exploring the Conserved Role of MANF in the Unfolded Protein Response in Drosophila melanogaster. PLoS ONE 2016, 11, e0151550. [Google Scholar] [CrossRef] [Green Version]
- Tadimalla, A.; Belmont, P.J.; Thuerauf, D.J.; Glassy, M.S.; Martindale, J.J.; Gude, N.; Sussman, M.A.; Glembotski, C.C. Mesencephalic astrocyte-derived neurotrophic factor is an ischemia-inducible secreted endoplasmic reticulum stress response protein in the heart. Circ. Res. 2008, 103, 1249–1258. [Google Scholar] [CrossRef] [Green Version]
- Bai, M.; Vozdek, R.; Hnízda, A.; Jiang, C.; Wang, B.; Kuchar, L.; Li, T.; Zhang, Y.; Wood, C.; Feng, L.; et al. Conserved roles of C. elegans and human MANFs in sulfatide binding and cytoprotection. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Araujo, N.; Hebbar, N.; Rangnekar, V.M. GRP78 Is a Targetable Receptor on Cancer and Stromal Cells. EBioMedicine 2018, 33, 2–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Li, Z. Glucose regulated protein 78: A critical link between tumor microenvironment and cancer hallmarks. Biochim. Biophys. Acta Rev. Cancer 2012, 1826, 13–22. [Google Scholar] [CrossRef] [PubMed]
- La, X.; Zhang, L.; Yang, Y.; Li, H.; Song, G.; Li, Z. Tumor-secreted GRP78 facilitates the migration of macrophages into tumors by promoting cytoskeleton remodeling. Cell. Signal. 2019, 60, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Louessard, M.; Bardou, I.; Lemarchand, E.; Thiebaut, A.M.; Parcq, J.; Leprince, J.; Terrisse, A.; Carraro, V.; Fafournoux, P.; Bruhat, A.; et al. Activation of cell surface GRP78 decreases endoplasmic reticulum stress and neuronal death. Cell Death Differ. 2017, 24, 1518–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vig, S.; Buitinga, M.; Rondas, D.; Crèvecoeur, I.; van Zandvoort, M.; Waelkens, E.; Eizirik, D.L.; Gysemans, C.; Baatsen, P.; Mathieu, C.; et al. Cytokine-induced translocation of GRP78 to the plasma membrane triggers a pro-apoptotic feedback loop in pancreatic beta cells. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Panayi, G.S.; Corrigall, V.M. BiP regulates autoimmune inflammation and tissue damage. Autoimmun. Rev. 2006, 5, 140–142. [Google Scholar] [CrossRef]
- Qin, K.; Ma, S.; Li, H.; Wu, M.; Sun, Y.; Fu, M.; Guo, Z.; Zhu, H.; Gong, F.; Lei, P.; et al. GRP78 impairs production of lipopolysaccharide-induced cytokines by interaction with CD14. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Triantafilou, K.; Fradelizi, D.; Wilson, K.; Triantafilou, M. GRP78, a Coreceptor for Coxsackievirus A9, Interacts with Major Histocompatibility Complex Class I Molecules Which Mediate Virus Internalization. J. Virol. 2002, 76, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Jindadamrongwech, S.; Thepparit, C.; Smith, D.R. Identification of GRP 78 (BiP) as a liver cell expressed receptor element for dengue virus serotype 2. Arch. Virol. 2004, 149, 915–927. [Google Scholar] [CrossRef]
- Honda, T.; Horie, M.; Daito, T.; Ikuta, K.; Tomonaga, K. Molecular Chaperone BiP Interacts with Borna Disease Virus Glycoprotein at the Cell Surface. J. Virol. 2009, 83, 12622–12625. [Google Scholar] [CrossRef] [Green Version]
- Nain, M.; Mukherjee, S.; Karmakar, S.P.; Paton, A.W.; Paton, J.C.; Abdin, M.Z.; Basu, A.; Kalia, M.; Vrati, S. GRP78 Is an Important Host Factor for Japanese Encephalitis Virus Entry and Replication in Mammalian Cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, H.; Chan, C.M.; Zhang, X.; Wang, Y.; Yuan, S.; Zhou, J.; Au-Yeung, R.K.H.; Sze, K.H.; Yang, D.; Shuai, H.; et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 2018, 293, 11709–11726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef]
- Somogyi, E.; Petersson, U.; Hultenby, K.; Wendel, M. Calreticulin—An endoplasmic reticulum protein with calcium-binding activity is also found in the extracellular matrix. Matrix Biol. 2003, 22, 179–191. [Google Scholar] [CrossRef]
- Tufi, R.; Panaretakis, T.; Bianchi, K.; Criollo, A.; Fazi, B.; Di Sano, F.; Tesniere, A.; Kepp, O.; Paterlini-Brechot, P.; Zitvogel, L.; et al. Reduction of endoplasmic reticulum Ca2+ levels favors plasma membrane surface exposure of calreticulin. Cell Death Differ. 2008, 15, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Greives, M.R.; Samra, F.; Pavlides, S.C.; Blechman, K.M.; Naylor, S.M.; Woodrell, C.D.; Cadacio, C.; Levine, J.P.; Bancroft, T.A.; Michalak, M.; et al. Exogenous calreticulin improves diabetic wound healing. Wound Repair Regen. 2012, 20, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef] [Green Version]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Owusu, B.Y.; Zimmerman, K.A.; Murphy-Ullrich, J.E. The role of the endoplasmic reticulum protein calreticulin in mediating TGF-β-stimulated extracellular matrix production in fibrotic disease. J. Cell Commun. Signal. 2018, 12, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Han, A.; Li, C.; Zahed, T.; Wong, M.; Smith, I.; Hoedel, K.; Green, D.; Boiko, A.D. Calreticulin is a critical cell survival factor in malignant neoplasms. PLoS Biol. 2019, 17. [Google Scholar] [CrossRef] [PubMed]
- Schcolnik-Cabrera, A.; Oldak, B.; Juárez, M.; Cruz-Rivera, M.; Flisser, A.; Mendlovic, F. Calreticulin in phagocytosis and cancer: Opposite roles in immune response outcomes. Apoptosis 2019. [Google Scholar] [CrossRef] [PubMed]
- Jockheck-Clark, A.R.; Bowers, E.V.; Totonchy, M.B.; Neubauer, J.; Pizzo, S.V.; Nicchitta, C.V. Re-Examination of CD91 Function in GRP94 (Glycoprotein 96) Surface Binding, Uptake, and Peptide Cross-Presentation. J. Immunol. 2010, 185, 6819–6830. [Google Scholar] [CrossRef] [Green Version]
- Bruneau, N.; Lombarde, D.; Bendayan, M. Participation of GRP94-related protein in secretion of pancreatic bile salt-dependent lipase and in its internalization by the intestinal epithelium. J. Cell Sci. 1998, 111, 2665–2679. [Google Scholar] [PubMed]
- Liu, B.; Dai, J.; Zheng, H.; Stoilova, D.; Sun, S.; Li, Z. Cell surface expression of an endoplasmic reticulum resident heat shock protein gp96 triggers MyD88-dependent systemic autoimmune diseases. Proc. Natl. Acad. Sci. USA 2003, 100, 15824–15829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danilova, T.; Lindahl, M. Emerging roles for mesencephalic astrocyte-derived neurotrophic factor (MANF) in pancreatic beta cells and diabetes. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef]
- 127. Kuhn, T.C.; Knobel, J.; Burkert-Rettenmaier, S.; Li, X.; Meyer, I.S.; Jungmann, A.; Sicklinger, F.; Backs, J.; Lasitschka, F.; Müller, O.J.; et al. Secretome Analysis of Cardiomyocytes Identifies PCSK6 (Proprotein Convertase Subtilisin/Kexin Type 6) as a Novel Player in Cardiac Remodeling After Myocardial Infarction. Circulation 2020, 141, 1628–1644. [Google Scholar] [CrossRef]
- Shen, Y.; Hendershot, L.M. ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP’s interactions with unfolded substrates. Mol. Biol. Cell 2005, 16, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Nizard, P.; Tetley, S.; Le Dréan, Y.; Watrin, T.; Le Goff, P.; Wilson, M.R.; Michel, D. Stress-induced retrotranslocation of clusterin/ApoJ into the cytosol. Traffic 2007, 8, 554–565. [Google Scholar] [CrossRef] [Green Version]
- St Clair, D.K.; Rybak, S.M.; Riordan, J.F.; Vallee, B.L. Angiogenin abolishes cell-free protein synthesis by specific ribonucleolytic inactivation of ribosomes. Proc. Natl. Acad. Sci. USA 1987, 84, 8330–8334. [Google Scholar] [CrossRef] [Green Version]
- Saxena, S.K.; Rybak, S.M.; Davey, R.T.; Youle, R.J.; Ackerman, E.J. Angiogenin is a cytotoxic, tRNA-specific ribonuclease in the RNase A superfamily. J. Biol. Chem. 1992, 267, 21982–21986. [Google Scholar] [PubMed]
- Greenway, M.J.; Andersen, P.M.; Russ, C.; Ennis, S.; Cashman, S.; Donaghy, C.; Patterson, V.; Swingler, R.; Kieran, D.; Prehn, J.; et al. ANG mutations segregate with familial and “sporadic” amyotrophic lateral sclerosis. Nat. Genet. 2006, 38, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Kieran, D.; Sebastia, J.; Greenway, M.J.; King, M.A.; Connaughton, D.; Concannon, C.G.; Fenner, B.; Hardiman, O.; Prehn, J.H.M. Control of motoneuron survival by angiogenin. J. Neurosci. 2008, 28, 14056–14061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastià, J.; Kieran, D.; Breen, B.; King, M.A.; Netteland, D.F.; Joyce, D.; Fitzpatrick, S.F.; Taylor, C.T.; Prehn, J.H.M. Angiogenin protects motoneurons against hypoxic injury. Cell Death Differ. 2009, 16, 1238–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skorupa, A.; King, M.A.; Aparicio, I.M.; Dussmann, H.; Coughlan, K.; Breen, B.; Kieran, D.; Concannon, C.G.; Marin, P.; Prehn, J.H.M. Motoneurons secrete angiogenin to induce RNA cleavage in astroglia. J. Neurosci. 2012, 32, 5024–5038. [Google Scholar] [CrossRef]
- Yamasaki, S.; Ivanov, P.; Hu, G.F.; Anderson, P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J. Cell Biol. 2009, 185, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Mami, I.; Tavernier, Q.; Bouvier, N.; Aboukamis, R.; Desbuissons, G.; Rabant, M.; Poindessous, V.; Laurent-Puig, P.; Beaune, P.; Tharaux, P.L.; et al. A novel extrinsic pathway for the unfolded protein response in the kidney. J. Am. Soc. Nephrol. 2016, 27, 2670–2683. [Google Scholar] [CrossRef] [Green Version]
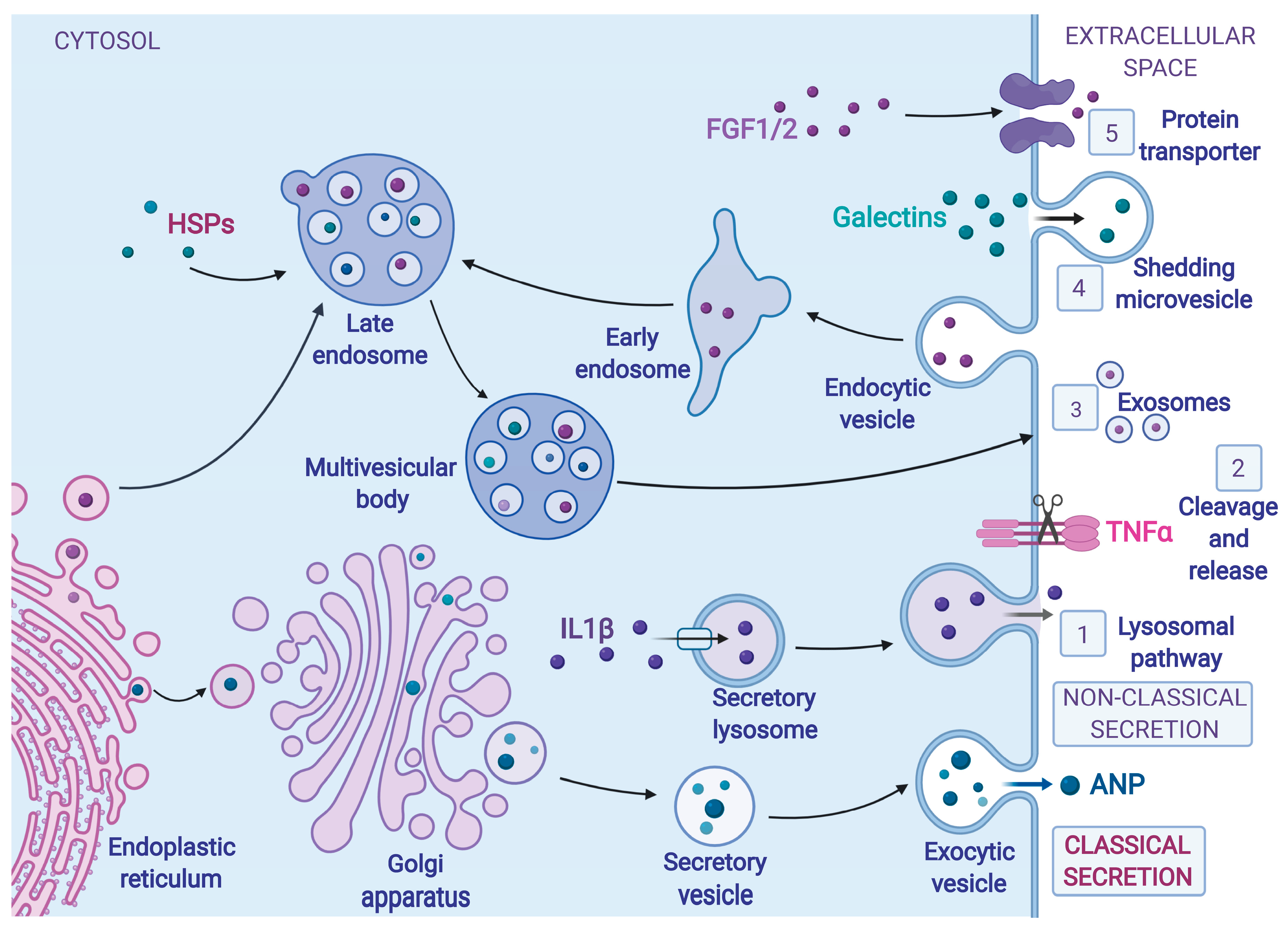
Figure 1.
Mechanisms of Cardiokine Secretion. Cardiokines (secreted proteins from cardiac cells), such as atrial natriuretic protein (ANP), made at the endoplasmic reticulum (ER), are conventionally transported to the Golgi apparatus, where they are modified and packaged into secretory vesicles and then to the plasma membrane, where they are released into the extracellular space, in what is known as classical protein secretion. Some cardiokines that enter the ER bypass the Golgi apparatus, in some cases entering membrane-bound organelles such as endosomes, on their way to the plasma membrane. Cardiokines secreted via the non-classical secretory pathway are synthesized on cytosolic ribosomes and traffic through various kinds of either vesicle- or nonvesicle-mediated pathways, without transiting through the ER and the Golgi apparatus. Non-classical protein secretion pathways include (1) incorporation into the lysosomal pathway, for example, interleukin 1β (IL1β), (2) cleavage and release from a membrane-bound precursor, for example, tumor necrosis factor α (TNFα), (3) release via exosomes derived from multivesicular bodies, for example, heat shock proteins (HSPs), (4) membrane blebbing and microvesicle shedding, for example, galectins, and (5) direct translocation of proteins across the plasma membrane through membrane transporters, for example fibroblast growth factor 1 and 2. Figures were created with BioRender.com.
Figure 1.
Mechanisms of Cardiokine Secretion. Cardiokines (secreted proteins from cardiac cells), such as atrial natriuretic protein (ANP), made at the endoplasmic reticulum (ER), are conventionally transported to the Golgi apparatus, where they are modified and packaged into secretory vesicles and then to the plasma membrane, where they are released into the extracellular space, in what is known as classical protein secretion. Some cardiokines that enter the ER bypass the Golgi apparatus, in some cases entering membrane-bound organelles such as endosomes, on their way to the plasma membrane. Cardiokines secreted via the non-classical secretory pathway are synthesized on cytosolic ribosomes and traffic through various kinds of either vesicle- or nonvesicle-mediated pathways, without transiting through the ER and the Golgi apparatus. Non-classical protein secretion pathways include (1) incorporation into the lysosomal pathway, for example, interleukin 1β (IL1β), (2) cleavage and release from a membrane-bound precursor, for example, tumor necrosis factor α (TNFα), (3) release via exosomes derived from multivesicular bodies, for example, heat shock proteins (HSPs), (4) membrane blebbing and microvesicle shedding, for example, galectins, and (5) direct translocation of proteins across the plasma membrane through membrane transporters, for example fibroblast growth factor 1 and 2. Figures were created with BioRender.com.
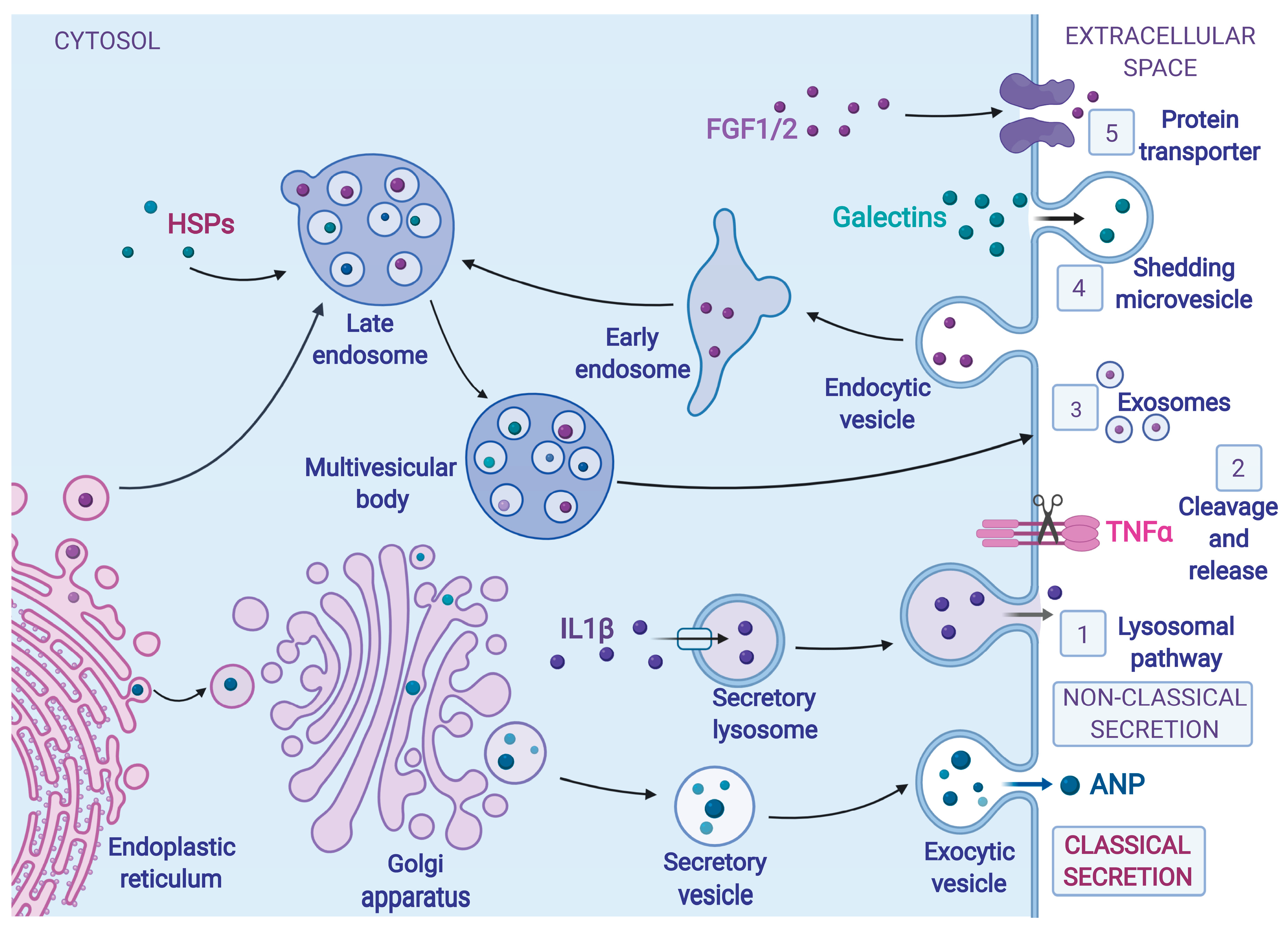
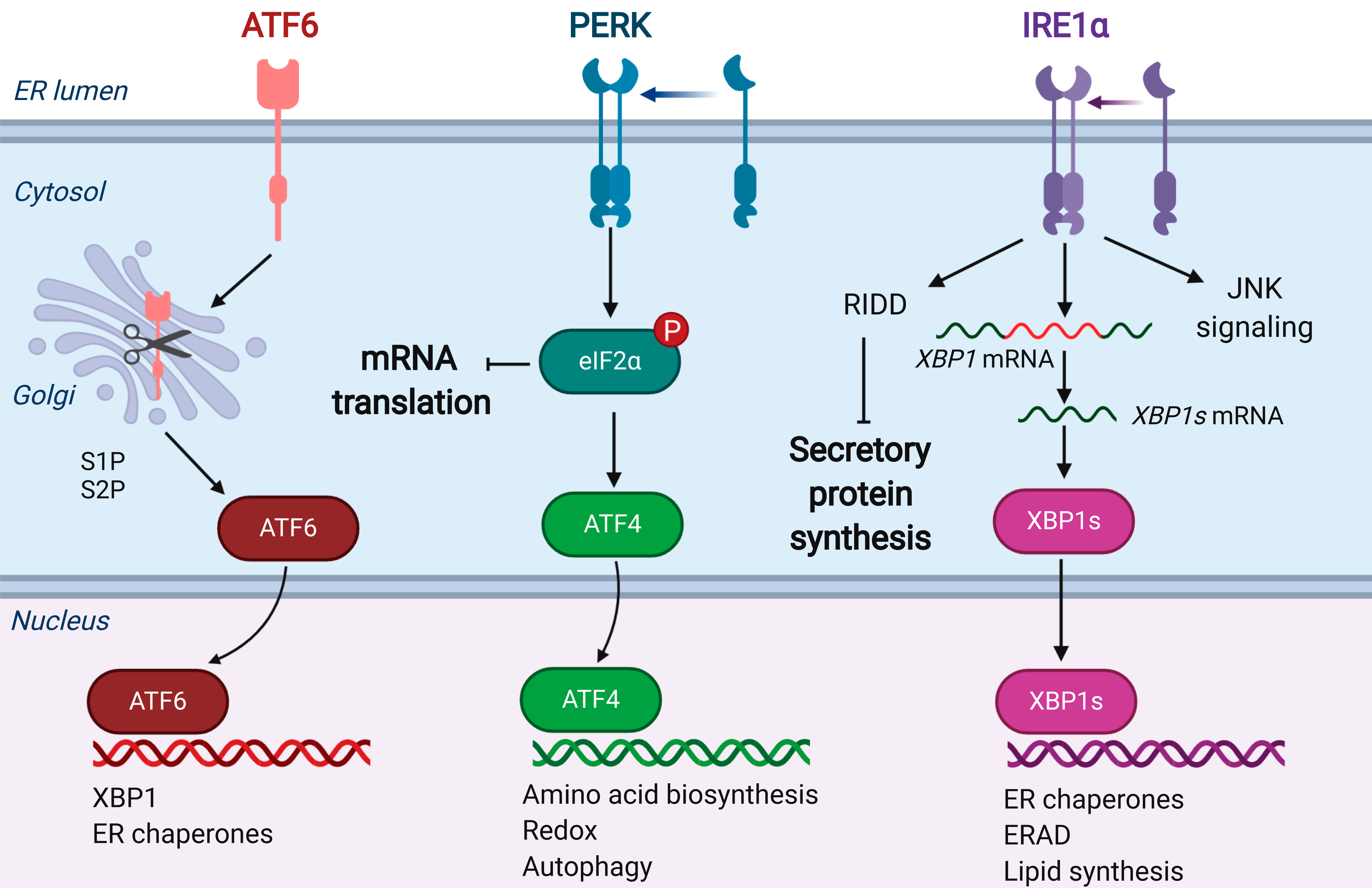
Figure 2.
Regulation of ER Stress by the Unfolded Protein Response and Effects on Cardiokine Secretion. Perturbations of the ER environment result in the activation of the unfolded protein response, mediated by ER-resident integral membrane proteins ATF6 (activating transcription factor 6), PERK (protein kinase RNA (PKR)-like ER kinase), and IRE1α (inositol requiring enzyme 1). Upon ER stress, ATF6 translocates to the Golgi apparatus, where it is cleaved by site-1 and site-2 proteases (S1P, S2P), liberating an N-terminal fragment to translocate to the nucleus, where it is a potent and labile transcription factor that activates the transcription of ER chaperones and the transcription factor X-box binding protein 1 (XBP1). Activation of ATF6 may increase ER secretory capacity [35]. PERK dimerization activates autophosphorylation of its kinase domain, leading to phosphorylation of the α-subunit of translation initiation factor 2 (eIF2α) at Ser51. This inhibits global translational initiation, including translation of mRNAs encoding secretory proteins, reducing the load of unfolded proteins entering the ER. PERK activation also results in the induction of translation of selective mRNAs, including activating transcription factor 4 (ATF4), which activates the transcription of a wide range of genes involved in adaptation to stress conditions. Upon activation, IRE1α oligomerizes and carries out regulated IRE1-dependent decay (RIDD) of ER-targeted transcripts, as well as unconventional RNA splicing, by removing an intron from the XBP1 mRNA, allowing it to become translated into the functional transcription factor, XBP1s. XBP1s activates the transcription of ER chaperones, endoplasmic reticulum associated degradation (ERAD) genes, and genes involved in lipid synthesis.
Figure 2.
Regulation of ER Stress by the Unfolded Protein Response and Effects on Cardiokine Secretion. Perturbations of the ER environment result in the activation of the unfolded protein response, mediated by ER-resident integral membrane proteins ATF6 (activating transcription factor 6), PERK (protein kinase RNA (PKR)-like ER kinase), and IRE1α (inositol requiring enzyme 1). Upon ER stress, ATF6 translocates to the Golgi apparatus, where it is cleaved by site-1 and site-2 proteases (S1P, S2P), liberating an N-terminal fragment to translocate to the nucleus, where it is a potent and labile transcription factor that activates the transcription of ER chaperones and the transcription factor X-box binding protein 1 (XBP1). Activation of ATF6 may increase ER secretory capacity [35]. PERK dimerization activates autophosphorylation of its kinase domain, leading to phosphorylation of the α-subunit of translation initiation factor 2 (eIF2α) at Ser51. This inhibits global translational initiation, including translation of mRNAs encoding secretory proteins, reducing the load of unfolded proteins entering the ER. PERK activation also results in the induction of translation of selective mRNAs, including activating transcription factor 4 (ATF4), which activates the transcription of a wide range of genes involved in adaptation to stress conditions. Upon activation, IRE1α oligomerizes and carries out regulated IRE1-dependent decay (RIDD) of ER-targeted transcripts, as well as unconventional RNA splicing, by removing an intron from the XBP1 mRNA, allowing it to become translated into the functional transcription factor, XBP1s. XBP1s activates the transcription of ER chaperones, endoplasmic reticulum associated degradation (ERAD) genes, and genes involved in lipid synthesis.
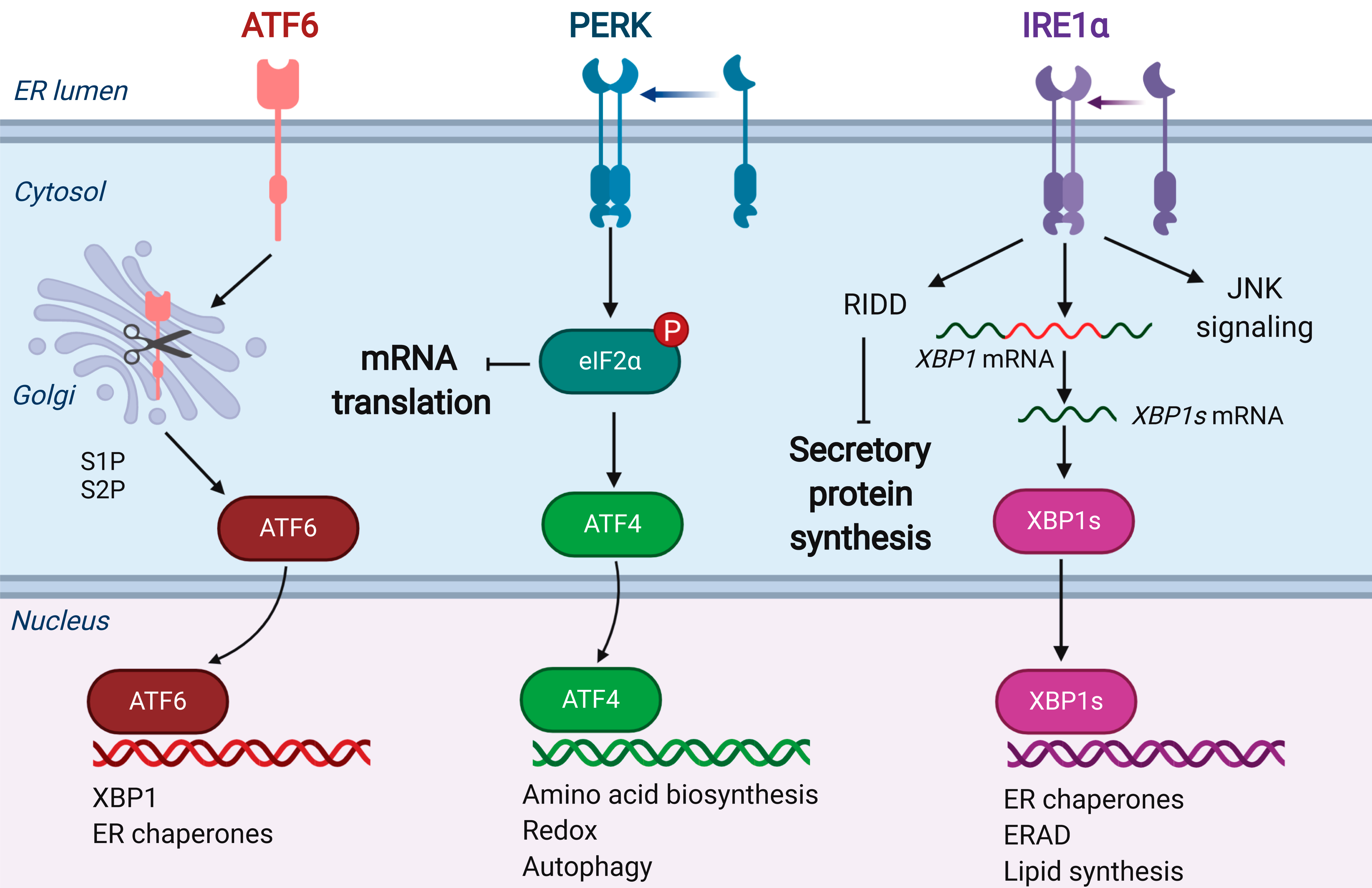
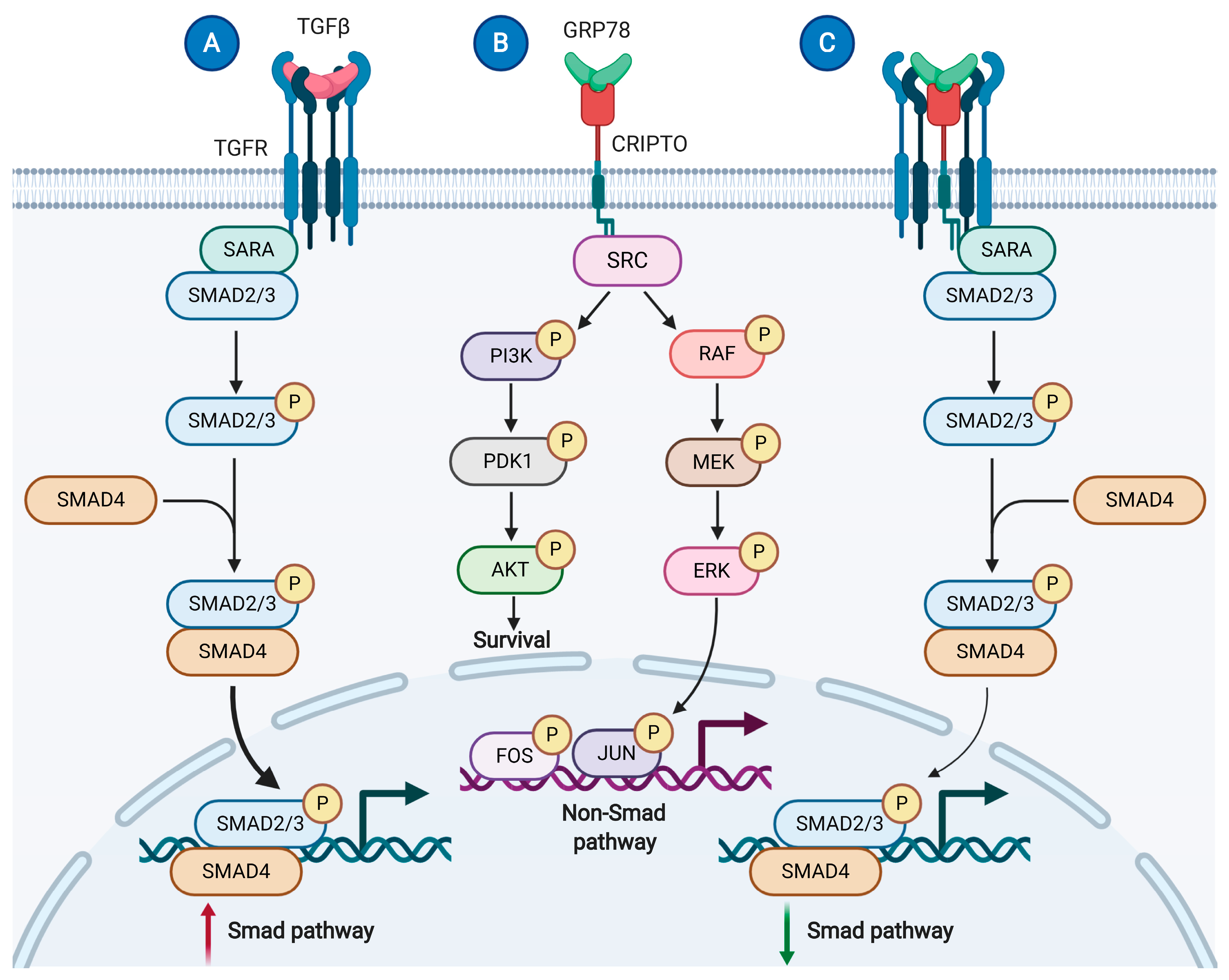
Figure 3.
Model of extracellular GRP78 and interacting protein, Cripto, modulation of TGFβ signaling. (A) Transforming growth factor beta (TGFβ) ligand binding to TGFβ receptors (TGFR) leads to activation of genes responsive to Smad2/3 signaling. (B) The 78 kDa glucose regulated protein (GRP78), an ER luminal Ca2+-binding chaperone, is secreted upon ER Ca2+ depletion. Secreted GRP78 is a co-factor required for Cripto-1 growth factor (Cripto) activation. Cripto activates Src, Ras/Raf/MAPK and PI3K/Akt pathways via a mechanism that remains unknown. (C) Cell surface Cripto/GRP78 interaction is required for Cripto modulation of TGFβ signaling.
Figure 3.
Model of extracellular GRP78 and interacting protein, Cripto, modulation of TGFβ signaling. (A) Transforming growth factor beta (TGFβ) ligand binding to TGFβ receptors (TGFR) leads to activation of genes responsive to Smad2/3 signaling. (B) The 78 kDa glucose regulated protein (GRP78), an ER luminal Ca2+-binding chaperone, is secreted upon ER Ca2+ depletion. Secreted GRP78 is a co-factor required for Cripto-1 growth factor (Cripto) activation. Cripto activates Src, Ras/Raf/MAPK and PI3K/Akt pathways via a mechanism that remains unknown. (C) Cell surface Cripto/GRP78 interaction is required for Cripto modulation of TGFβ signaling.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Meyer, B.A.; Doroudgar, S. ER Stress-Induced Secretion of Proteins and Their Extracellular Functions in the Heart. Cells 2020, 9, 2066. https://doi.org/10.3390/cells9092066
AMA Style
Meyer BA, Doroudgar S. ER Stress-Induced Secretion of Proteins and Their Extracellular Functions in the Heart. Cells. 2020; 9(9):2066. https://doi.org/10.3390/cells9092066
Chicago/Turabian StyleMeyer, Bianca A., and Shirin Doroudgar. 2020. "ER Stress-Induced Secretion of Proteins and Their Extracellular Functions in the Heart" Cells 9, no. 9: 2066. https://doi.org/10.3390/cells9092066
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.