ER Stress-Induced Secretion of Proteins and Their Extracellular Functions in the Heart



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Protein Secretion in the Heart
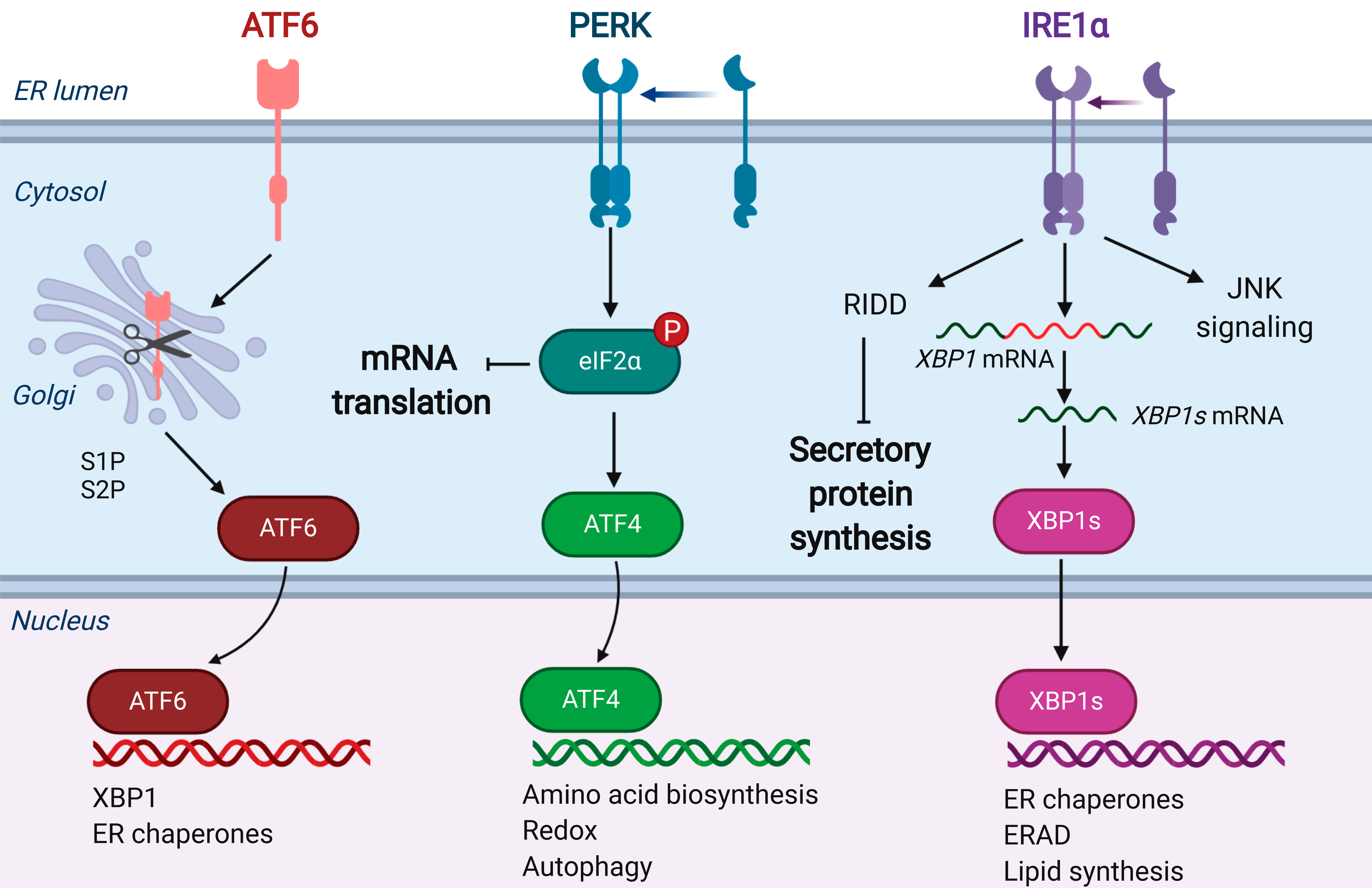
2. ER Stress in the Heart
2.1. Cardiac Proteostasis at the ER
2.2. ER Stress and Heart Disease
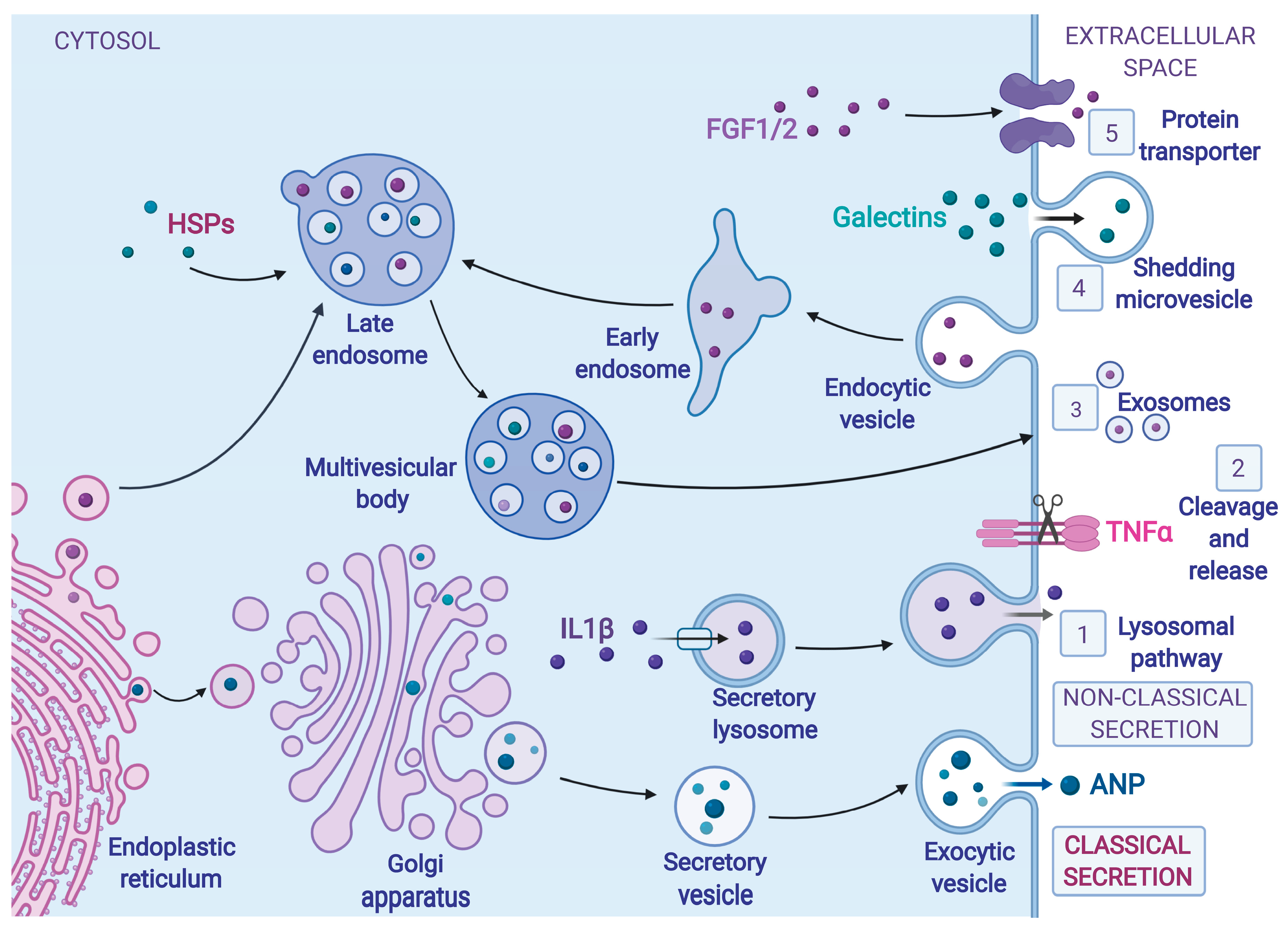
3. Protein Secretion during ER Stress
4. Secretion of Ca2+-Binding ER Chaperones
4.1. Secretion during Ca2+-Mediated ER Stress
4.2. Extracellular Functions of ER Chaperones in the Cardiovascular System
4.3. Extracellular Functions of ER Chaperones in Non-Cardiovascular Cells and Tissues
4.3.1. 78 kDa Glucose-Regulated Protein (GRP78; Binding Immunoglobulin Protein)
4.3.2. Other Ca2+-Binding ER Chaperones: Calreticulin, 94 kDa Glucose-Regulated Protein (GRP94; Endoplasmin; Gp96), MANF
5. Secretion of Other Proteins during ER Stress
5.1. Other Cardiokines
5.2. ERdj3 (Endoplasmic Reticulum DNA J Domain-Containing Protein 3; DnaJ Homolog Subfamily B Member 11)
5.3. Angiogenin
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kakkar, R.; Lee, R.T. Intramyocardial fibroblast myocyte communication. Circ. Res. 2010, 106, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Pu, W.T. Recounting Cardiac Cellular Composition. Circ. Res. 2016, 118, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Doroudgar, S.; Glembotski, C.C. The cardiokine story unfolds: Ischemic stress-induced protein secretion in the heart. Trends Mol. Med. 2011, 17, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Shimano, M.; Ouchi, N.; Walsh, K. Cardiokines: Recent progress in elucidating the cardiac secretome. Circulation 2012, 126, e327–e332. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.S.; Miller, E.A.; Goldberg, J.; Orci, L.; Schekman, R. Bi-Directional Protein Transport Between the Er and Golgi. Annu. Rev. Cell Dev. Biol. 2004, 20, 87–123. [Google Scholar] [CrossRef] [PubMed]
- Nickel, W.; Rabouille, C. Mechanisms of regulated unconventional protein secretion. Nat. Rev. Mol. Cell Biol. 2009, 10, 148–155. [Google Scholar] [CrossRef]
- Elvekrog, M.M.; Walter, P. Dynamics of co-translational protein targeting. Curr. Opin. Chem. Biol. 2015, 29, 79–86. [Google Scholar] [CrossRef]
- Cabrita, L.D.; Dobson, C.M.; Christodoulou, J. Protein folding on the ribosome. Curr. Opin. Struct. Biol. 2010, 20, 33–45. [Google Scholar] [CrossRef]
- Kraut-Cohen, J.; Gerst, J.E. Addressing mRNAs to the ER: Cis sequences act up! Trends Biochem. Sci. 2010, 35, 459–469. [Google Scholar] [CrossRef]
- Weis, B.L.; Schleiff, E.; Zerges, W. Protein targeting to subcellular organelles via mRNA localization. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 260–273. [Google Scholar] [CrossRef]
- Cohen-Zontag, O.; Baez, C.; Lim, L.Q.J.; Olender, T.; Schirman, D.; Dahary, D.; Pilpel, Y.; Gerst, J.E. A secretion-enhancing cis regulatory targeting element (SECReTE) involved in mRNA localization and protein synthesis. PLoS Genet. 2019, 15, e1008248. [Google Scholar] [CrossRef] [PubMed]
- Palade, G. Intracellular Aspects of the Process of Protein Synthesis. Science 1975, 189, 867. [Google Scholar] [CrossRef] [PubMed]
- Bloch, K.D.; Seidman, J.G.; Naftilan, J.D.; Fallon, J.T.; Seidman, C.E. Neonatal atria and ventricles secrete atrial natriuretic factor via tissue-specific secretory pathways. Cell 1986, 47, 695–702. [Google Scholar] [CrossRef]
- Sei, C.A.; Hand, G.L.; Murray, S.F.; Glembotski, C.C. The cosecretional maturation of atrial natriuretic factor by primary atrial myocytes. Mol. Endocrinol. 1992, 6, 309–319. [Google Scholar] [CrossRef]
- Maack, T. Role of atrial natriuretic factor in volume control. Kidney Int. 1996, 49, 1732–1737. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, J.; Gee, H.Y.; Lee, M.G. Unconventional protein secretion— New insights into the pathogenesis and therapeutic targets of human diseases. J. Cell Sci. 2018, 131, jcs213686. [Google Scholar] [CrossRef] [PubMed]
- Nickel, W.; Seedorf, M. Unconventional Mechanisms of Protein Transport to the Cell Surface of Eukaryotic Cells. Annu. Rev. Cell Dev. Biol. 2008, 24, 287–308. [Google Scholar] [CrossRef]
- Most, P.; Boerries, M.; Eicher, C.; Schweda, C.; Völkers, M.; Wedel, T.; Söllner, S.; Katus, H.A.; Remppis, A.; Aebi, U.; et al. Distinct subcellular location of the Ca2+-binding protein S100A1 differentially modulates Ca2+-cycling in ventricular rat cardiomyocytes. J. Cell Sci. 2005, 118, 421–431. [Google Scholar] [CrossRef][Green Version]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Treaster, S.B.; Ridgway, I.D.; Richardson, C.A.; Gaspar, M.B.; Chaudhuri, A.R.; Austad, S.N. Superior proteome stability in the longest lived animal. Age (Omaha) 2014. [Google Scholar] [CrossRef] [PubMed]
- Henning, R.H.; Brundel, B.J.J.M. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637. [Google Scholar] [CrossRef] [PubMed]
- Christis, C.; Lubsen, N.H.; Braakman, I. Protein folding includes oligomerization—Examples from the endoplasmic reticulum and cytosol. FEBS J. 2008, 275, 4700–4727. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef]
- Mohorko, E.; Glockshuber, R.; Aebi, M. Oligosaccharyltransferase: The central enzyme of N-linked protein glycosylation. J. Inherit. Metab. Dis. 2011, 34, 869–878. [Google Scholar] [CrossRef]
- Hudson, D.A.; Gannon, S.A.; Thorpe, C. Oxidative protein folding: From thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic. Biol. Med. 2015, 80, 171–182. [Google Scholar] [CrossRef]
- Gidalevitz, T.; Stevens, F.; Argon, Y. Orchestration of secretory protein folding by ER chaperones. Biochim. Biophys. Acta 2013, 1833, 2410–2424. [Google Scholar] [CrossRef]
- Brodsky, J.L. Cleaning Up: ER-associated degradation to the rescue. Cell 2012, 151, 1163–1167. [Google Scholar] [CrossRef]
- Claessen, J.H.L.; Kundrat, L.; Ploegh, H.L. Protein quality control in the ER: Balancing the ubiquitin checkbook. Trends Cell Biol. 2012, 22, 22–32. [Google Scholar] [CrossRef]
- Chyn, T.L.; Martonosi, A.N.; Morimoto, T.; Sabatini, D.D. In vitro synthesis of the Ca2+ transport ATPase by ribosomes bound to sarcoplasmic reticulum membranes. Proc. Natl. Acad. Sci. USA 1979, 76, 1241–1245. [Google Scholar] [CrossRef]
- Rossi, A.E.; Dirksen, R.T. Sarcoplasmic reticulum: The dynamic calcium governor of muscle. Muscle Nerve 2006, 33, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. The Biology of Proteostasis in Aging and Disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- McLendon, P.M.; Robbins, J. Proteotoxicity and Cardiac Dysfunction. Circ. Res. 2015, 116, 1863–1882. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Paxman, R.J.; Plate, L.; Kelly, J.W.; Wiseman, R.L.; Glembotski, C.C. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat. Commun. 2019, 10, 187. [Google Scholar] [CrossRef]
- Kilberg, M.S.; Shan, J.; Su, N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009, 20, 436–443. [Google Scholar] [CrossRef]
- Plemper, R.K.; Wolf, D.H. Endoplasmic reticulum degradation. Reverse protein transport and its end in the proteasome. Mol. Biol. Rep. 1999, 26, 125–130. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic- reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and Characterization of Pancreatic Eukaryotic Initiation Factor 2 α-Subunit Kinase, PEK, Involved in Translational Control. Mol. Cell. Biol. 1998, 18, 7499–7509. [Google Scholar] [CrossRef]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Tirasophon, W.; Welihinda, A.A.; Kaufman, R.J. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998, 12, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Harding, H.P.; Zhang, Y.; Jolicoeur, E.M.; Kuroda, M.; Ron, D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998, 17, 5708–5717. [Google Scholar] [CrossRef] [PubMed]
- Niwa, M.; Sidrauski, C.; Kaufman, R.J.; Walter, P. A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell 1999, 99, 691–702. [Google Scholar] [CrossRef]
- Zhu, C.; Johansen, F.E.; Prywes, R. Interaction of ATF6 and serum response factor. Mol. Cell. Biol. 1997, 17, 4957–4966. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose- regulated proteins: Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxidants Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front. Mol. Biosci. 2019, 6. [Google Scholar] [CrossRef]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.K.; Lawrence, D.A.; Lu, M.; Tan, J.; Harnoss, J.M.; Marsters, S.A.; Liu, P.; Sandoval, W.; Martin, S.E.; Ashkenazi, A. Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Mol. Cell 2018, 71, 629.e5–636.e5. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Katus, H.A.; Doroudgar, S. Protein Misfolding in Cardiac Disease. Circulation 2019, 139, 2085–2088. [Google Scholar] [CrossRef]
- Thuerauf, D.J.; Marcinko, M.; Gude, N.; Rubio, M.; Sussman, M.A.; Glembotski, C.C. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ. Res. 2006, 99, 275–282. [Google Scholar] [CrossRef]
- Doroudgar, S.; Thuerauf, D.J.; Marcinko, M.C.; Belmont, P.J.; Glembotski, C.C. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J. Biol. Chem. 2009, 284, 29735–29745. [Google Scholar] [CrossRef]
- Groenendyk, J.; Sreenivasaiah, P.K.; Kim, D.H.; Agellon, L.B.; Michalak, M. Biology of endoplasmic reticulum stress in the heart. Circ. Res. 2010, 107, 1185–1197. [Google Scholar] [CrossRef]
- Buja, L.M. Myocardial ischemia and reperfusion injury. Cardiovasc. Pathol. 2005, 14, 170–175. [Google Scholar] [CrossRef]
- Doroudgar, S.; Glembotski, C.C. New concepts of endoplasmic reticulum function in the heart: Programmed to conserve. J. Mol. Cell. Cardiol. 2013, 55, 85–91. [Google Scholar] [CrossRef]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Engl. J. Med. 2008, 358, 1370. [Google Scholar] [CrossRef]
- Blackwood, E.A.; Hofmann, C.; Santo Domingo, M.; Bilal, A.S.; Sarakki, A.; Stauffer, W.; Arrieta, A.; Thuerauf, D.J.; Kolkhorst, F.W.; Müller, O.J.; et al. ATF6 Regulates Cardiac Hypertrophy by Transcriptional Induction of the mTORC1 Activator, Rheb. Circ. Res. 2019, 124, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.A.; Thuerauf, D.J.; Stastna, M.; Stephens, H.; Sand, Z.; Pentoney, A.; Azizi, K.; Jakobi, T.; Van Eyk, J.E.; Katus, H.A.; et al. Proteomic analysis of the cardiac myocyte secretome reveals extracellular protective functions for the ER stress response. J. Mol. Cell. Cardiol. 2020, 143, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.W.; Chen, Q.; Tay, A.S.L.; Shenolikar, S.; Nicchitta, C.V. The unfolded protein response triggers selective mRNA release from the endoplasmic reticulum. Cell 2014, 158, 1362–1374. [Google Scholar] [CrossRef]
- Peters, L.R.; Raghavan, M. Endoplasmic Reticulum Calcium Depletion Impacts Chaperone Secretion, Innate Immunity, and Phagocytic Uptake of Cells. J. Immunol. 2011, 187, 919–931. [Google Scholar] [CrossRef]
- Genereux, J.C.; Qu, S.; Zhou, M.; Ryno, L.M.; Wang, S.; Shoulders, M.D.; Kaufman, R.J.; Lasmézas, C.I.; Kelly, J.W.; Wiseman, R.L. Unfolded protein response-induced ER dj3 secretion links ER stress to extracellular proteostasis. EMBO J. 2015, 34, 4–19. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium Cycling and Signaling in Cardiac Myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef]
- Davlouros, P.A.; Gkizas, V.; Vogiatzi, C.; Giannopoulos, G.; Alexopoulos, D.; Deftereos, S. Calcium Homeostasis and Kinetics in Heart Failure. Med. Chem. 2016, 12, 151–161. [Google Scholar] [CrossRef]
- da Silva, D.C.; Valentão, P.; Andrade, P.B.; Pereira, D.M. Endoplasmic reticulum stress signaling in cancer and neurodegenerative disorders: Tools and strategies to understand its complexity. Pharmacol. Res. 2020, 155. [Google Scholar] [CrossRef]
- Wetmore, D.R.; Hardman, K.D. Roles of the propeptide and metal ions in the folding and stability of the catalytic domain of stromelysin (matrix metalloproteinase 3). Biochemistry 1996, 35, 6549–6558. [Google Scholar] [CrossRef]
- Halperin, L.; Jung, J.; Michalak, M. The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 2014, 66, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Corbett, E.F.; Oikawa, K.; Francois, P.; Tessier, D.C.; Kay, C.; Bergeron, J.J.M.; Thomas, D.Y.; Krause, K.-H.; Michalak, M. Ca2+ Regulation of Interactions between Endoplasmic Reticulum Chaperones. J. Biol. Chem. 1999, 274, 6203–6211. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.-J.; Bergeron, J.J.M.; Li, Y.; Kang, C.Y.; Thomas, D.Y. Conformational Changes Induced in the Endoplasmic Reticulum Luminal Domain of Calnexin by Mg-ATP and Ca. J. Biol. Chem. 1995, 270, 18051–18059. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, C.K.; Bonifacino, J.S.; Lin, A.Y.; Davis, M.M.; Klausner, R.D. Regulating the retention of T-cell receptor alpha chain variants within the endoplasmic reticulum: Ca(2+)-dependent association with BiP. J. Cell Biol. 1991, 114, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Klappa, P.; Kietzmann, D.T. The Endoplasmic Reticulum: Folding, Calcium Homeostasis, Signaling, and Redox Control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef]
- Glembotski, C.C.; Thuerauf, D.J.; Huang, C.; Vekich, J.A.; Gottlieb, R.A.; Doroudgar, S. Mesencephalic astrocyte-derived neurotrophic factor protects the heart from ischemic damage and is selectively secreted upon sarco/endoplasmic reticulum calcium depletion. J. Biol. Chem. 2012, 287, 25893–25904. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef]
- Gold, L.I.; Eggleton, P.; Sweetwyne, M.T.; Van Duyn, L.B.; Greives, M.R.; Naylor, S.; Michalak, M.; Murphy-Ullrich, J.E. Calreticulin: Non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010, 24, 665–683. [Google Scholar] [CrossRef]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 774–787. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, F.; Liu, Z.; Su, Q.; Liu, Y.; Liu, Z.; Li, Y. The ER-localized Ca2+-binding protein calreticulin couples ER stress to autophagy by associating with microtubule-associated protein 1A/1B light chain 3. J. Biol. Chem. 2019, 294, 772–782. [Google Scholar] [CrossRef]
- Takemoto, H.; Yoshimori, T.; Yamamoto, A.; Miyata, Y.; Yahara, I.; Inoue, K.; Tashiro, Y. Heavy chain binding protein (BiP/GRP78) and endoplasmin are exported from the endoplasmic reticulum in rat exocrine pancreatic cells, similar to protein disulfide-isomerase. Arch. Biochem. Biophys. 1992, 296, 129–136. [Google Scholar] [CrossRef]
- Capitani, M.; Sallese, M. The KDEL receptor: New functions for an old protein. FEBS Lett. 2009, 583, 3863–3871. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R. Unsolved Mysteries in Membrane Traffic. Annu. Rev. Biochem. 2007, 76, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, R.; Ni, M.; Gill, P.; Lee, A.S.S. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J. Biol. Chem. 2010, 285, 15065–15075. [Google Scholar] [CrossRef] [PubMed]
- Glembotski, C.C. Functions for the cardiomyokine, MANF, in cardioprotection, hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 512–517. [Google Scholar] [CrossRef] [PubMed]
- De Castro, N.P.; Rangel, M.C.; Nagaoka, T.; Salomon, D.S.; Bianco, C. Cripto-1: An embryonic gene that promotes tumorigenesis. Future Oncol. 2010, 6, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Gray, P.C.; Vale, W. Cripto/GRP78 modulation of the TGF-β pathway in development and oncogenesis. FEBS Lett. 2012, 586, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Shani, G.; Fischer, W.H.; Justice, N.J.; Kelber, J.A.; Vale, W.; Gray, P.C. GRP78 and Cripto Form a Complex at the Cell Surface and Collaborate To Inhibit Transforming Growth Factor β Signaling and Enhance Cell Growth. Mol. Cell. Biol. 2008, 28, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Kelber, J.A.; Panopoulos, A.D.; Shani, G.; Booker, E.C.; Belmonte, J.C.; Vale, W.W.; Gray, P.C. Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways. Oncogene 2009, 28, 2324–2336. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.L.; Zhang, Y.; Tseng, C.C.; Stanciauskas, R.; Pinaud, F.; Lee, A.S. Characterization and mechanism of stress-induced translocation of 78-kilodalton glucose-regulated protein (GRP78) to the cell surface. J. Biol. Chem. 2015, 290, 8049–8064. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gronow, M.; Selim, M.A.; Papalas, J.; Pizzo, S.V. GRP78: A multifunctional receptor on the cell surface. Antioxidants Redox Signal. 2009, 11, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Zhang, G.; Wang, X.; Nguyen, C.; May, H.I.; Li, X.; Al-Hashimi, A.A.; Austin, R.C.; Gillette, T.G.; Fu, G.; et al. Endoplasmic Reticulum Chaperone GRP78 Protects Heart From Ischemia/Reperfusion Injury Through Akt Activation. Circ. Res. 2018, 122, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, G.; Ahamed, J.; Pedersen, B.; El-Sheikh, A.; Mackman, N.; Ruf, W.; Liu, C.; Edgington, T.S. Regulation of tissue factor-mediated initiation of the coagulatiion cascade by cell surface Grp78. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1737–1743. [Google Scholar] [CrossRef]
- Zhao, D.; Feng, P.; Sun, Y.; Qin, Z.; Zhang, Z.; Tan, Y.; Gao, E.; Lau, W.B.; Ma, X.; Yang, J.; et al. Cardiac-derived CTRP9 protects against myocardial ischemia/reperfusion injury via calreticulin-dependent inhibition of apoptosis article. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Petrova, P.S.; Raibekas, A.; Pevsner, J.; Vigo, N.; Anafi, M.; Moore, M.K.; Peaire, A.E.; Shridhar, V.; Smith, D.I.; Kelly, J.; et al. MANF. J. Mol. Neurosci. 2003, 20, 173–187. [Google Scholar] [CrossRef]
- Palgi, M.; Lindström, R.; Peränen, J.; Piepponen, T.P.; Saarma, M.; Heino, T.I. Evidence that DmMANF is an invertebrate neurotrophic factor supporting dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2009, 106, 2429–2434. [Google Scholar] [CrossRef] [PubMed]
- Neves, J.; Zhu, J.; Sousa-Victor, P.; Konjikusic, M.; Riley, R.; Chew, S.; Qi, Y.; Jasper, H.; Lamba, D.A. Immune modulation by MANF promotes tissue repair and regenerative success in the retina. Science 2016, 353. [Google Scholar] [CrossRef]
- Lindström, R.; Lindholm, P.; Kallijärvi, J.; Palgi, M.; Saarma, M.; Heino, T.I. Exploring the Conserved Role of MANF in the Unfolded Protein Response in Drosophila melanogaster. PLoS ONE 2016, 11, e0151550. [Google Scholar] [CrossRef]
- Tadimalla, A.; Belmont, P.J.; Thuerauf, D.J.; Glassy, M.S.; Martindale, J.J.; Gude, N.; Sussman, M.A.; Glembotski, C.C. Mesencephalic astrocyte-derived neurotrophic factor is an ischemia-inducible secreted endoplasmic reticulum stress response protein in the heart. Circ. Res. 2008, 103, 1249–1258. [Google Scholar] [CrossRef]
- Bai, M.; Vozdek, R.; Hnízda, A.; Jiang, C.; Wang, B.; Kuchar, L.; Li, T.; Zhang, Y.; Wood, C.; Feng, L.; et al. Conserved roles of C. elegans and human MANFs in sulfatide binding and cytoprotection. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Araujo, N.; Hebbar, N.; Rangnekar, V.M. GRP78 Is a Targetable Receptor on Cancer and Stromal Cells. EBioMedicine 2018, 33, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Z. Glucose regulated protein 78: A critical link between tumor microenvironment and cancer hallmarks. Biochim. Biophys. Acta Rev. Cancer 2012, 1826, 13–22. [Google Scholar] [CrossRef] [PubMed]
- La, X.; Zhang, L.; Yang, Y.; Li, H.; Song, G.; Li, Z. Tumor-secreted GRP78 facilitates the migration of macrophages into tumors by promoting cytoskeleton remodeling. Cell. Signal. 2019, 60, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Louessard, M.; Bardou, I.; Lemarchand, E.; Thiebaut, A.M.; Parcq, J.; Leprince, J.; Terrisse, A.; Carraro, V.; Fafournoux, P.; Bruhat, A.; et al. Activation of cell surface GRP78 decreases endoplasmic reticulum stress and neuronal death. Cell Death Differ. 2017, 24, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Vig, S.; Buitinga, M.; Rondas, D.; Crèvecoeur, I.; van Zandvoort, M.; Waelkens, E.; Eizirik, D.L.; Gysemans, C.; Baatsen, P.; Mathieu, C.; et al. Cytokine-induced translocation of GRP78 to the plasma membrane triggers a pro-apoptotic feedback loop in pancreatic beta cells. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Panayi, G.S.; Corrigall, V.M. BiP regulates autoimmune inflammation and tissue damage. Autoimmun. Rev. 2006, 5, 140–142. [Google Scholar] [CrossRef]
- Qin, K.; Ma, S.; Li, H.; Wu, M.; Sun, Y.; Fu, M.; Guo, Z.; Zhu, H.; Gong, F.; Lei, P.; et al. GRP78 impairs production of lipopolysaccharide-induced cytokines by interaction with CD14. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef]
- Triantafilou, K.; Fradelizi, D.; Wilson, K.; Triantafilou, M. GRP78, a Coreceptor for Coxsackievirus A9, Interacts with Major Histocompatibility Complex Class I Molecules Which Mediate Virus Internalization. J. Virol. 2002, 76, 633–643. [Google Scholar] [CrossRef]
- Jindadamrongwech, S.; Thepparit, C.; Smith, D.R. Identification of GRP 78 (BiP) as a liver cell expressed receptor element for dengue virus serotype 2. Arch. Virol. 2004, 149, 915–927. [Google Scholar] [CrossRef]
- Honda, T.; Horie, M.; Daito, T.; Ikuta, K.; Tomonaga, K. Molecular Chaperone BiP Interacts with Borna Disease Virus Glycoprotein at the Cell Surface. J. Virol. 2009, 83, 12622–12625. [Google Scholar] [CrossRef]
- Nain, M.; Mukherjee, S.; Karmakar, S.P.; Paton, A.W.; Paton, J.C.; Abdin, M.Z.; Basu, A.; Kalia, M.; Vrati, S. GRP78 Is an Important Host Factor for Japanese Encephalitis Virus Entry and Replication in Mammalian Cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, C.M.; Zhang, X.; Wang, Y.; Yuan, S.; Zhou, J.; Au-Yeung, R.K.H.; Sze, K.H.; Yang, D.; Shuai, H.; et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 2018, 293, 11709–11726. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef]
- Somogyi, E.; Petersson, U.; Hultenby, K.; Wendel, M. Calreticulin—An endoplasmic reticulum protein with calcium-binding activity is also found in the extracellular matrix. Matrix Biol. 2003, 22, 179–191. [Google Scholar] [CrossRef]
- Tufi, R.; Panaretakis, T.; Bianchi, K.; Criollo, A.; Fazi, B.; Di Sano, F.; Tesniere, A.; Kepp, O.; Paterlini-Brechot, P.; Zitvogel, L.; et al. Reduction of endoplasmic reticulum Ca2+ levels favors plasma membrane surface exposure of calreticulin. Cell Death Differ. 2008, 15, 274–282. [Google Scholar] [CrossRef]
- Greives, M.R.; Samra, F.; Pavlides, S.C.; Blechman, K.M.; Naylor, S.M.; Woodrell, C.D.; Cadacio, C.; Levine, J.P.; Bancroft, T.A.; Michalak, M.; et al. Exogenous calreticulin improves diabetic wound healing. Wound Repair Regen. 2012, 20, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef]
- Owusu, B.Y.; Zimmerman, K.A.; Murphy-Ullrich, J.E. The role of the endoplasmic reticulum protein calreticulin in mediating TGF-β-stimulated extracellular matrix production in fibrotic disease. J. Cell Commun. Signal. 2018, 12, 289–299. [Google Scholar] [CrossRef]
- Han, A.; Li, C.; Zahed, T.; Wong, M.; Smith, I.; Hoedel, K.; Green, D.; Boiko, A.D. Calreticulin is a critical cell survival factor in malignant neoplasms. PLoS Biol. 2019, 17. [Google Scholar] [CrossRef] [PubMed]
- Schcolnik-Cabrera, A.; Oldak, B.; Juárez, M.; Cruz-Rivera, M.; Flisser, A.; Mendlovic, F. Calreticulin in phagocytosis and cancer: Opposite roles in immune response outcomes. Apoptosis 2019. [Google Scholar] [CrossRef] [PubMed]
- Jockheck-Clark, A.R.; Bowers, E.V.; Totonchy, M.B.; Neubauer, J.; Pizzo, S.V.; Nicchitta, C.V. Re-Examination of CD91 Function in GRP94 (Glycoprotein 96) Surface Binding, Uptake, and Peptide Cross-Presentation. J. Immunol. 2010, 185, 6819–6830. [Google Scholar] [CrossRef]
- Bruneau, N.; Lombarde, D.; Bendayan, M. Participation of GRP94-related protein in secretion of pancreatic bile salt-dependent lipase and in its internalization by the intestinal epithelium. J. Cell Sci. 1998, 111, 2665–2679. [Google Scholar] [PubMed]
- Liu, B.; Dai, J.; Zheng, H.; Stoilova, D.; Sun, S.; Li, Z. Cell surface expression of an endoplasmic reticulum resident heat shock protein gp96 triggers MyD88-dependent systemic autoimmune diseases. Proc. Natl. Acad. Sci. USA 2003, 100, 15824–15829. [Google Scholar] [CrossRef] [PubMed]
- Danilova, T.; Lindahl, M. Emerging roles for mesencephalic astrocyte-derived neurotrophic factor (MANF) in pancreatic beta cells and diabetes. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef]
- 127. Kuhn, T.C.; Knobel, J.; Burkert-Rettenmaier, S.; Li, X.; Meyer, I.S.; Jungmann, A.; Sicklinger, F.; Backs, J.; Lasitschka, F.; Müller, O.J.; et al. Secretome Analysis of Cardiomyocytes Identifies PCSK6 (Proprotein Convertase Subtilisin/Kexin Type 6) as a Novel Player in Cardiac Remodeling After Myocardial Infarction. Circulation 2020, 141, 1628–1644. [Google Scholar] [CrossRef]
- Shen, Y.; Hendershot, L.M. ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP’s interactions with unfolded substrates. Mol. Biol. Cell 2005, 16, 40–50. [Google Scholar] [CrossRef]
- Nizard, P.; Tetley, S.; Le Dréan, Y.; Watrin, T.; Le Goff, P.; Wilson, M.R.; Michel, D. Stress-induced retrotranslocation of clusterin/ApoJ into the cytosol. Traffic 2007, 8, 554–565. [Google Scholar] [CrossRef]
- St Clair, D.K.; Rybak, S.M.; Riordan, J.F.; Vallee, B.L. Angiogenin abolishes cell-free protein synthesis by specific ribonucleolytic inactivation of ribosomes. Proc. Natl. Acad. Sci. USA 1987, 84, 8330–8334. [Google Scholar] [CrossRef]
- Saxena, S.K.; Rybak, S.M.; Davey, R.T.; Youle, R.J.; Ackerman, E.J. Angiogenin is a cytotoxic, tRNA-specific ribonuclease in the RNase A superfamily. J. Biol. Chem. 1992, 267, 21982–21986. [Google Scholar] [PubMed]
- Greenway, M.J.; Andersen, P.M.; Russ, C.; Ennis, S.; Cashman, S.; Donaghy, C.; Patterson, V.; Swingler, R.; Kieran, D.; Prehn, J.; et al. ANG mutations segregate with familial and “sporadic” amyotrophic lateral sclerosis. Nat. Genet. 2006, 38, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Kieran, D.; Sebastia, J.; Greenway, M.J.; King, M.A.; Connaughton, D.; Concannon, C.G.; Fenner, B.; Hardiman, O.; Prehn, J.H.M. Control of motoneuron survival by angiogenin. J. Neurosci. 2008, 28, 14056–14061. [Google Scholar] [CrossRef] [PubMed]
- Sebastià, J.; Kieran, D.; Breen, B.; King, M.A.; Netteland, D.F.; Joyce, D.; Fitzpatrick, S.F.; Taylor, C.T.; Prehn, J.H.M. Angiogenin protects motoneurons against hypoxic injury. Cell Death Differ. 2009, 16, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Skorupa, A.; King, M.A.; Aparicio, I.M.; Dussmann, H.; Coughlan, K.; Breen, B.; Kieran, D.; Concannon, C.G.; Marin, P.; Prehn, J.H.M. Motoneurons secrete angiogenin to induce RNA cleavage in astroglia. J. Neurosci. 2012, 32, 5024–5038. [Google Scholar] [CrossRef]
- Yamasaki, S.; Ivanov, P.; Hu, G.F.; Anderson, P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J. Cell Biol. 2009, 185, 35–42. [Google Scholar] [CrossRef]
- Mami, I.; Tavernier, Q.; Bouvier, N.; Aboukamis, R.; Desbuissons, G.; Rabant, M.; Poindessous, V.; Laurent-Puig, P.; Beaune, P.; Tharaux, P.L.; et al. A novel extrinsic pathway for the unfolded protein response in the kidney. J. Am. Soc. Nephrol. 2016, 27, 2670–2683. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer, B.A.; Doroudgar, S. ER Stress-Induced Secretion of Proteins and Their Extracellular Functions in the Heart. Cells 2020, 9, 2066. https://doi.org/10.3390/cells9092066
Meyer BA, Doroudgar S. ER Stress-Induced Secretion of Proteins and Their Extracellular Functions in the Heart. Cells. 2020; 9(9):2066. https://doi.org/10.3390/cells9092066
Chicago/Turabian StyleMeyer, Bianca A., and Shirin Doroudgar. 2020. "ER Stress-Induced Secretion of Proteins and Their Extracellular Functions in the Heart" Cells 9, no. 9: 2066. https://doi.org/10.3390/cells9092066
APA StyleMeyer, B. A., & Doroudgar, S. (2020). ER Stress-Induced Secretion of Proteins and Their Extracellular Functions in the Heart. Cells, 9(9), 2066. https://doi.org/10.3390/cells9092066