Targeting Oxidative Phosphorylation Reverses Drug Resistance in Cancer Cells by Blocking Autophagy Recycling


 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Sulforhodamine B-Cell Proliferation Assay
2.3. Western Blot Analysis
2.4. Mitochondrial Function Analysis
2.5. Measurement of Mitochondrial Membrane Potential (∆ψm)
2.6. Determination of Autophagy
2.7. Immunohistochemistry
2.8. Xenograft Tumor Models
2.9. Establishment of Resistant Cell
2.10. Statistical Analysis
3. Results
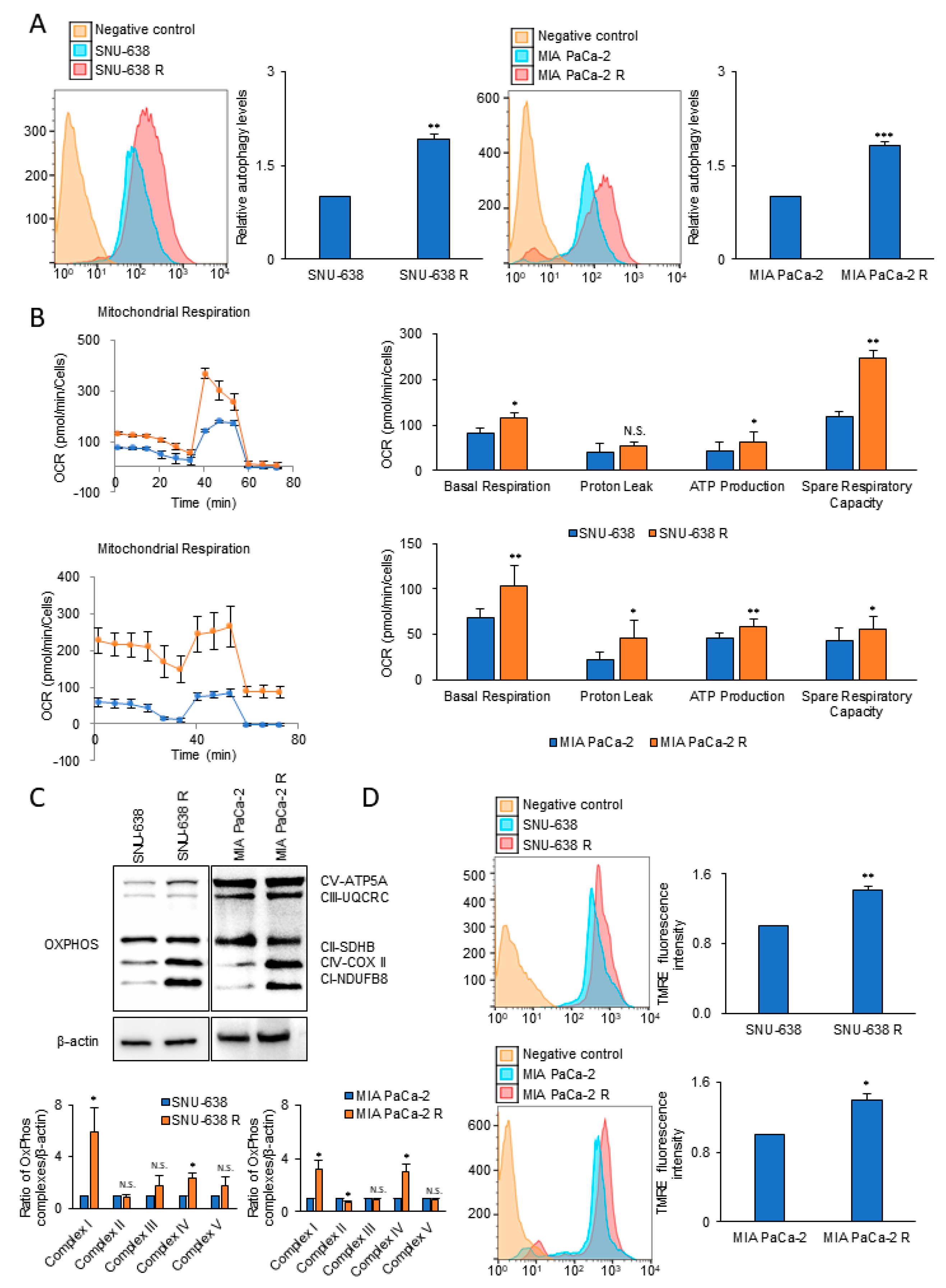
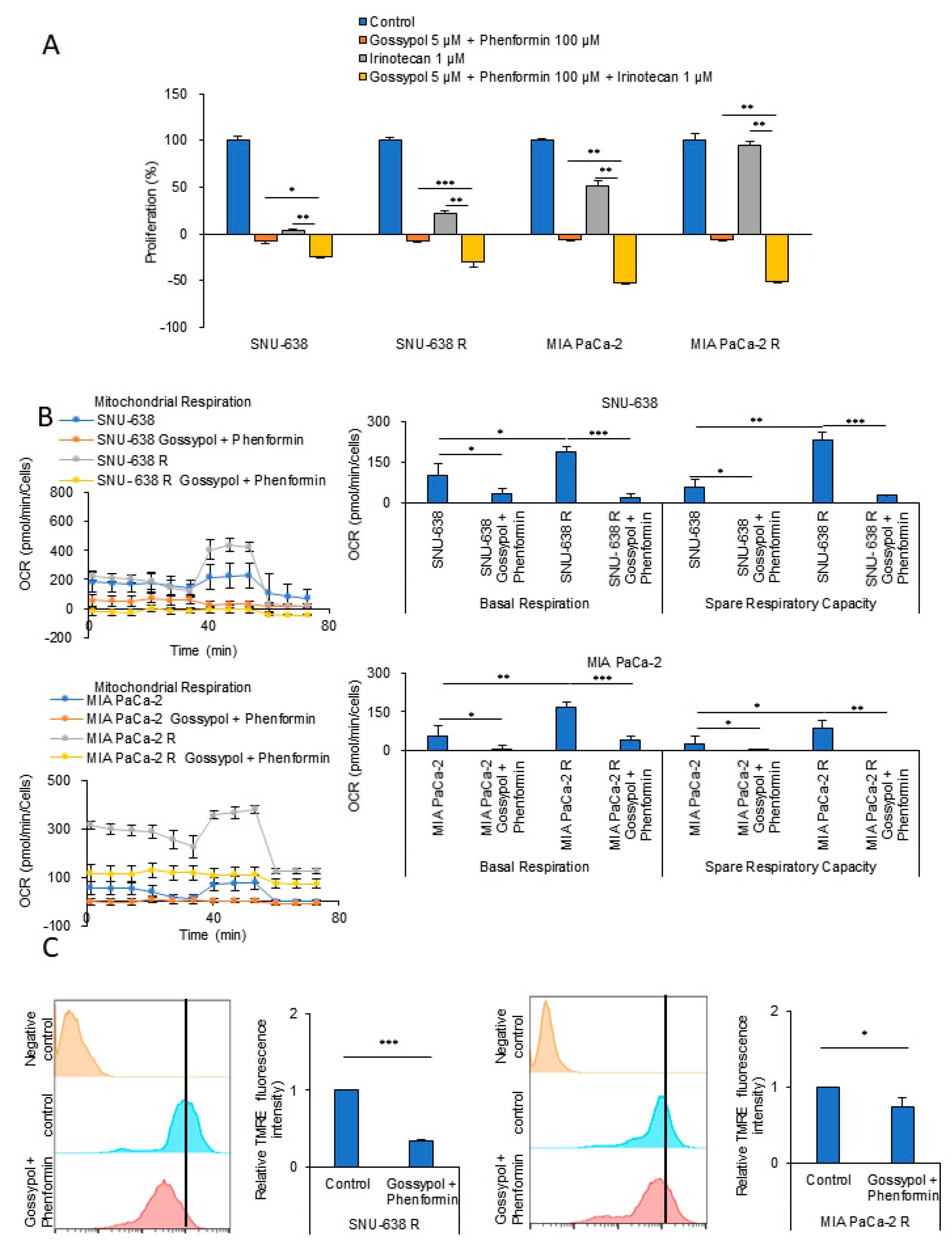
3.1. Autophagy and Mitochondrial OxPhos Markedly Increases in Drug-Resistant Cancer Cells
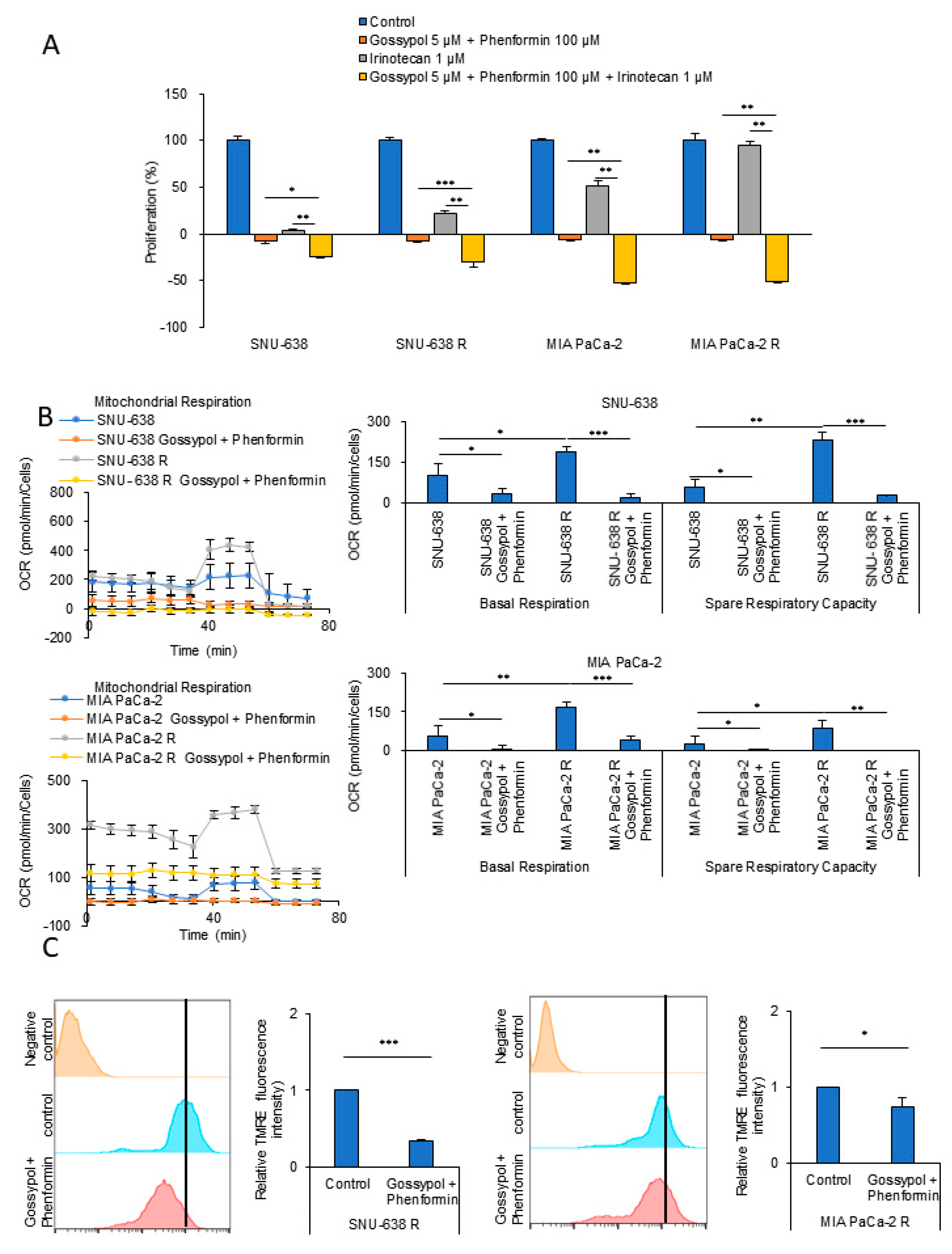
3.2. OxPhos Inhibition by Gossypol and Phenformin Reverses Anticancer Drug Resistance
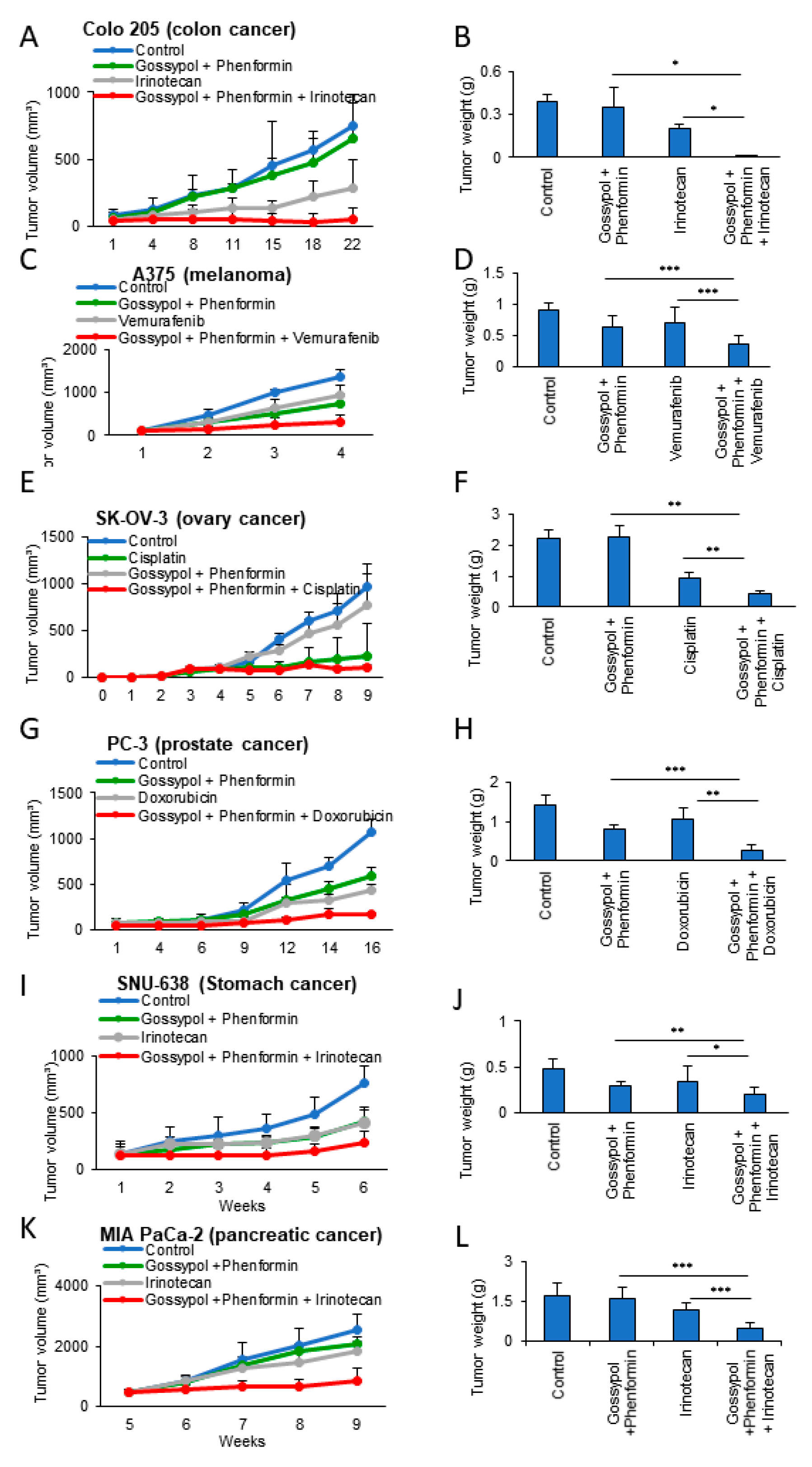
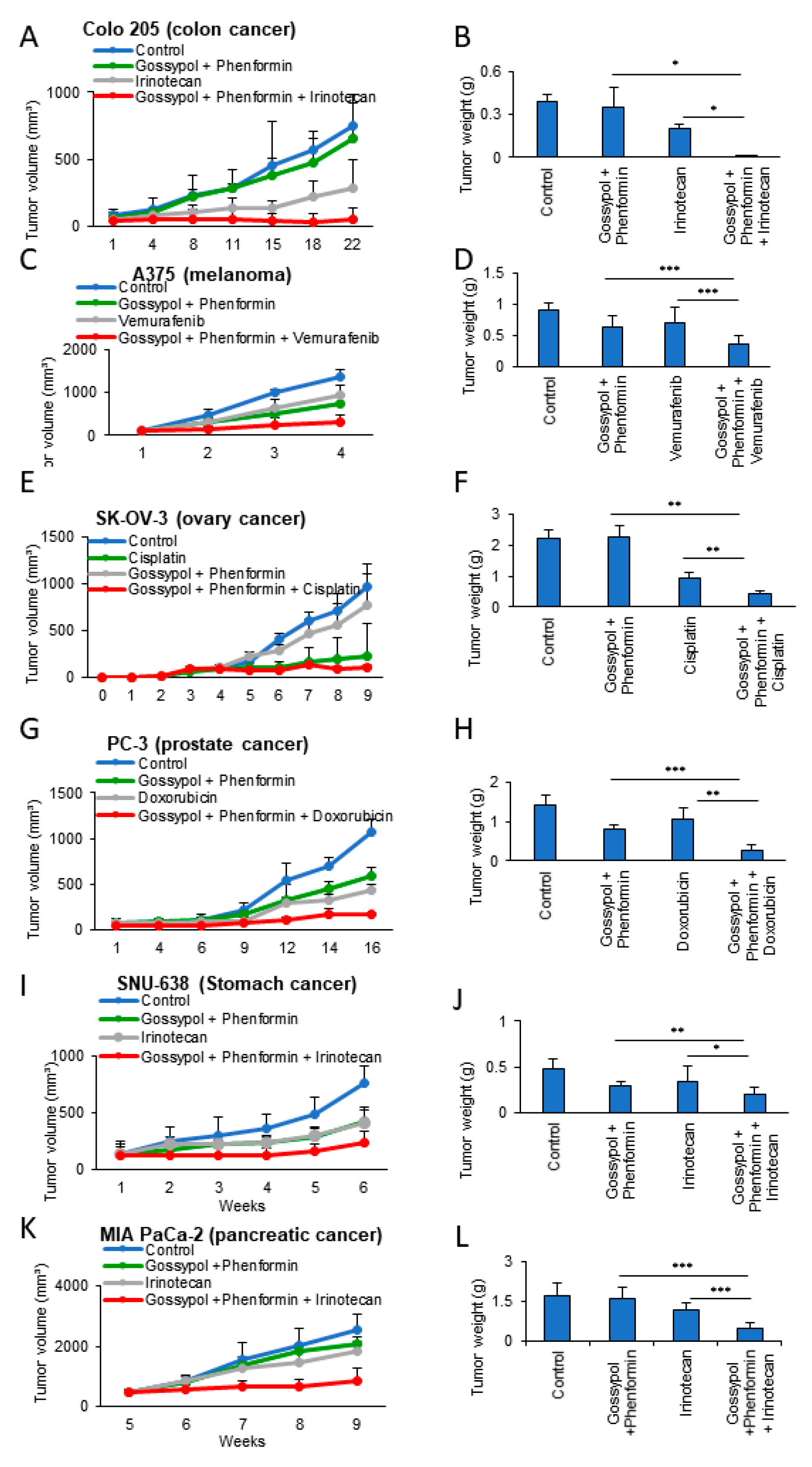
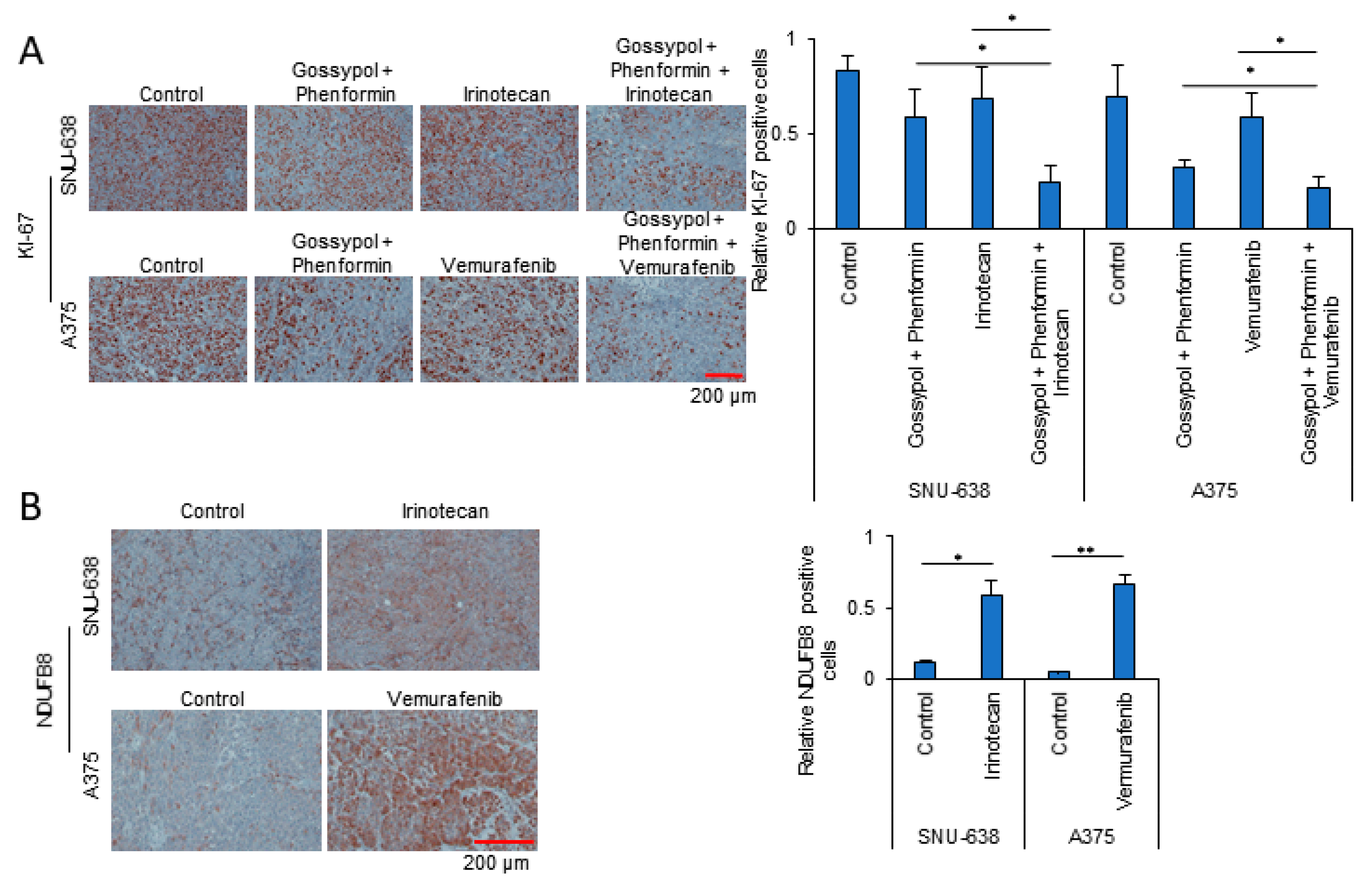
3.3. OxPhos Inhibition Promotes Chemotherapy Efficacy in Various Cancer Cells
3.4. Anticancer Drug Treatment Combined with Phenformin and Gossypol Suppress the Growth of Tumors in a Mouse Xenograft Model
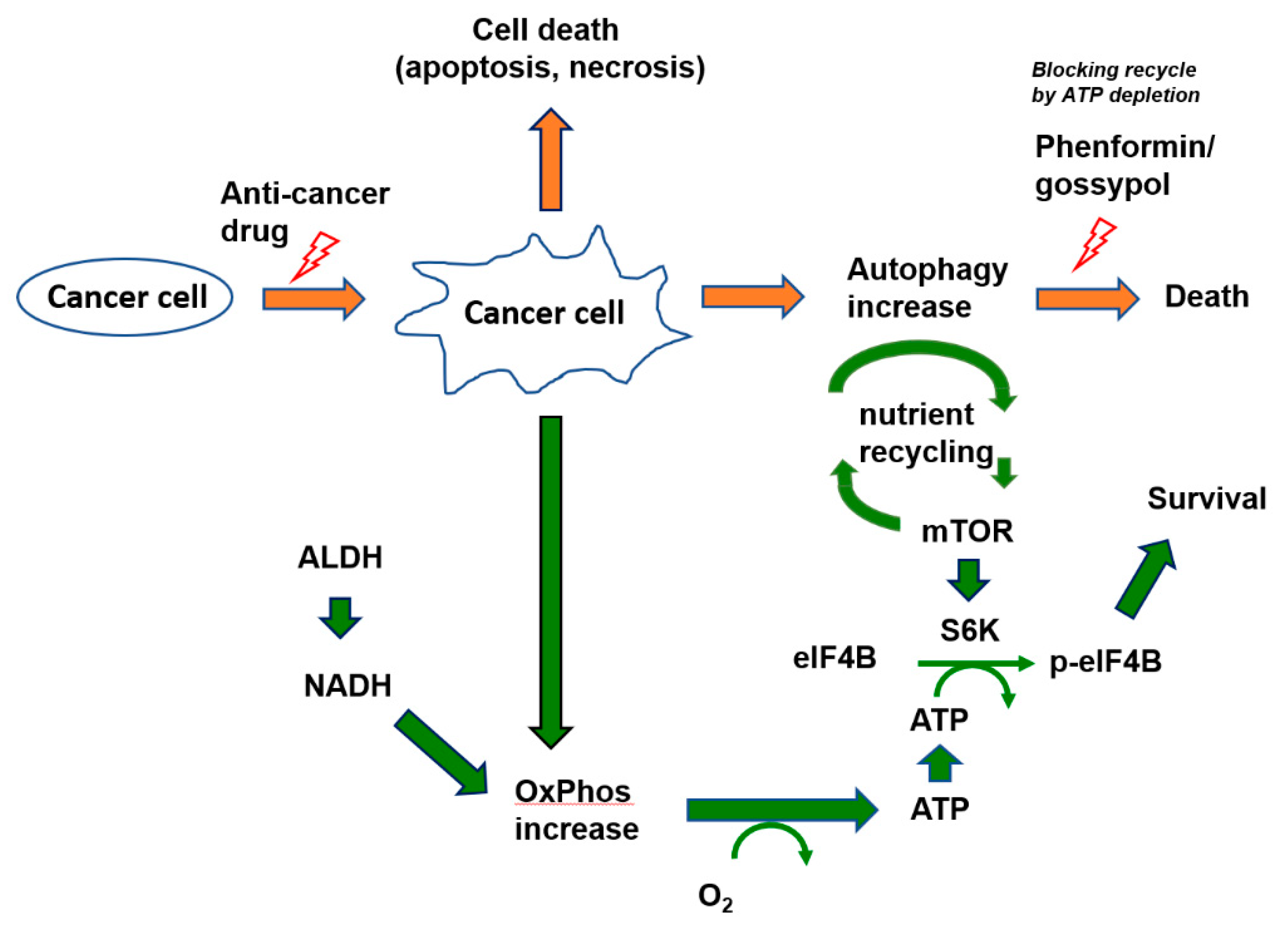
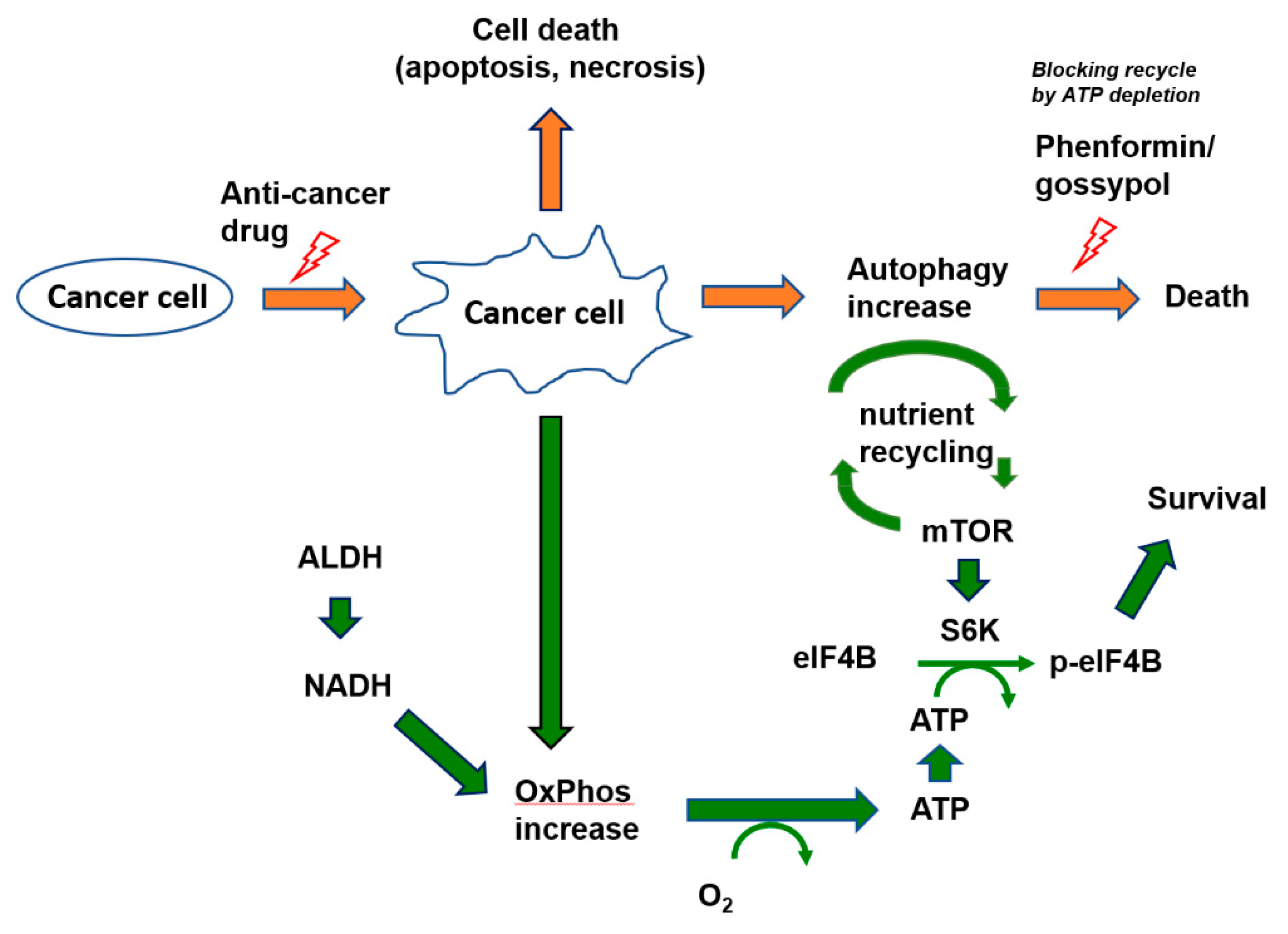
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. Resistance to Targeted Therapies: Refining Anticancer Therapy in the Era of Molecular Oncology. Clin. Cancer Res. 2009, 15, 7471–7478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groenendijk, F.H.; Bernards, R. Drug resistance to targeted therapies: Déjà vu all over again. Mol. Oncol. 2014, 8, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.J.; Gorski, S.M. Molecular Mechanisms Underlying Autophagy-Mediated Treatment Resistance in Cancer. Cancers 2019, 11, 1775. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Buszczak, M.; Kramer, H. Autophagy Keeps the Balance in Tissue Homeostasis. Dev. Cell 2019, 49, 499–500. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Y.; Li, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Liu, J.; Li, H. Autophagy: A novel mechanism of chemoresistance in cancers. Biomed. Pharmacother. 2019, 119, 109415. [Google Scholar] [CrossRef]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.-D.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. et Biophys. Acta (BBA) - Rev. Cancer 2010, 1806, 220–229. [Google Scholar] [CrossRef]
- Hsiang, Y.H.; Liu, L.F. Identification of mammalian DNA topoisomerase I as an intracellular target of the anticancer drug camptothecin. Cancer Res. 1988, 48, 1722–1726. [Google Scholar]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, J.-S.; Seo, J.; Lee, S.-H.; Kang, J.H.; Song, J.; Kim, S.-Y. Targeting Mitochondrial Oxidative Phosphorylation Abrogated Irinotecan Resistance in NSCLC. Sci. Rep. 2018, 8, 15707. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y. Targeting cancer energy metabolism: A potential systemic cure for cancer. Arch. Pharmacal Res. 2019, 42, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y. Cancer Energy Metabolism: Shutting Power off Cancer Factory. Biomol. Ther. 2018, 26, 39–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-S.; Lee, H.; Lee, S.; Kang, J.H.; Lee, S.-H.; Kim, S.-G.; Cho, E.S.; Kim, N.H.; Yook, J.I.; Kim, S.-Y. Loss of SLC25A11 causes suppression of NSCLC and melanoma tumor formation. EBioMedicine 2019, 40, 184–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, M.; Gray, S.; Hoffmann, A.-C.; Hilger, R.A.; Thomale, J.; O’Flaherty, J.D.; Fennell, D.A.; Richard, D.J.; O’Leary, J.J.; O’Byrne, K.J. Generation and Characterisation of Cisplatin-Resistant Non-Small Cell Lung Cancer Cell Lines Displaying a Stem-Like Signature. PLoS ONE 2013, 8, e54193. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Lee, S.-H.; Hong, D.; Lee, J.-S.; Ahn, H.-S.; Ahn, J.-H.; Seong, T.W.; Lee, C.-H.; Jang, H.; Hong, K.M.; et al. Aldehyde dehydrogenase is used by cancer cells for energy metabolism. Exp. Mol. Med. 2016, 48, e272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.H.; Lee, S.-H.; Lee, J.-S.; Nam, B.; Seong, T.W.; Son, J.; Jang, H.; Hong, K.M.; Lee, C.; Kim, S.-Y. Aldehyde dehydrogenase inhibition combined with phenformin treatment reversed NSCLC through ATP depletion. Oncotarget 2016, 7, 49397–49410. [Google Scholar] [CrossRef]
- Lee, S.-H.; Jeon, Y.; Kang, J.H.; Jang, H.; Lee, H.; Kim, S.-Y. The Combination of Loss of ALDH1L1 Function and Phenformin Treatment Decreases Tumor Growth in KRAS-Driven Lung Cancer. Cancers 2020, 12, 1382. [Google Scholar] [CrossRef]
- Sweet, S.; Singh, G. Accumulation of human promyelocytic leukemic (HL-60) cells at two energetic cell cycle checkpoints. Cancer Res. 1995, 55, 5164–5167. [Google Scholar]
- Lieberthal, W.; Menza, S.A.; Levine, J.S. Graded ATP depletion can cause necrosis or apoptosis of cultured mouse proximal tubular cells. Am. J. Physiol. Content 1998, 274, F315–F327. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Kim, S.H.; Lee, S.; Kang, J.H.; Lee, S.-H.; Cheong, J.-H.; Kim, S.-Y. Gastric cancer depends on aldehyde dehydrogenase 3A1 for fatty acid oxidation. Sci. Rep. 2019, 9, 16313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgos, C.; De Burgos, N.M.; Rovai, L.E.; Blanco, A. In vitro inhibition by gossypol of oxidoreductases from human tissues. Biochem. Pharmacol. 1986, 35, 801–804. [Google Scholar] [CrossRef]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde Dehydrogenase Inhibitors: A Comprehensive Review of the Pharmacology, Mechanism of Action, Substrate Specificity, and Clinical Application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, S.; Humphries, K.M. Selective Inhibition of Deactivated Mitochondrial Complex I by Biguanides. Biochemistry 2015, 54, 2011–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Shim, J.-K.; Kang, J.H.; Choi, J.; Chang, J.H.; Kim, S.-Y.; Kang, S.-G. Regulation of bioenergetics through dual inhibition of aldehyde dehydrogenase and mitochondrial complex I suppresses glioblastoma tumorspheres. Neuro Oncol. 2017, 20, 954–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, P.B.; Stuart, H.R. Mammalian TOR: A Homeostatic ATP Sensor. Science 2001, 294, 1102–1105. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Otten, E.G. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-S.; Lee, H.; Jang, H.; Woo, S.M.; Park, J.B.; Lee, S.-H.; Kang, J.H.; Kim, H.Y.; Song, J.; Kim, S.-Y. Targeting Oxidative Phosphorylation Reverses Drug Resistance in Cancer Cells by Blocking Autophagy Recycling. Cells 2020, 9, 2013. https://doi.org/10.3390/cells9092013
Lee J-S, Lee H, Jang H, Woo SM, Park JB, Lee S-H, Kang JH, Kim HY, Song J, Kim S-Y. Targeting Oxidative Phosphorylation Reverses Drug Resistance in Cancer Cells by Blocking Autophagy Recycling. Cells. 2020; 9(9):2013. https://doi.org/10.3390/cells9092013
Chicago/Turabian StyleLee, Jae-Seon, Ho Lee, Hyonchol Jang, Sang Myung Woo, Jong Bae Park, Seon-Hyeong Lee, Joon Hee Kang, Hee Yeon Kim, Jaewhan Song, and Soo-Youl Kim. 2020. "Targeting Oxidative Phosphorylation Reverses Drug Resistance in Cancer Cells by Blocking Autophagy Recycling" Cells 9, no. 9: 2013. https://doi.org/10.3390/cells9092013
APA StyleLee, J.-S., Lee, H., Jang, H., Woo, S. M., Park, J. B., Lee, S.-H., Kang, J. H., Kim, H. Y., Song, J., & Kim, S.-Y. (2020). Targeting Oxidative Phosphorylation Reverses Drug Resistance in Cancer Cells by Blocking Autophagy Recycling. Cells, 9(9), 2013. https://doi.org/10.3390/cells9092013