The Impact of Natural Dietary Compounds and Food-Borne Mycotoxins on DNA Methylation and Cancer


Abstract
1. Introduction
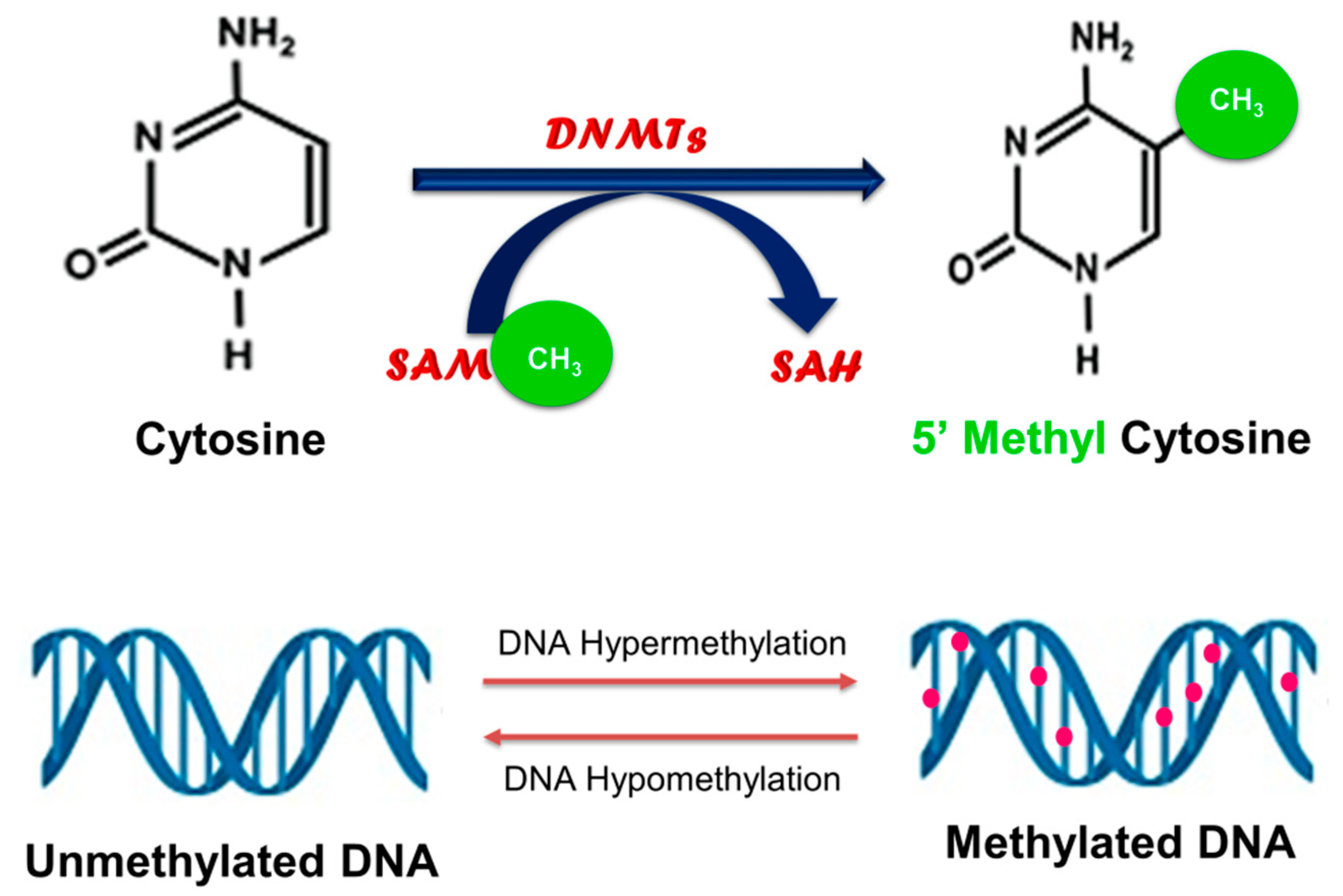
2. Cancer and DNA Methylation
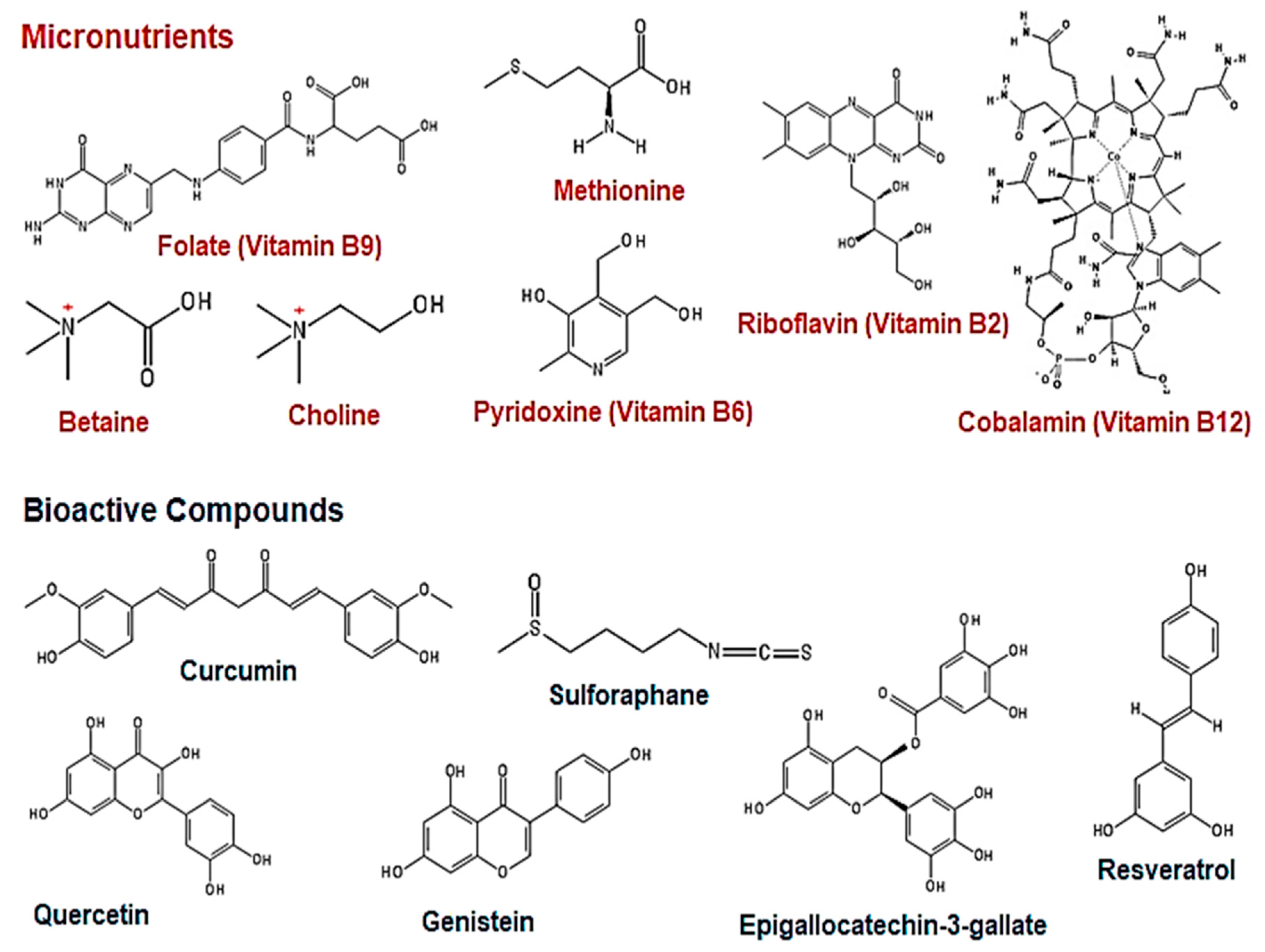
3. The Effect of Natural Dietary Micronutrients and Bioactive Compounds on DNA Methylation in Cancer
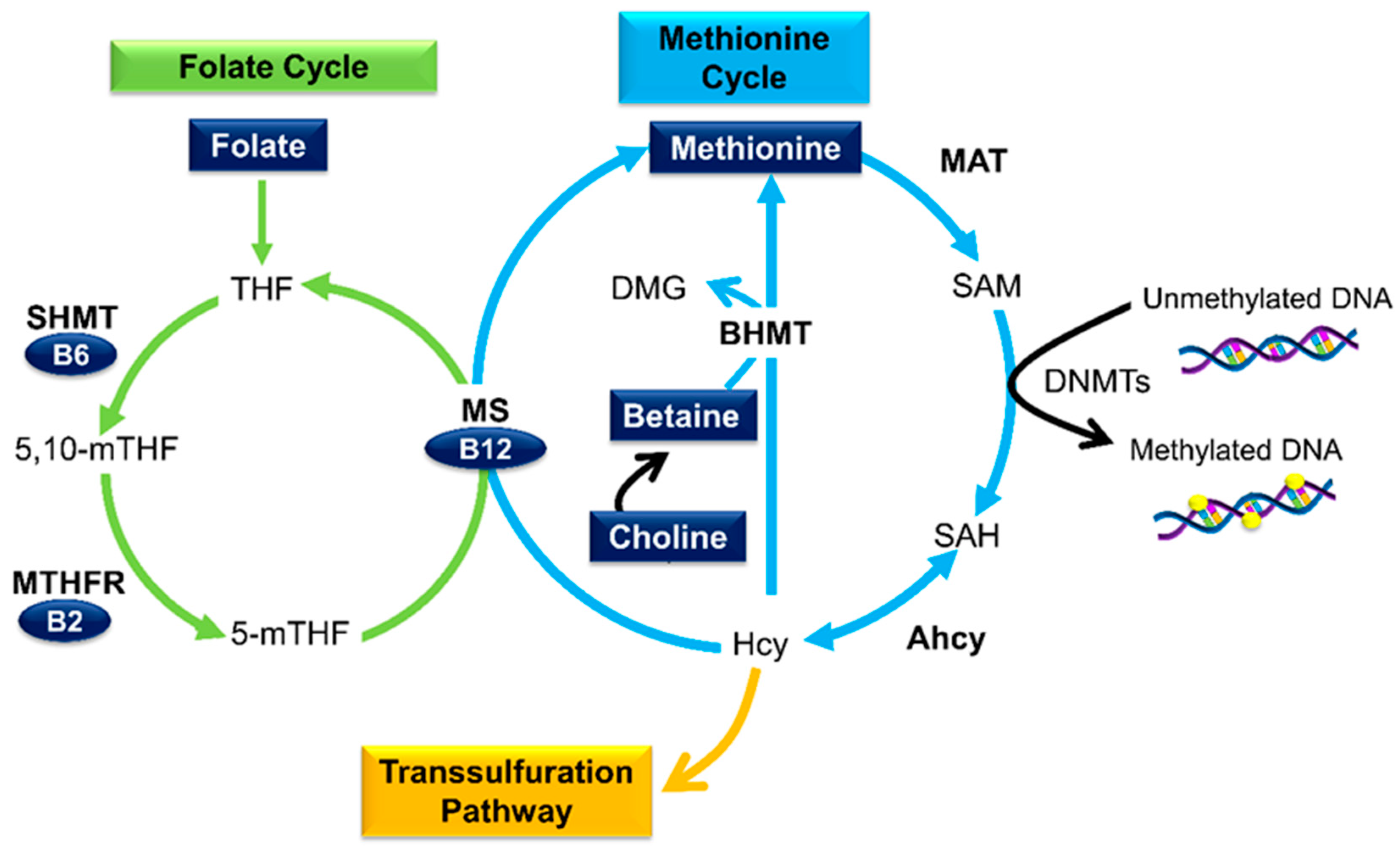
3.1. Micronutrients as Methyl Donors
3.2. Folate
3.3. Other B Vitamins
3.4. Betaine and Choline
3.5. Methionine
3.6. Curcumin
3.7. Quercetin
3.8. Resveratrol
3.9. Sulforaphane
3.10. Genistein
3.11. Epigallocatechin-3-Gallate
3.12. Combinational Effects of Dietary Compounds on DNA Methylation in Cancer
3.13. Clinical Trials with Bioactive Dietary Compounds and DNA Methylation in Cancer
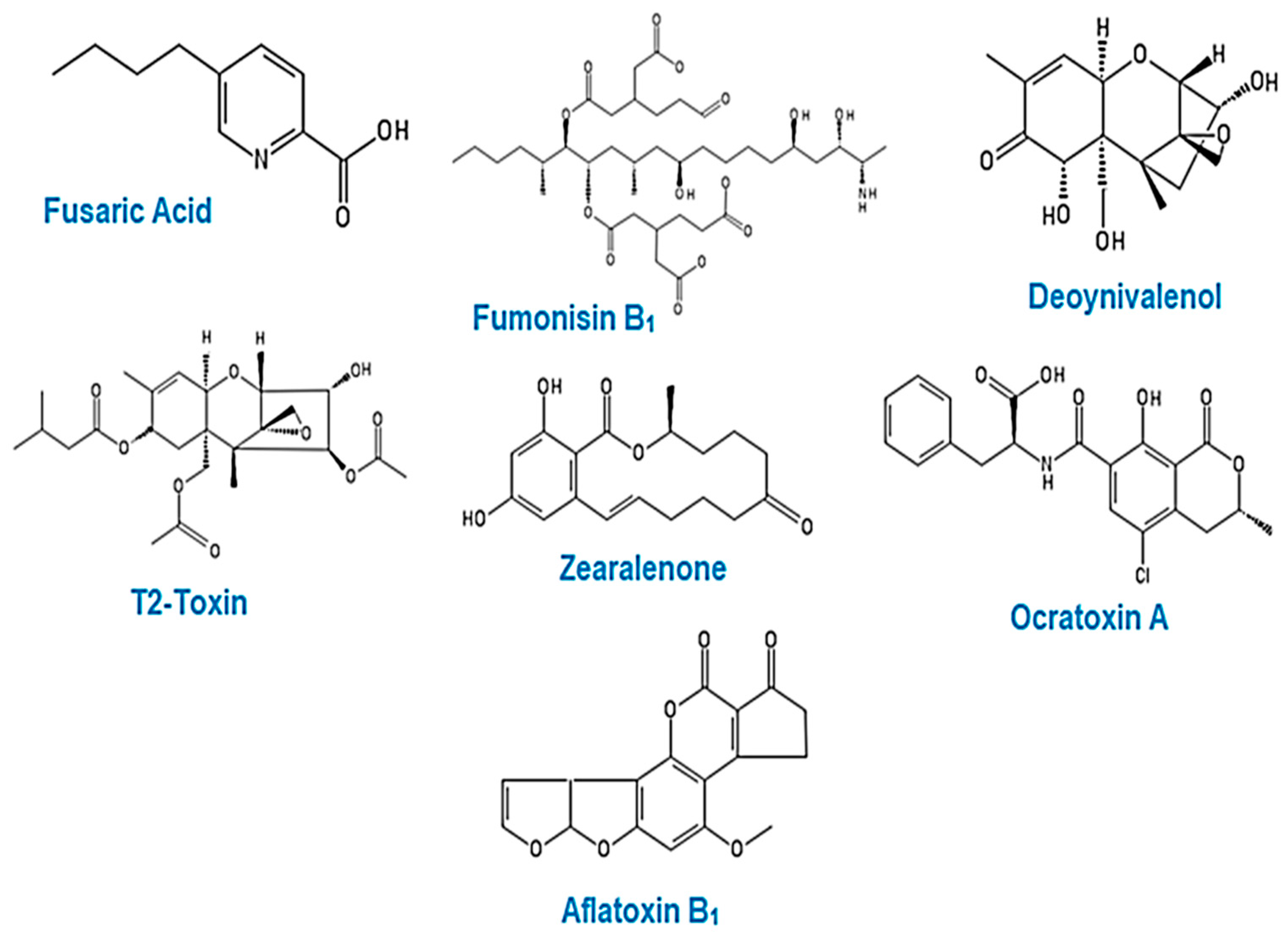
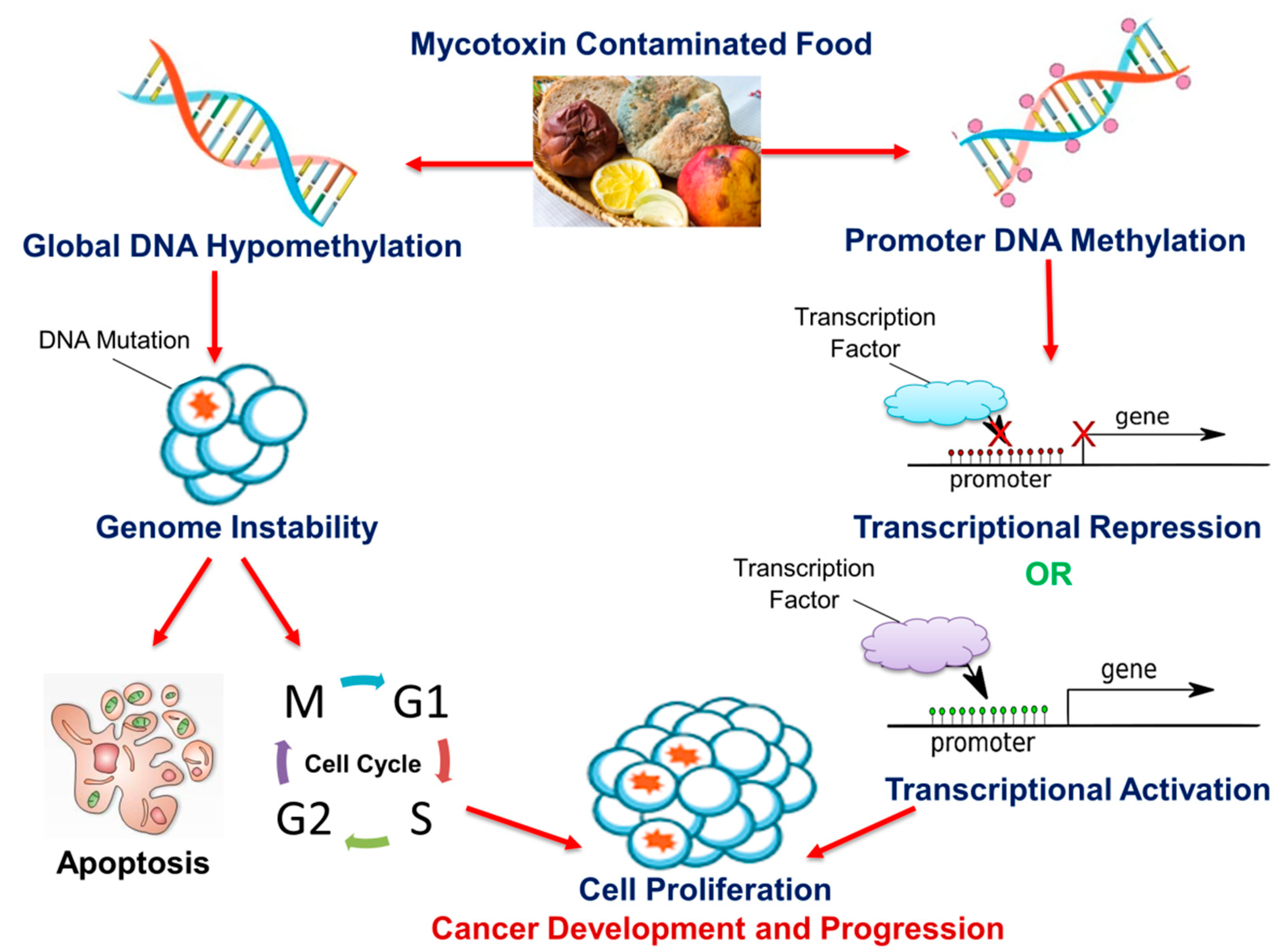
4. Mycotoxins
4.1. Fusaric Acid
4.2. Fumonisin B1
4.3. Ochratoxin A
4.4. Aflatoxin B1
4.5. Zearalenone
4.6. Deoxynivalenol
4.7. T-2 Toxin
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. WHO Report on Cancer: Setting Priorities, Investing Wisely and Providing Care for all. Available online: https://apps.who.int/iris/handle/10665/330745 (accessed on 27 August 2020).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Adlercreutz, H.; Mousavi, Y.; Höckerstedt, K. Diet and breast cancer. Acta Oncol. 1992, 31, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.; Potter, J.; Ma, K.-N.; Caan, B.; Leppert, M.; Samowitz, W. Western diet, family history of colorectal cancer, NAT2, GSTM-1 and risk of colon cancer. Cancer Causes Control 2000, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Stoll, B. Breast cancer and the western diet: Role of fatty acids and antioxidant vitamins. Eur. J. Cancer 1998, 34, 1852–1856. [Google Scholar] [CrossRef]
- Stoll, B.A. Western diet, early puberty, and breast cancer risk. Breast Cancer Res. Treat. 1998, 49, 187–193. [Google Scholar] [CrossRef]
- Peers, F.; Linsell, C. Dietary aflatoxins and liver cancer—A population based study in Kenya. Br. J. Cancer 1973, 27, 473. [Google Scholar] [CrossRef]
- Yoshizawa, T.; Yamashita, A.; Luo, Y. Fumonisin occurrence in corn from high-and low-risk areas for human esophageal cancer in China. Appl. Environ. Microbiol. 1994, 60, 1626–1629. [Google Scholar] [CrossRef]
- Gelderblom, W.; Abel, S.; Smuts, C.M.; Marnewick, J.; Marasas, W.; Lemmer, E.R.; Ramljak, D. Fumonisin-induced hepatocarcinogenesis: Mechanisms related to cancer initiation and promotion. Environ. Health Perspect. 2001, 109, 291–300. [Google Scholar]
- Castegnaro, M.; Mohr, U.; Pfohl-Leszkowicz, A.; Estève, J.; Steinmann, J.; Tillmann, T.; Michelon, J.; Bartsch, H. Sex-and strain-specific induction of renal tumors by ochratoxin A in rats correlates with DNA adduction. Int. J. Cancer 1998, 77, 70–75. [Google Scholar] [CrossRef]
- Belhassen, H.; Jiménez-Díaz, I.; Arrebola, J.; Ghali, R.; Ghorbel, H.; Olea, N.; Hedili, A. Zearalenone and its metabolites in urine and breast cancer risk: A case-control study in Tunisia. Chemosphere 2015, 128, 1–6. [Google Scholar] [CrossRef]
- Khan, N.; Afaq, F.; Mukhtar, H. Cancer chemoprevention through dietary antioxidants: Progress and promise. Antioxid. Redox Signal. 2008, 10, 475–510. [Google Scholar] [CrossRef] [PubMed]
- Ali Khan, M.; Kedhari Sundaram, M.; Hamza, A.; Quraishi, U.; Gunasekera, D.; Ramesh, L.; Goala, P.; Al Alami, U.; Ansari, M.Z.; Rizvi, T.A. Sulforaphane reverses the expression of various tumor suppressor genes by targeting DNMT3B and HDAC1 in human cervical cancer cells. Evid.-Based Complement. Altern. Med. 2015, 2015, 412149. [Google Scholar] [CrossRef] [PubMed]
- Al-Yousef, N.; Shinwari, Z.; Al-Shahrani, B.; Al-Showimi, M.; Al-Moghrabi, N. Curcumin induces re-expression of BRCA1 and suppression of γ synuclein by modulating DNA promoter methylation in breast cancer cell lines. Oncol. Rep. 2020, 43, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of hypermethylation and reactivation of p16INK4a, RARβ, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res. 2005, 11, 7033–7041. [Google Scholar] [CrossRef]
- Fudhaili, A.; Yoon, N.; Kang, S.; Ryu, J.; Jeong, J.Y.; Lee, D.H.; Kang, S.S. Resveratrol epigenetically regulates the expression of zinc finger protein 36 in non-small cell lung cancer cell lines. Oncol. Rep. 2019, 41, 1377–1386. [Google Scholar] [CrossRef]
- Gamet-Payrastre, L.; Li, P.; Lumeau, S.; Cassar, G.; Dupont, M.-A.; Chevolleau, S.; Gasc, N.; Tulliez, J.; Tercé, F. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res. 2000, 60, 1426–1433. [Google Scholar]
- Ji, Z.; Huo, C.; Yang, P. Genistein inhibited the proliferation of kidney cancer cells via CDKN2a hypomethylation: Role of abnormal apoptosis. Int. Urol. Nephrol. 2020, 1–7. [Google Scholar] [CrossRef]
- Hassanpour, S.H.; Dehghani, M. Review of cancer from perspective of molecular. J. Cancer Res. Pract. 2017, 4, 127–129. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef]
- Detich, N.; Theberge, J.; Szyf, M. Promoter-specific activation and demethylation by MBD2/demethylase. J. Biol. Chem. 2002, 277, 35791–35794. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Esteller, M.; Rountree, M.R.; Bachman, K.E.; Schuebel, K.; Herman, J.G. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet. 2001, 10, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Jones, P.A. DNA methylation and breast carcinogenesis. Oncogene 2002, 21, 5462–5482. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, B.; Huang, J.; Bhattacharyya, B.; Suderman, M.; Hallett, M.; Han, Z.-G.; Szyf, M. Definition of the landscape of promoter DNA hypomethylation in liver cancer. Cancer Res. 2011, 71, 5891–5903. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.I.; Giuliano, A.; Hatch, K.D.; Schneider, A.; Nour, M.A.; Dallal, G.E.; Selhub, J.; Mason, J.B. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer 1994, 74, 893–899. [Google Scholar] [CrossRef]
- Choi, J.-Y.; James, S.R.; Link, P.A.; McCann, S.E.; Hong, C.-C.; Davis, W.; Nesline, M.K.; Ambrosone, C.B.; Karpf, A.R. Association between global DNA hypomethylation in leukocytes and risk of breast cancer. Carcinogenesis 2009, 30, 1889–1897. [Google Scholar] [CrossRef]
- Dodge, J.E.; Okano, M.; Dick, F.; Tsujimoto, N.; Chen, T.; Wang, S.; Ueda, Y.; Dyson, N.; Li, E. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 2005, 280, 17986–17991. [Google Scholar] [CrossRef]
- Esteller, M.; Sanchez-Cespedes, M.; Rosell, R.; Sidransky, D.; Baylin, S.B.; Herman, J.G. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999, 59, 67–70. [Google Scholar]
- Sanchez-Cespedes, M.; Esteller, M.; Wu, L.; Nawroz-Danish, H.; Yoo, G.H.; Koch, W.M.; Jen, J.; Herman, J.G.; Sidransky, D. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res. 2000, 60, 892–895. [Google Scholar]
- Liu, Z.; Xie, Z.; Jones, W.; Pavlovicz, R.E.; Liu, S.; Yu, J.; Li, P.-k.; Lin, J.; Fuchs, J.R.; Marcucci, G. Curcumin is a potent DNA hypomethylation agent. Bioorganic Med. Chem. Lett. 2009, 19, 706–709. [Google Scholar] [CrossRef]
- Kedhari Sundaram, M.; Hussain, A.; Haque, S.; Raina, R.; Afroze, N. Quercetin modifies 5′ CpG promoter methylation and reactivates various tumor suppressor genes by modulating epigenetic marks in human cervical cancer cells. J. Cell. Biochem. 2019, 120, 18357–18369. [Google Scholar] [CrossRef] [PubMed]
- Lubecka-Pietruszewska, K.; Kaufman-Szymczyk, A.; Stefanska, B.; Fabianowska-Majewska, K. Folic acid enforces DNA methylation-mediated transcriptional silencing of PTEN, APC and RARbeta2 tumour suppressor genes in breast cancer. Biochem. Biophys. Res. Commun. 2013, 430, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Farias, N.; Ho, N.; Butler, S.; Delaney, L.; Morrison, J.; Shahrzad, S.; Coomber, B.L. The effects of folic acid on global DNA methylation and colonosphere formation in colon cancer cell lines. J. Nutr. Biochem. 2015, 26, 818–826. [Google Scholar] [CrossRef]
- Obeid, R. The metabolic burden of methyl donor deficiency with focus on the betaine homocysteine methyltransferase pathway. Nutrients 2013, 5, 3481–3495. [Google Scholar] [CrossRef] [PubMed]
- Clare, C.E.; Brassington, A.H.; Kwong, W.Y.; Sinclair, K.D. One-Carbon Metabolism: Linking Nutritional Biochemistry to Epigenetic Programming of Long-Term Development. Annu. Rev. Anim. Biosci. 2019, 7, 263–287. [Google Scholar] [CrossRef]
- Anderson, O.S.; Sant, K.E.; Dolinoy, D.C. Nutrition and epigenetics: An interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr Biochem 2012, 23, 853–859. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Ali, M.M. Methyl Donor Micronutrients that Modify DNA Methylation and Cancer Outcome. Nutrients 2019, 11, 608. [Google Scholar] [CrossRef]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hine, R.J.; James, S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J. Biol. Chem. 2000, 275, 29318–29323. [Google Scholar] [CrossRef]
- Rowling, M.J.; McMullen, M.H.; Chipman, D.C.; Schalinske, K.L. Hepatic glycine N-methyltransferase is up-regulated by excess dietary methionine in rats. J. Nutr. 2002, 132, 2545–2550. [Google Scholar] [CrossRef]
- Zhang, N. Role of methionine on epigenetic modification of DNA methylation and gene expression in animals. Anim. Nutr. 2018, 4, 11–16. [Google Scholar] [CrossRef]
- Rowling, M.J.; Schalinske, K.L. Retinoid compounds activate and induce hepatic glycine N-methyltransferase in rats. J. Nutr. 2001, 131, 1914–1917. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA methylation: A review of molecular mechanisms and the evidence for folate’s role. Adv. Nutr 2012, 3, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Crider, K.S.; Bailey, L.B.; Berry, R.J. Folic acid food fortification-its history, effect, concerns, and future directions. Nutrients 2011, 3, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.A.; Bell, S.J.; Guan, Y.; Yu, Y.-H. Folic Acid supplementation and pregnancy: More than just neural tube defect prevention. Rev. Obs. Gynecol. 2011, 4, 52–59. [Google Scholar]
- Stempak, J.M.; Sohn, K.-J.; Chiang, E.-P.; Shane, B.; Kim, Y.-I. Cell and stage of transformation-specific effects of folate deficiency on methionine cycle intermediates and DNA methylation in an in vitro model. Carcinogenesis 2005, 26, 981–990. [Google Scholar] [CrossRef]
- Wasson, G.R.; McGlynn, A.P.; McNulty, H.; O’Reilly, S.L.; McKelvey-Martin, V.J.; McKerr, G.; Strain, J.; Scott, J.; Downes, C.S. Global DNA and p53 region-specific hypomethylation in human colonic cells is induced by folate depletion and reversed by folate supplementation. J. Nutr. 2006, 136, 2748–2753. [Google Scholar] [CrossRef]
- Wang, T.-P.; Hsu, S.-H.; Feng, H.-C.; Huang, R.-F.S. Folate deprivation enhances invasiveness of human colon cancer cells mediated by activation of sonic hedgehog signaling through promoter hypomethylation and cross action with transcription nuclear factor-kappa B pathway. Carcinogenesis 2012, 33, 1158–1168. [Google Scholar] [CrossRef]
- Feng, H.C.; Lin, J.Y.; Hsu, S.H.; Lan, W.Y.; Kuo, C.S.; Tian, Y.F.; Sun, D.P.; Huang, R.F.S. Low folate metabolic stress reprograms DNA methylation-activated sonic hedgehog signaling to mediate cancer stem cell-like signatures and invasive tumour stage-specific malignancy of human colorectal cancers. Int. J. Cancer 2017, 141, 2537–2550. [Google Scholar] [CrossRef]
- Berner, C.; Aumüller, E.; Gnauck, A.; Nestelberger, M.; Just, A.; Haslberger, A.G. Epigenetic control of estrogen receptor expression and tumor suppressor genes is modulated by bioactive food compounds. Ann. Nutr. Metab. 2010, 57, 183–189. [Google Scholar] [CrossRef]
- Jhaveri, M.S.; Wagner, C.; Trepel, J.B. Impact of extracellular folate levels on global gene expression. Mol. Pharmacol. 2001, 60, 1288–1295. [Google Scholar] [CrossRef]
- Sie, K.K.; Medline, A.; van Weel, J.; Sohn, K.-J.; Choi, S.-W.; Croxford, R.; Kim, Y.-I. Effect of maternal and postweaning folic acid supplementation on colorectal cancer risk in the offspring. Gut 2011, 60, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Ly, A.; Lee, H.; Chen, J.; Sie, K.K.; Renlund, R.; Medline, A.; Sohn, K.-J.; Croxford, R.; Thompson, L.U.; Kim, Y.-I. Effect of maternal and postweaning folic acid supplementation on mammary tumor risk in the offspring. Cancer Res. 2011, 71, 988–997. [Google Scholar] [CrossRef]
- Kim, Y.I.; Pogribny, I.P.; Basnakian, A.G.; Miller, J.W.; Selhub, J.; James, S.J.; Mason, J.B. Folate deficiency in rats induces DNA strand breaks and hypomethylation within the p53 tumor suppressor gene. Am. J. Clin. Nutr. 1997, 65, 46–52. [Google Scholar] [CrossRef]
- Mckay, J.A.; Williams, E.A.; Mathers, J.C. Effect of maternal and post-weaning folate supply on gene-specific DNA methylation in the small intestine of weaning and adult apc+/min and wild type mice. Front. Genet. 2011, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.; Grau, M.V.; Levine, A.J.; Shen, L.; Hamdan, R.; Chen, X.; Gui, J.; Haile, R.W.; Barry, E.L.; Ahnen, D.; et al. Association between folate levels and CpG Island hypermethylation in normal colorectal mucosa. Cancer Prev. Res. 2010, 3, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Cole, B.F.; Baron, J.A.; Sandler, R.S.; Haile, R.W.; Ahnen, D.J.; Bresalier, R.S.; McKeown-Eyssen, G.; Summers, R.W.; Rothstein, R.I.; Burke, C.A. Folic acid for the prevention of colorectal adenomas: A randomized clinical trial. JAMA 2007, 297, 2351–2359. [Google Scholar] [CrossRef]
- Oliai Araghi, S.; Kiefte-de Jong, J.C.; van Dijk, S.C.; Swart, K.M.A.; van Laarhoven, H.W.; van Schoor, N.M.; de Groot, L.; Lemmens, V.; Stricker, B.H.; Uitterlinden, A.G.; et al. Folic Acid and Vitamin B12 Supplementation and the Risk of Cancer: Long-term Follow-up of the B Vitamins for the Prevention of Osteoporotic Fractures (B-PROOF) Trial. Cancer Epidemiol. Biomark. Prev. 2019, 28, 275–282. [Google Scholar] [CrossRef]
- Ebbing, M.; Bønaa, K.H.; Nygård, O.; Arnesen, E.; Ueland, P.M.; Nordrehaug, J.E.; Rasmussen, K.; Njølstad, I.; Refsum, H.; Nilsen, D.W.; et al. Cancer incidence and mortality after treatment with folic acid and vitamin B12. JAMA 2009, 302, 2119–2126. [Google Scholar] [CrossRef]
- Cravo, M.; Fidalgo, P.; Pereira, A.; Gouveia-Oliveira, A.; Chaves, P.; Selhub, J.; Mason, J.; Mira, F.; Leitao, C. DNA methylation as an intermediate biomarker in colorectal cancer: Modulation by folic acid supplementation. Eur. J. Cancer Prev. 1994, 3, 473–480. [Google Scholar] [CrossRef]
- Kim, Y.-I.; Baik, H.W.; Fawaz, K.; Knox, T.; Lee, Y.M.; Norton, R.; Libby, E.; Mason, J.B. Effects of folate supplementation on two provisional molecular markers of colon cancer: A prospective, randomized trial. Am. J. Gastroenterol. 2001, 96, 184–195. [Google Scholar] [CrossRef]
- Pufulete, M.; Al-Ghnaniem, R.; Khushal, A.; Appleby, P.; Harris, N.; Gout, S.; Emery, P.; Sanders, T. Effect of folic acid supplementation on genomic DNA methylation in patients with colorectal adenoma. Gut 2005, 54, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.C.; Kelsey, K.T.; Zheng, S.; Houseman, E.A.; Marsit, C.J.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Kushi, L.H.; et al. Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 2010, 6, e1001043. [Google Scholar] [CrossRef]
- Bastid, J.; Dejou, C.; Docquier, A.; Bonnefoy, N. The Emerging Role of the IL-17B/IL-17RB Pathway in Cancer. Front. Immunol 2020, 11, 718. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; Chuang, S.C.; Vaissière, T.; Cuenin, C.; Ricceri, F.; Johansson, M.; Ueland, P.; Brennan, P.; Herceg, Z. DNA methylation changes associated with cancer risk factors and blood levels of vitamin metabolites in a prospective study. Epigenetics 2011, 6, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Ospina, C.A.; Nava-Mesa, M.O. B Vitamins in the nervous system: Current knowledge of the biochemical modes of action and synergies of thiamine, pyridoxine, and cobalamin. Cns Neurosci. Ther. 2020, 26, 5–13. [Google Scholar] [CrossRef]
- Otani, T.; Iwasaki, M.; Hanaoka, T.; Kobayashi, M.; Ishihara, J.; Natsukawa, S.; Shaura, K.; Koizumi, Y.; Kasuga, Y.; Yoshimura, K.; et al. Folate, vitamin B6, vitamin B12, and vitamin B2 intake, genetic polymorphisms of related enzymes, and risk of colorectal cancer in a hospital-based case-control study in Japan. Nutr. Cancer 2005, 53, 42–50. [Google Scholar] [CrossRef]
- Singh, S.M.; Murphy, B.; O’Reilly, R.L. Involvement of gene-diet/drug interaction in DNA methylation and its contribution to complex diseases: From cancer to schizophrenia. Clin. Genet. 2003, 64, 451–460. [Google Scholar] [CrossRef]
- Maruti, S.S.; Ulrich, C.M.; White, E. Folate and one-carbon metabolism nutrients from supplements and diet in relation to breast cancer risk. Am. J. Clin. Nutr. 2009, 89, 624–633. [Google Scholar] [CrossRef]
- Wei, E.K.; Giovannucci, E.; Selhub, J.; Fuchs, C.S.; Hankinson, S.E.; Ma, J. Plasma vitamin B6 and the risk of colorectal cancer and adenoma in women. J. Natl. Cancer Inst. 2005, 97, 684–692. [Google Scholar] [CrossRef]
- Qiang, Y.; Li, Q.; Xin, Y.; Fang, X.; Tian, Y.; Ma, J.; Wang, J.; Wang, Q.; Zhang, R.; Wang, J.; et al. Intake of Dietary One-Carbon Metabolism-Related B Vitamins and the Risk of Esophageal Cancer: A Dose-Response Meta-Analysis. Nutrients 2018, 10, 835. [Google Scholar] [CrossRef]
- Amaral, C.L.D.; Bueno, R.d.B.e.L.; Burim, R.V.; Queiroz, R.H.C.; Bianchi, M.d.L.P.; Antunes, L.M.G. The effects of dietary supplementation of methionine on genomic stability and p53 gene promoter methylation in rats. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2011, 722, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Piyathilake, C.; Johanning, G.; Macaluso, M.; Whiteside, M.; Oelschlager, D.; Heimburger, D.; Grizzle, W. Localized Folate and Vitamin B-12 Deficiency in Squamous Cell Lung Cancer Is Associated With Global DNA Hypomethylation. Nutr. Cancer 2000, 37, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Johanning, G.L.; Heimburger, D.C.; Piyathilake, C.J. DNA methylation and diet in cancer. J. Nutr. 2002, 132, 3814S–3818S. [Google Scholar] [CrossRef]
- Piyathilake, C.J.; Macaluso, M.; Chambers, M.M.; Badiga, S.; Siddiqui, N.R.; Bell, W.C.; Edberg, J.C.; Partridge, E.E.; Alvarez, R.D.; Johanning, G.L. Folate and vitamin B12 may play a critical role in lowering the HPV 16 methylation-associated risk of developing higher grades of CIN. Cancer Prev. Res. 2014, 7, 1128–1137. [Google Scholar] [CrossRef]
- Gylling, B.; Van Guelpen, B.; Schneede, J.; Hultdin, J.; Ueland, P.M.; Hallmans, G.; Johansson, I.; Palmqvist, R. Low folate levels are associated with reduced risk of colorectal cancer in a population with low folate status. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2136–2144. [Google Scholar] [CrossRef]
- Newberne, P.M.; Rogers, A.E. Labile methyl groups and the promotion of cancer. Annu. Rev. Nutr. 1986, 6, 407–432. [Google Scholar] [CrossRef]
- Kanduc, D.; Ghoshal, A.; Quagliariello, E.; Farber, E. DNA hypomethylation in ethionine-induced rat preneoplastic hepatocyte nodules. Biochem. Biophys. Res. Commun. 1988, 150, 739–744. [Google Scholar] [CrossRef]
- Asada, K.; Kotake, Y.; Asada, R.; Saunders, D.; Broyles, R.H.; Towner, R.A.; Fukui, H.; Floyd, R.A. LINE-1 hypomethylation in a choline-deficiency-induced liver cancer in rats: Dependence on feeding period. J. Biomed. Biotechnol. 2006, 2006, 17142. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Simile, M.M.; Ladu, S.; Pellegrino, R.; De Murtas, V.; Pinna, F.; Tomasi, M.L.; Frau, M.; Virdis, P.; De Miglio, M.R.; et al. Altered methionine metabolism and global DNA methylation in liver cancer: Relationship with genomic instability and prognosis. Int. J. Cancer 2007, 121, 2410–2420. [Google Scholar] [CrossRef]
- Hultdin, J.; Van Guelpen, B.; Bergh, A.; Hallmans, G.; Stattin, P. Plasma folate, vitamin B12, and homocysteine and prostate cancer risk: A prospective study. Int. J. Cancer 2005, 113, 819–824. [Google Scholar] [CrossRef]
- Tao, M.-H.; Mason, J.B.; Marian, C.; McCann, S.E.; Platek, M.E.; Millen, A.; Ambrosone, C.; Edge, S.B.; Krishnan, S.S.; Trevisan, M.; et al. Promoter methylation of E-cadherin, p16, and RAR-β(2) genes in breast tumors and dietary intake of nutrients important in one-carbon metabolism. Nutr. Cancer 2011, 63, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B(6), Folate, Vitamin B(12), Pantothenic Acid, Biotin, and Choline; National Academy of Sciences: Washington, DC, USA, 1998. [Google Scholar]
- Ueland, P.M. Choline and betaine in health and disease. J. Inherit. Metab. Dis. 2011, 34, 3–15. [Google Scholar] [CrossRef]
- Vennemann, F.B.C.; Ioannidou, S.; Valsta, L.M.; Dumas, C.; Ocké, M.C.; Mensink, G.B.M.; Lindtner, O.; Virtanen, S.M.; Tlustos, C.; D’Addezio, L.; et al. Dietary intake and food sources of choline in European populations. Br. J. Nutr. 2015, 114, 2046–2055. [Google Scholar] [CrossRef]
- Hollenbeck, C.B. An introduction to the nutrition and metabolism of choline. Cent. Nerv. Syst. Agents Med. Chem. 2012, 12, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Craig, S.A. Betaine in human nutrition. Am. J. Clin. Nutr. 2004, 80, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, M.D.; Craciunescu, C.N.; Zeisel, S.H. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. FASEB J. 2006, 20, 43–49. [Google Scholar] [CrossRef]
- Mehedint, M.G.; Niculescu, M.D.; Craciunescu, C.N.; Zeisel, S.H. Choline deficiency alters global histone methylation and epigenetic marking at the Re1 site of the calbindin 1 gene. FASEB J. 2010, 24, 184–195. [Google Scholar] [CrossRef]
- Mehedint, M.G.; Craciunescu, C.N.; Zeisel, S.H. Maternal dietary choline deficiency alters angiogenesis in fetal mouse hippocampus. Proc. Natl. Acad. Sci. USA 2010, 107, 12834–12839. [Google Scholar] [CrossRef]
- Kovacheva, V.P.; Mellott, T.J.; Davison, J.M.; Wagner, N.; Lopez-Coviella, I.; Schnitzler, A.C.; Blusztajn, J.K. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J. Biol. Chem. 2007, 282, 31777–31788. [Google Scholar] [CrossRef]
- Da Costa, K.A.; Cochary, E.F.; Blusztajn, J.K.; Garner, S.C.; Zeisel, S.H. Accumulation of 1,2-sn-diradylglycerol with increased membrane-associated protein kinase C may be the mechanism for spontaneous hepatocarcinogenesis in choline-deficient rats. J. Biol. Chem. 1993, 268, 2100–2105. [Google Scholar]
- Da Costa, K.A.; Garner, S.C.; Chang, J.; Zeisel, S.H. Effects of prolonged (1 year) choline deficiency and subsequent re-feeding of choline on 1,2-sn-diradylglycerol, fatty acids and protein kinase C in rat liver. Carcinogenesis 1995, 16, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Shivapurkar, N.; Poirier, L.A. Tissue levels of S-adenosylmethionine and S-adenosylhomocysteine in rats fed methyl-deficient, amino acid-defined diets for one to five weeks. Carcinogenesis 1983, 4, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Tsujiuchi, T.; Tsutsumi, M.; Sasaki, Y.; Takahama, M.; Konishi, Y. Hypomethylation of CpG sites and c-myc gene overexpression in hepatocellular carcinomas, but not hyperplastic nodules, induced by a choline-deficient L-amino acid-defined diet in rats. Jpn. J. Cancer Res. 1999, 90, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Tryndyak, V.P.; Han, T.; Muskhelishvili, L.; Fuscoe, J.C.; Ross, S.A.; Beland, F.A.; Pogribny, I.P. Coupling global methylation and gene expression profiles reveal key pathophysiological events in liver injury induced by a methyl-deficient diet. Mol. Nutr. Food Res. 2011, 55, 411–418. [Google Scholar] [CrossRef]
- Ravanel, S.; Gakière, B.; Job, D.; Douce, R. The specific features of methionine biosynthesis and metabolism in plants. Proc. Natl. Acad. Sci. USA 1998, 95, 7805–7812. [Google Scholar] [CrossRef]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Drabkin, H.J.; RajBhandary, U.L. Initiation of protein synthesis in mammalian cells with codons other than AUG and amino acids other than methionine. Mol. Cell Biol. 1998, 18, 5140–5147. [Google Scholar] [CrossRef]
- Lee, J.U.; Sul, H.J.; Son, J.W. Promoter Methylation of CDKN2A, RARβ, and RASSF1A in Non-Small Cell Lung Carcinoma: Quantitative Evaluation Using Pyrosequencing. Tuberc. Respir. Dis. 2012, 73, 11–21. [Google Scholar] [CrossRef]
- Vaissière, T.; Hung, R.J.; Zaridze, D.; Moukeria, A.; Cuenin, C.; Fasolo, V.; Ferro, G.; Paliwal, A.; Hainaut, P.; Brennan, P.; et al. Quantitative Analysis of DNA Methylation Profiles in Lung Cancer Identifies Aberrant DNA Methylation of Specific Genes and Its Association with Gender and Cancer Risk Factors. Cancer Res. 2009, 69, 243–252. [Google Scholar] [CrossRef]
- Fanidi, A.; Muller, D.C.; Yuan, J.M.; Stevens, V.L.; Weinstein, S.J.; Albanes, D.; Prentice, R.; Thomsen, C.A.; Pettinger, M.; Cai, Q.; et al. Circulating Folate, Vitamin B6, and Methionine in Relation to Lung Cancer Risk in the Lung Cancer Cohort Consortium (LC3). J. Natl. Cancer Inst. 2018, 110, 57–67. [Google Scholar] [CrossRef]
- Bassett, J.K.; Hodge, A.M.; English, D.R.; Baglietto, L.; Hopper, J.L.; Giles, G.G.; Severi, G. Dietary intake of B vitamins and methionine and risk of lung cancer. Eur. J. Clin. Nutr. 2012, 66, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Feigelson, H.; Jonas, C.; Richardson, A.; McCullough, M.; Thun, M.; Calle, E. Alcohol, folate, methionine, and risk of incident breast cancer in the American Cancer Society Cancer Prevention Study II Nutrition Cohort. Cancer Epidemiol. Biomark. Prev. 2003, 12, 161–164. [Google Scholar]
- Wu, W.; Kang, S.; Zhang, D. Association of vitamin B6, vitamin B12 and methionine with risk of breast cancer: A dose-response meta-analysis. Br. J. Cancer 2013, 109, 1926–1944. [Google Scholar] [CrossRef] [PubMed]
- Nitter, M.; Norgård, B.; de Vogel, S.; Eussen, S.J.P.M.; Meyer, K.; Ulvik, A.; Ueland, P.M.; Nygård, O.; Vollset, S.E.; Bjørge, T.; et al. Plasma methionine, choline, betaine, and dimethylglycine in relation to colorectal cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC). Ann. Oncol. 2014, 25, 1609–1615. [Google Scholar] [CrossRef]
- Kune, G.; Watson, L. Colorectal Cancer Protective Effects and the Dietary Micronutrients Folate, Methionine, Vitamins B6, B12, C, E, Selenium, and Lycopene. Nutr. Cancer 2006, 56, 11–21. [Google Scholar] [CrossRef]
- Su, L.J.; Arab, L. Nutritional status of folate and colon cancer risk: Evidence from NHANES I epidemiologic follow-up study. Ann. Epidemiol. 2001, 11, 65–72. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Willett, W.C.; Colditz, G.A.; Hunter, D.J.; Stampfer, M.J.; Speizer, F.E.; Giovannucci, E.L. The Influence of Folate and Multivitamin Use on the Familial Risk of Colon Cancer in Women. Cancer Epidemiol. Biomark. Prev. 2002, 11, 227–234. [Google Scholar]
- Lee, J.E.; Willett, W.C.; Fuchs, C.S.; Smith-Warner, S.A.; Wu, K.; Ma, J.; Giovannucci, E. Folate intake and risk of colorectal cancer and adenoma: Modification by time. Am. J. Clin. Nutr. 2011, 93, 817–825. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Hoffman, R.M.; Bertino, J.R. Exploiting methionine restriction for cancer treatment. Biochem. Pharmacol. 2018, 154, 170–173. [Google Scholar] [CrossRef]
- Hoffman, R.M. Methionine dependence in cancer cells—A review. In Vitr. 1982, 18, 421–428. [Google Scholar] [CrossRef]
- Hoffman, R.M. Altered methionine metabolism, DNA methylation and oncogene expression in carcinogenesis. A review and synthesis. Biochim. Et Biophys. Acta 1984, 738, 49–87. [Google Scholar] [CrossRef]
- Birnbaum, S.M.; Greenstein, J.P.; Winitz, M. Quantitative nutritional studies with water-soluble, chemically defined diets. II. Nitrogen balance and metabolism. Arch. Biochem. Biophys. 1957, 72, 417–427. [Google Scholar] [CrossRef]
- Komninou, D.; Leutzinger, Y.; Reddy, B.S.; Richie, J.P., Jr. Methionine restriction inhibits colon carcinogenesis. Nutr. Cancer 2006, 54, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Cooper, T.K.; Rogers, C.J.; Sinha, I.; Turbitt, W.J.; Calcagnotto, A.; Perrone, C.E.; Richie, J.P., Jr. Dietary methionine restriction inhibits prostatic intraepithelial neoplasia in TRAMP mice. Prostate 2014, 74, 1663–1673. [Google Scholar] [CrossRef]
- Thivat, E.; Durando, X.; Demidem, A.; Farges, M.C.; Rapp, M.; Cellarier, E.; Guenin, S.; D’Incan, M.; Vasson, M.P.; Chollet, P. A methionine-free diet associated with nitrosourea treatment down-regulates methylguanine-DNA methyl transferase activity in patients with metastatic cancer. Anticancer Res. 2007, 27, 2779–2783. [Google Scholar]
- Link, A.; Balaguer, F.; Shen, Y.; Lozano, J.J.; Leung, H.-C.E.; Boland, C.R.; Goel, A. Curcumin modulates DNA methylation in colorectal cancer cells. PLoS ONE 2013, 8, e57709. [Google Scholar] [CrossRef]
- Lao, C.D.; Ruffin, M.T.; Normolle, D.; Heath, D.D.; Murray, S.I.; Bailey, J.M.; Boggs, M.E.; Crowell, J.; Rock, C.L.; Brenner, D.E. Dose escalation of a curcuminoid formulation. BMC Complementary Altern. Med. 2006, 6, 1–4. [Google Scholar] [CrossRef]
- Motterlini, R.; Foresti, R.; Bassi, R.; Green, C.J. Curcumin, an antioxidant and anti-inflammatory agent, induces heme oxygenase-1 and protects endothelial cells against oxidative stress. Free Radic. Biol. Med. 2000, 28, 1303–1312. [Google Scholar] [CrossRef]
- Bachmeier, B.E.; Mohrenz, I.V.; Mirisola, V.; Schleicher, E.; Romeo, F.; Höhneke, C.; Jochum, M.; Nerlich, A.G.; Pfeffer, U. Curcumin downregulates the inflammatory cytokines CXCL1 and-2 in breast cancer cells via NFκB. Carcinogenesis 2008, 29, 779–789. [Google Scholar] [CrossRef]
- Yu, J.; Peng, Y.; Wu, L.-C.; Xie, Z.; Deng, Y.; Hughes, T.; He, S.; Mo, X.; Chiu, M.; Wang, Q.-E. Curcumin down-regulates DNA methyltransferase 1 and plays an anti-leukemic role in acute myeloid leukemia. PLoS ONE 2013, 8, e55934. [Google Scholar] [CrossRef]
- Jha, A.; Nikbakht, M.; Parashar, G.; Shrivastava, A.; Capalash, N.; Kaur, J. Reversal of hypermethylation and reactivation of the RARbeta2 gene by natural compounds in cervical cancer cell lines. Folia Biol. 2010, 56, 195–200. [Google Scholar]
- Jiang, A.; Wang, X.; Shan, X.; Li, Y.; Wang, P.; Jiang, P.; Feng, Q. Curcumin reactivates silenced tumor suppressor gene RARβ by reducing DNA methylation. Phytother. Res. 2015, 29, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Jha, A.; Kumar, A.; Bhatnagar, A.; Narayan, G.; Kaur, I. Curcumin-mediated reversal of p15 gene promoter methylation: Implication in anti-neoplastic action against acute lymphoid leukaemia cell line. Folia Biol. 2015, 61, 81. [Google Scholar]
- Du, L.; Xie, Z.; Wu, L.-c.; Chiu, M.; Lin, J.; Chan, K.K.; Liu, S.; Liu, Z. Reactivation of RASSF1A in breast cancer cells by curcumin. Nutr. Cancer 2012, 64, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Kumar, U.; Sharma, U.; Rathi, G. Reversal of hypermethylation and reactivation of glutathione S-transferase pi 1 gene by curcumin in breast cancer cell line. Tumor Biol. 2017, 39, 1010428317692258. [Google Scholar] [CrossRef]
- Khor, T.O.; Huang, Y.; Wu, T.-Y.; Shu, L.; Lee, J.; Kong, A.-N.T. Pharmacodynamics of curcumin as DNA hypomethylation agent in restoring the expression of Nrf2 via promoter CpGs demethylation. Biochem. Pharmacol. 2011, 82, 1073–1078. [Google Scholar] [CrossRef]
- Shu, L.; Khor, T.O.; Lee, J.-H.; Boyanapalli, S.S.; Huang, Y.; Wu, T.-Y.; Saw, C.L.-L.; Cheung, K.-L.; Kong, A.-N.T. Epigenetic CpG demethylation of the promoter and reactivation of the expression of Neurog1 by curcumin in prostate LNCaP cells. AAPS J. 2011, 13, 606–614. [Google Scholar] [CrossRef]
- Lewinska, A.; Adamczyk, J.; Pajak, J.; Stoklosa, S.; Kubis, B.; Pastuszek, P.; Slota, E.; Wnuk, M. Curcumin-mediated decrease in the expression of nucleolar organizer regions in cervical cancer (HeLa) cells. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2014, 771, 43–52. [Google Scholar] [CrossRef]
- Zheng, J.; Wu, C.; Lin, Z.; Guo, Y.; Shi, L.; Dong, P.; Lu, Z.; Gao, S.; Liao, Y.; Chen, B. Curcumin up-regulates phosphatase and tensin homologue deleted on chromosome 10 through micro RNA-mediated control of DNA methylation–a novel mechanism suppressing liver fibrosis. FEBS J. 2014, 281, 88–103. [Google Scholar] [CrossRef]
- Wu, R.; Wang, L.; Yin, R.; Hudlikar, R.; Li, S.; Kuo, H.C.D.; Peter, R.; Sargsyan, D.; Guo, Y.; Liu, X. Epigenetics/epigenomics and prevention by curcumin of early stages of inflammatory-driven colon cancer. Mol. Carcinog. 2020. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; López-Vallejo, F.; Kuck, D.; Lyko, F. Natural products as DNA methyltransferase inhibitors: A computer-aided discovery approach. Mol. Divers. 2011, 15, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.E.; Carlson, S.; Abdallah, I.; Buttolph, T.; Glass, K.C.; Fandy, T.E. Curcumin and dimethoxycurcumin induced epigenetic changes in leukemia cells. Pharm. Res. 2015, 32, 863–875. [Google Scholar] [CrossRef]
- Bentz, A.B. A Review of Quercetin: Chemistry, Antioxident Properties, and Bioavailability. J. Young Investig. 2017. [Google Scholar]
- Richter, M.; Ebermann, R.; Marian, B. Quercetin-induced apoptosis in colorectal tumor cells: Possible role of EGF receptor signaling. Nutr. Cancer 1999, 34, 88–99. [Google Scholar] [CrossRef]
- Deschner, E.E.; Ruperto, J.; Wong, G.; Newmark, H.L. Quercetin and rutin as inhibitors of azoxymethanol-induced colonic neoplasia. Carcinogenesis 1991, 12, 1193–1196. [Google Scholar] [CrossRef]
- Mahmoud, N.N.; Carothers, A.M.; Grunberger, D.; Bilinski, R.T.; Churchill, M.R.; Martucci, C.; Newmark, H.L.; Bertagnolli, M.M. Plant phenolics decrease intestinal tumors in an animal model of familial adenomatous polyposis. Carcinogenesis 2000, 21, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Avila, M.A.; Velasco, J.A.; Cansado, J.; Notario, V. Quercetin mediates the down-regulation of mutant p53 in the human breast cancer cell line MDA-MB468. Cancer Res. 1994, 54, 2424–2428. [Google Scholar] [PubMed]
- Ferrandina, G.; Almadori, G.; Maggiano, N.; Lanza, P.; Ferlini, C.; Cattani, P.; Piantelli, M.; Scambia, G.; Ranelletti, F.O. Growth-inhibitory effect of tamoxifen and quercetin and presence of type II estrogen binding sites in human laryngeal cancer cell lines and primary laryngeal tumors. Int. J. Cancer 1998, 77, 747–754. [Google Scholar] [CrossRef]
- Ranelletti, F.O.; Ricci, R.; Larocca, L.M.; Maggiano, N.; Capelli, A.; Scambia, G.; Benedetti-Panici, P.; Mancuso, S.; Rumi, C.; Piantelli, M. Growth-inhibitory effect of quercetin and presence of type-II estrogen-binding sites in human colon-cancer cell lines and primary colorectal tumors. Int. J. Cancer 1992, 50, 486–492. [Google Scholar] [CrossRef]
- Yoshida, M.; Yamamoto, M.; Nikaido, T. Quercetin arrests human leukemic T-cells in late G1 phase of the cell cycle. Cancer Res. 1992, 52, 6676–6681. [Google Scholar]
- Ma, L.; Feugang, J.M.; Konarski, P.; Wang, J.; Lu, J.; Fu, S.; Ma, B.; Tian, B.; Zou, C.; Wang, Z. Growth inhibitory effects of quercetin on bladder cancer cell. Front. Biosci. 2006, 11, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.C.; Maso, V.; Torello, C.O.; Ferro, K.P.; Saad, S.T.O. The polyphenol quercetin induces cell death in leukemia by targeting epigenetic regulators of pro-apoptotic genes. Clin. Epigenetics 2018, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Wang, C.; Lu, C.; Zhao, B.; Cui, Y.; Shi, X.; Ma, X. Quercetin is able to demethylate the p16INK4a gene promoter. Chemotherapy 2009, 55, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Farhan, M.; Ullah, M.F.; Faisal, M.; Farooqi, A.A.; Sabitaliyevich, U.Y.; Biersack, B.; Ahmad, A. Differential methylation and acetylation as the epigenetic basis of resveratrol’s anticancer activity. Medicines 2019, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Medina-Aguilar, R.; Pérez-Plasencia, C.; Marchat, L.A.; Gariglio, P.; Mena, J.G.; Cuevas, S.R.; Ruíz-García, E.; Astudillo-De La Vega, H.; Juárez, J.H.; Flores-Perez, A. Methylation landscape of human breast cancer cells in response to dietary compound resveratrol. PLoS ONE 2016, 11, e0157866. [Google Scholar] [CrossRef]
- Qin, W.; Zhu, W.; Sauter, E. Resveratrol Induced DNA Methylation in ER+ Breast Cancer; AACR: Philadelphia, PA, USA, 2005. [Google Scholar]
- Qin, W.; Zhang, K.; Clarke, K.; Weiland, T.; Sauter, E.R. Methylation and miRNA effects of resveratrol on mammary tumors vs. normal tissue. Nutr. Cancer 2014, 66, 270–277. [Google Scholar] [CrossRef]
- Kala, R.; Shah, H.N.; Martin, S.L.; Tollefsbol, T.O. Epigenetic-based combinatorial resveratrol and pterostilbene alters DNA damage response by affecting SIRT1 and DNMT enzyme expression, including SIRT1-dependent γ-H2AX and telomerase regulation in triple-negative breast cancer. BMC Cancer 2015, 15, 672. [Google Scholar] [CrossRef]
- Papoutsis, A.J.; Borg, J.L.; Selmin, O.I.; Romagnolo, D.F. BRCA-1 promoter hypermethylation and silencing induced by the aromatic hydrocarbon receptor-ligand TCDD are prevented by resveratrol in MCF-7 cells. J. Nutr. Biochem. 2012, 23, 1324–1332. [Google Scholar] [CrossRef]
- Kala, R.; Tollefsbol, T.O. A novel combinatorial epigenetic therapy using resveratrol and pterostilbene for restoring estrogen receptor-α (ERα) expression in ERα-negative breast cancer cells. PLoS ONE 2016, 11, e0155057. [Google Scholar] [CrossRef]
- Mirza, S.; Sharma, G.; Parshad, R.; Gupta, S.D.; Pandya, P.; Ralhan, R. Expression of DNA methyltransferases in breast cancer patients and to analyze the effect of natural compounds on DNA methyltransferases and associated proteins. J. Breast Cancer 2013, 16, 23–31. [Google Scholar] [CrossRef]
- Stefanska, B.; Salamé, P.; Bednarek, A.; Fabianowska-Majewska, K. Comparative effects of retinoic acid, vitamin D and resveratrol alone and in combination with adenosine analogues on methylation and expression of phosphatase and tensin homologue tumour suppressor gene in breast cancer cells. Br. J. Nutr. 2012, 107, 781–790. [Google Scholar] [CrossRef]
- Lubecka, K.; Kurzava, L.; Flower, K.; Buvala, H.; Zhang, H.; Teegarden, D.; Camarillo, I.; Suderman, M.; Kuang, S.; Andrisani, O. Stilbenoids remodel the DNA methylation patterns in breast cancer cells and inhibit oncogenic NOTCH signaling through epigenetic regulation of MAML2 transcriptional activity. Carcinogenesis 2016, 37, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Torres, E.; Hernández-Oliveras, A.; Meneses-Morales, I.; Rodríguez, G.; Fuentes-García, G.; Zarain-Herzberg, Á. Resveratrol up-regulates ATP2A3 gene expression in breast cancer cell lines through epigenetic mechanisms. Int. J. Biochem. Cell Biol. 2019, 113, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Lin, H.; Zhang, X.; Li, J. Resveratrol reverses temozolomide resistance by downregulation of MGMT in T98G glioblastoma cells by the NF-κB-dependent pathway. Oncol. Rep. 2012, 27, 2050–2056. [Google Scholar]
- Yang, H.C.; Wang, J.Y.; Bu, X.Y.; Yang, B.; Wang, B.Q.; Hu, S.; Yan, Z.Y.; Gao, Y.S.; Han, S.Y.; Qu, M.Q. Resveratrol restores sensitivity of glioma cells to temozolamide through inhibiting the activation of Wnt signaling pathway. J. Cell. Physiol. 2019, 234, 6783–6800. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhou, Y.; Tian, H.; Yang, G.; Li, C.; Geng, Y.; Wu, S.; Wu, W. Sulforaphane inhibits invasion by phosphorylating ERK1/2 to regulate E-cadherin and CD44v6 in human prostate cancer DU145 cells. Oncol. Rep. 2015, 34, 1565–1572. [Google Scholar] [CrossRef]
- Cheng, Y.-M.; Tsai, C.-C.; Hsu, Y.-C. Sulforaphane, a dietary isothiocyanate, induces G2/M arrest in cervical cancer cells through cyclinB1 downregulation and GADD45β/CDC2 association. Int. J. Mol. Sci. 2016, 17, 1530. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-X.; Zou, Y.-J.; Zhuang, X.-B.; Chen, S.-X.; Lin, Y.; Li, W.-L.; Lin, J.-J.; Lin, Z.-Q. Sulforaphane suppresses EMT and metastasis in human lung cancer through miR-616-5p-mediated GSK3β/β-catenin signaling pathways. Acta Pharmacol. Sin. 2017, 38, 241–251. [Google Scholar] [CrossRef]
- Meeran, S.M.; Patel, S.N.; Tollefsbol, T.O. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS ONE 2010, 5, e11457. [Google Scholar] [CrossRef]
- Hsu, A.; Wong, C.P.; Yu, Z.; Williams, D.E.; Dashwood, R.H.; Ho, E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin. Epigenetics 2011, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Lewinska, A.; Adamczyk-Grochala, J.; Deregowska, A.; Wnuk, M. Sulforaphane-induced cell cycle arrest and senescence are accompanied by DNA hypomethylation and changes in microRNA profile in breast cancer cells. Theranostics 2017, 7, 3461. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.P.; Hsu, A.; Buchanan, A.; Palomera-Sanchez, Z.; Beaver, L.M.; Houseman, E.A.; Williams, D.E.; Dashwood, R.H.; Ho, E. Effects of sulforaphane and 3, 3′-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PLoS ONE 2014, 9, e86787. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Su, Z.-Y.; Khor, T.O.; Shu, L.; Kong, A.-N.T. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem. Pharmacol. 2013, 85, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.W.; Wang, M.; Sun, N.X.; Qing, Y.; Yin, T.F.; Li, C.; Wu, D. Sulforaphane-induced epigenetic regulation of Nrf2 expression by DNA methyltransferase in human Caco-2 cells. Oncol. Lett. 2019, 18, 2639–2647. [Google Scholar] [CrossRef] [PubMed]
- Lubecka-Pietruszewska, K.; Kaufman-Szymczyk, A.; Stefanska, B.; Cebula-Obrzut, B.; Smolewski, P.; Fabianowska-Majewska, K. Sulforaphane alone and in combination with clofarabine epigenetically regulates the expression of DNA methylation-silenced tumour suppressor genes in human breast cancer cells. Lifestyle Genom. 2015, 8, 91–101. [Google Scholar] [CrossRef]
- Wu, Q.; Odwin-Dacosta, S.; Cao, S.; Yager, J.D.; Tang, W.-y. Estrogen down regulates COMT transcription via promoter DNA methylation in human breast cancer cells. Toxicol. Appl. Pharmacol. 2019, 367, 12–22. [Google Scholar] [CrossRef]
- Abbas, A.; Hall, J.A.; Patterson III, W.L.; Ho, E.; Hsu, A.; Al-Mulla, F.; Georgel, P.T. Sulforaphane modulates telomerase activity via epigenetic regulation in prostate cancer cell lines. Biochem. Cell Biol. 2016, 94, 71–81. [Google Scholar] [CrossRef]
- Chen, L.; Chan, L.S.; Lung, H.L.; Yip, T.T.C.; Ngan, R.K.C.; Wong, J.W.C.; Lo, K.W.; Ng, W.T.; Lee, A.W.M.; Tsao, G.S.W. Crucifera sulforaphane (SFN) inhibits the growth of nasopharyngeal carcinoma through DNA methyltransferase 1 (DNMT1)/Wnt inhibitory factor 1 (WIF1) axis. Phytomedicine 2019, 63, 153058. [Google Scholar] [CrossRef]
- Dos Santos, P.W.d.S.; Machado, A.R.T.; De Grandis, R.A.; Ribeiro, D.L.; Tuttis, K.; Morselli, M.; Aissa, A.F.; Pellegrini, M.; Antunes, L.M.G. Transcriptome and DNA methylation changes modulated by sulforaphane induce cell cycle arrest, apoptosis, DNA damage, and suppression of proliferation in human liver cancer cells. Food Chem. Toxicol. 2020, 136, 111047. [Google Scholar] [CrossRef]
- Royston, K.J.; Paul, B.; Nozell, S.; Rajbhandari, R.; Tollefsbol, T.O. Withaferin A and sulforaphane regulate breast cancer cell cycle progression through epigenetic mechanisms. Exp. Cell Res. 2018, 368, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Chiang, T.-c.; Koss, B.; Su, L.J.; Washam, C.L.; Byrum, S.D.; Storey, A.; Tackett, A.J. Effect of sulforaphane and 5-aza-2’-deoxycytidine on melanoma cell growth. Medicines 2019, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, M.; Xu, L.; Ananthanarayanan, V.; Cooper, J.; Takimoto, C.H.; Helenowski, I.; Pelling, J.C.; Bergan, R.C. Dietary genistein inhibits metastasis of human prostate cancer in mice. Cancer Res. 2008, 68, 2024–2032. [Google Scholar] [CrossRef]
- Gu, Y.; Zhu, C.-F.; Iwamoto, H.; Chen, J.-S. Genistein inhibits invasive potential of human hepatocellular carcinoma by altering cell cycle, apoptosis, and angiogenesis. World J. Gastroenterol. 2005, 11, 6512. [Google Scholar] [CrossRef] [PubMed]
- Fotsis, T.; Pepper, M.; Adlercreutz, H.; Hase, T.; Montesano, R.; Schweigerer, L. Genistein, a dietary ingested isoflavonoid, inhibits cell proliferation and in vitro angiogenesis. J. Nutr. 1995, 125, 790S–797S. [Google Scholar]
- Xie, Q.; Bai, Q.; Zou, L.Y.; Zhang, Q.Y.; Zhou, Y.; Chang, H.; Yi, L.; Zhu, J.D.; Mi, M.T. Genistein inhibits DNA methylation and increases expression of tumor suppressor genes in human breast cancer cells. Genes Chromosomes Cancer 2014, 53, 422–431. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Q.; Chen, H. DNA methylation and histone modifications of Wnt genes by genistein during colon cancer development. Carcinogenesis 2013, 34, 1756–1763. [Google Scholar] [CrossRef]
- Romagnolo, D.F.; Donovan, M.G.; Papoutsis, A.J.; Doetschman, T.C.; Selmin, O.I. Genistein prevents BRCA1 CpG methylation and proliferation in human breast cancer cells with activated aromatic hydrocarbon receptor. Curr. Dev. Nutr. 2017, 1, e000562. [Google Scholar] [CrossRef]
- Donovan, M.G.; Selmin, O.I.; Doetschman, T.C.; Romagnolo, D.F. Epigenetic Activation of BRCA1 by Genistein In Vivo and Triple Negative Breast Cancer Cells Linked to Antagonism toward Aryl Hydrocarbon Receptor. Nutrients 2019, 11, 2559. [Google Scholar] [CrossRef]
- King-Batoon, A.; Leszczynska, J.M.; Klein, C.B. Modulation of gene methylation by genistein or lycopene in breast cancer cells. Environ. Mol. Mutagen. 2008, 49, 36–45. [Google Scholar] [CrossRef]
- Vardi, A.; Bosviel, R.; Rabiau, N.; Adjakly, M.; Satih, S.; Dechelotte, P.; Boiteux, J.-P.; Fontana, L.; Bignon, Y.-J.; Guy, L. Soy phytoestrogens modify DNA methylation of GSTP1, RASSF1A, EPH2 and BRCA1 promoter in prostate cancer cells. Vivo 2010, 24, 393–400. [Google Scholar]
- Majid, S.; Kikuno, N.; Nelles, J.; Noonan, E.; Tanaka, Y.; Kawamoto, K.; Hirata, H.; Li, L.C.; Zhao, H.; Okino, S.T. Genistein induces the p21WAF1/CIP1 and p16INK4a tumor suppressor genes in prostate cancer cells by epigenetic mechanisms involving active chromatin modification. Cancer Res. 2008, 68, 2736–2744. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.K.; Ansari, M.Z.; Al Mutery, A.; Ashraf, M.; Nasab, R.; Rai, S.; Rais, N.; Hussain, A. Genistein induces alterations of epigenetic modulatory signatures in human cervical cancer cells. Anti-Cancer Agents Med. Chem. 2018, 18, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.K.; Unni, S.; Somvanshi, P.; Bhardwaj, T.; Mandal, R.K.; Hussain, A.; Haque, S. Genistein Modulates Signaling Pathways and Targets Several Epigenetic Markers in HeLa Cells. Genes 2019, 10, 955. [Google Scholar] [CrossRef] [PubMed]
- Majid, S.; Dar, A.A.; Shahryari, V.; Hirata, H.; Ahmad, A.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Dahiya, R. Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B-Cell translocation gene 3 in prostate cancer. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2010, 116, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Majid, S.; Dar, A.A.; Ahmad, A.E.; Hirata, H.; Kawakami, K.; Shahryari, V.; Saini, S.; Tanaka, Y.; Dahiya, A.V.; Khatri, G. BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis 2009, 30, 662–670. [Google Scholar] [CrossRef]
- Nejad, M.A.; Nikbakht, M.; Afsa, M.; Malekzadeh, K. Restraining the Proliferation of Acute Lymphoblastic Leukemia Cells by Genistein through Up-regulation of B-cell Translocation Gene-3 at Transcription Level. Galen Med. J. 2019, 8, 1229. [Google Scholar]
- Zhu, J.; Ren, J.; Tang, L. Genistein inhibits invasion and migration of colon cancer cells by recovering WIF1 expression. Mol. Med. Rep. 2018, 17, 7265–7273. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, H. Genistein increases gene expression by demethylation of WNT5a promoter in colon cancer cell line SW1116. Anticancer Res. 2010, 30, 4537–4545. [Google Scholar]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (−)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p 16 INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar]
- Khan, M.A.; Hussain, A.; Sundaram, M.K.; Alalami, U.; Gunasekera, D.; Ramesh, L.; Hamza, A.; Quraishi, U. (-)-Epigallocatechin-3-gallate reverses the expression of various tumor-suppressor genes by inhibiting DNA methyltransferases and histone deacetylases in human cervical cancer cells. Oncol. Rep. 2015, 33, 1976–1984. [Google Scholar] [CrossRef]
- Gao, Z.; Xu, Z.; Hung, M.-S.; Lin, Y.-C.; Wang, T.; Gong, M.; Zhi, X.; Jablon, D.M.; You, L. Promoter demethylation of WIF-1 by epigallocatechin-3-gallate in lung cancer cells. Anticancer Res. 2009, 29, 2025–2030. [Google Scholar] [PubMed]
- Fang, M.; Chen, D.; Yang, C.S. Dietary polyphenols may affect DNA methylation. J. Nutr. 2007, 137, 223S–228S. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Perán, E.; Cabezas-Herrera, J.; del Campo, L.S.; Rodríguez-López, J.N. Effects of folate cycle disruption by the green tea polyphenol epigallocatechin-3-gallate. Int. J. Biochem. Cell Biol. 2007, 39, 2215–2225. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tollefsbol, T.O. Impact on DNA methylation in cancer prevention and therapy by bioactive dietary components. Curr. Med. Chem. 2010, 17, 2141–2151. [Google Scholar] [CrossRef]
- Mittal, A.; Piyathilake, C.; Hara, Y.; Katiyar, S.K. Exceptionally high protection of photocarcinogenesis by topical application of (—)-epigallocatechin-3-gallate in hydrophilic cream in SKH-1 hairless mouse model: Relationship to inhibition of UVB-induced global DNA hypomethylation. Neoplasia 2003, 5, 555. [Google Scholar] [CrossRef]
- Shi, S.T.; Wang, Z.-Y.; Smith, T.J.; Hong, J.-Y.; Chen, W.-F.; Ho, C.-T.; Yang, C.S. Effects of green tea and black tea on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone bioactivation, DNA methylation, and lung tumorigenesis in A/J mice. Cancer Res. 1994, 54, 4641–4647. [Google Scholar]
- Morris, J.; Moseley, V.R.; Cabang, A.B.; Coleman, K.; Wei, W.; Garrett-Mayer, E.; Wargovich, M.J. Reduction in promotor methylation utilizing EGCG (epigallocatechin-3-gallate) restores RXRα expression in human colon cancer cells. Oncotarget 2016, 7, 35313. [Google Scholar] [CrossRef]
- Meng, J.; Tong, Q.; Liu, X.; Yu, Z.; Zhang, J.; Gao, B. Epigallocatechin-3-gallate inhibits growth and induces apoptosis in esophageal cancer cells through the demethylation and reactivation of the p16 gene. Oncol. Lett. 2017, 14, 1152–1156. [Google Scholar] [CrossRef][Green Version]
- Sheng, J.; Shi, W.; Guo, H.; Long, W.; Wang, Y.; Qi, J.; Liu, J.; Xu, Y. The Inhibitory Effect of (−)-Epigallocatechin-3-Gallate on Breast Cancer Progression via Reducing SCUBE2 Methylation and DNMT Activity. Molecules 2019, 24, 2899. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Long, N.; Makita, H.; Toida, M.; Yamashita, T.; Hatakeyama, D.; Hara, A.; Mori, H.; Shibata, T. Effects of green tea polyphenol on methylation status of RECK gene and cancer cell invasion in oral squamous cell carcinoma cells. Br. J. Cancer 2008, 99, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Patel, S.N.; Chan, T.-H.; Tollefsbol, T.O. A novel prodrug of epigallocatechin-3-gallate: Differential epigenetic hTERT repression in human breast cancer cells. Cancer Prev. Res. 2011, 4, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Kumar, L.; Mohanty, S.K.; Maikhuri, J.P.; Rajender, S.; Gupta, G. Sensitization of androgen refractory prostate cancer cells to anti-androgens through re-expression of epigenetically repressed androgen receptor–synergistic action of quercetin and curcumin. Mol. Cell. Endocrinol. 2016, 431, 12–23. [Google Scholar] [CrossRef]
- Meeran, S.M.; Patel, S.N.; Li, Y.; Shukla, S.; Tollefsbol, T.O. Bioactive dietary supplements reactivate ER expression in ER-negative breast cancer cells by active chromatin modifications. PLoS ONE 2012, 7, e37748. [Google Scholar] [CrossRef]
- Bilir, B.; Sharma, N.V.; Lee, J.; Hammarstrom, B.; Svindland, A.; Kucuk, O.; Moreno, C.S. Effects of genistein supplementation on genome-wide DNA methylation and gene expression in patients with localized prostate cancer. Int. J. Oncol. 2017, 51, 223–234. [Google Scholar] [CrossRef]
- Qin, W.; Zhu, W.; Shi, H.; Hewett, J.E.; Ruhlen, R.L.; MacDonald, R.S.; Rottinghaus, G.E.; Chen, Y.-C.; Sauter, E.R. Soy isoflavones have an antiestrogenic effect and alter mammary promoter hypermethylation in healthy premenopausal women. Nutr. Cancer 2009, 61, 238–244. [Google Scholar] [CrossRef]
- Zhu, W.; Qin, W.; Zhang, K.; Rottinghaus, G.E.; Chen, Y.-C.; Kliethermes, B.; Sauter, E.R. Trans-resveratrol alters mammary promoter hypermethylation in women at increased risk for breast cancer. Nutr. Cancer 2012, 64, 393–400. [Google Scholar] [CrossRef]
- Omotayo, O.P.; Omotayo, A.O.; Mwanza, M.; Babalola, O.O. Prevalence of mycotoxins and their consequences on human health. Toxicol. Res. 2019, 35, 1–7. [Google Scholar] [CrossRef]
- Ghazi, T.; Nagiah, S.; Naidoo, P.; Chuturgoon, A.A. Fusaric acid-induced promoter methylation of DNA methyltransferases triggers DNA hypomethylation in human hepatocellular carcinoma (HepG2) cells. Epigenetics 2019, 14, 804–817. [Google Scholar] [CrossRef]
- Chuturgoon, A.; Phulukdaree, A.; Moodley, D. Fumonisin B1 induces global DNA hypomethylation in HepG2 cells—An alternative mechanism of action. Toxicology 2014, 315, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, J.; Huang, K.; Qi, X.; Dai, Q.; Mei, X.; Xu, W. Dynamic changes of global DNA methylation and hypermethylation of cell adhesion-related genes in rat kidneys in response to ochratoxin A. World Mycotoxin J. 2015, 1, 1–12. [Google Scholar] [CrossRef]
- Rieswijk, L.; Claessen, S.M.; Bekers, O.; van Herwijnen, M.; Theunissen, D.H.; Jennen, D.G.; de Kok, T.M.; Kleinjans, J.C.; van Breda, S.G. Aflatoxin B1 induces persistent epigenomic effects in primary human hepatocytes associated with hepatocellular carcinoma. Toxicology 2016, 350, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Karaman, E.F.; Ozden, S. Alterations in global DNA methylation and metabolism-related genes caused by zearalenone in MCF7 and MCF10F cells. Mycotoxin Res. 2019, 35, 309–320. [Google Scholar] [CrossRef]
- Wang, H.; Zong, Q.; Wang, S.; Zhao, C.; Wu, S.; Bao, W. Genome-Wide DNA Methylome and Transcriptome Analysis of Porcine Intestinal Epithelial Cells upon Deoxynivalenol Exposure. J. Agric. Food Chem. 2019, 67, 6423–6431. [Google Scholar] [CrossRef]
- Liu, A.; Sun, Y.; Wang, X.; Ihsan, A.; Tao, Y.; Chen, D.; Peng, D.; Wu, Q.; Wang, X.; Yuan, Z. DNA methylation is involved in pro-inflammatory cytokines expression in T-2 toxin-induced liver injury. Food Chem. Toxicol. 2019, 132, 110661. [Google Scholar] [CrossRef]
- Bacon, C.W.; Porter, J.K.; Norred, W.P.; Leslie, J.F. Production of fusaric acid by Fusarium species. Appl. Environ. Microbiol. 1996, 62, 4039. [Google Scholar] [CrossRef]
- Devaraja, S.; Girish, K.S.; Santhosh, M.S.; Hemshekhar, M.; Nayaka, S.C.; Kemparaju, K. Fusaric acid, a mycotoxin, and its influence on blood coagulation and platelet function. Blood Coagul. Fibrinolysis 2013, 24, 419–423. [Google Scholar] [CrossRef]
- Reddy, R.V.; Larson, C.A.; Brimer, G.E.; Frappier, B.L.; Reddy, C.S. Developmental toxic effects of fusaric acid in CD1 mice. Bull. Environ. Contam. Toxicol. 1996, 57, 354–360. [Google Scholar] [CrossRef]
- Hidaka, H.; Nagatsu, T.; Takeya, K.; Takeuchi, T.; Suda, H. Fusaric acid, a hypotensive agent produced by fungi. J. Antibiot. 1969, 22, 228–230. [Google Scholar] [CrossRef]
- Terasawa, F.; Kameyama, M. The clinical trial of a new hypotensive agent, "fusaric acid (5-butylpicolinic acid)": The preliminary report. Jpn. Circ. J. 1971, 35, 339–357. [Google Scholar] [CrossRef] [PubMed]
- Yin, E.S.; Rakhmankulova, M.; Kucera, K.; de Sena Filho, J.G.; Portero, C.E.; Narváez-Trujillo, A.; Holley, S.A.; Strobel, S.A. Fusaric acid induces a notochord malformation in zebrafish via copper chelation. Biometals 2015, 28, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Sheik Abdul, N.; Nagiah, S.; Chuturgoon, A.A. Fusaric acid induces mitochondrial stress in human hepatocellular carcinoma (HepG2) cells. Toxicon 2016, 119, 336–344. [Google Scholar] [CrossRef] [PubMed]
- D’Alton, A.; Etherton, B. Effects of fusaric Acid on tomato root hair membrane potentials and ATP levels. Plant. Physiol. 1984, 74, 39–42. [Google Scholar] [CrossRef]
- Ghazi, T.; Nagiah, S.; Tiloke, C.; Sheik Abdul, N.; Chuturgoon, A.A. Fusaric Acid Induces DNA Damage and Post-Translational Modifications of p53 in Human Hepatocellular Carcinoma (HepG(2) ) Cells. J. Cell Biochem. 2017, 118, 3866–3874. [Google Scholar] [CrossRef]
- Dhani, S.; Nagiah, S.; Naidoo, D.B.; Chuturgoon, A.A. Fusaric Acid immunotoxicity and MAPK activation in normal peripheral blood mononuclear cells and Thp-1 cells. Sci. Rep. 2017, 7, 3051. [Google Scholar] [CrossRef]
- Domijan, A.-M.; Zeljezić, D.; Milić, M.; Peraica, M. Fumonisin B(1): Oxidative status and DNA damage in rats. Toxicology 2007, 232, 163–169. [Google Scholar] [CrossRef]
- Gelderblom, W.C.; Marasas, W.F. Controversies in fumonisin mycotoxicology and risk assessment. Hum. Exp. Toxicol. 2012, 31, 215–235. [Google Scholar] [CrossRef]
- Marasas, W.F. Discovery and occurrence of the fumonisins: A historical perspective. Environ. Health Perspect. 2001, 109, 239–243. [Google Scholar] [CrossRef]
- Cancer, I.A.f.R.o. Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. Working group on the evaluation of carcinogenic risks to humans. Iarc Monogr Eval. Carcinog. Risks Hum. 2002, 82, 1–556. [Google Scholar]
- Soriano, J.M.; González, L.; Catalá, A.I. Mechanism of action of sphingolipids and their metabolites in the toxicity of fumonisin B1. Prog. Lipid Res. 2005, 44, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Voss, K.A.; Howard, P.C.; Riley, R.T.; Sharma, R.P.; Bucci, T.J.; Lorentzen, R.J. Carcinogenicity and mechanism of action of fumonisin B1: A mycotoxin produced by Fusarium moniliforme (= F. verticillioides). Cancer Detect. Prev 2002, 26, 1–9. [Google Scholar] [CrossRef]
- Alizadeh, A.M.; Rohandel, G.; Roudbarmohammadi, S.; Roudbary, M.; Sohanaki, H.; Ghiasian, S.A.; Taherkhani, A.; Semnani, S.; Aghasi, M. Fumonisin B1 contamination of cereals and risk of esophageal cancer in a high risk area in northeastern Iran. Asian Pac. J. Cancer Prev. 2012, 13, 2625–2628. [Google Scholar] [CrossRef]
- Chu, F.S.; Li, G.Y. Simultaneous occurrence of fumonisin B1 and other mycotoxins in moldy corn collected from the People’s Republic of China in regions with high incidences of esophageal cancer. Appl. Environ. Microbiol. 1994, 60, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Shephard, G.S.; Marasas, W.F.; Burger, H.-M.; Somdyala, N.; Rheeder, J.; Van der Westhuizen, L.; Gatyeni, P.; Van Schalkwyk, D.J. Exposure assessment for fumonisins in the former Transkei region of South Africa. Food Addit. Contam. 2007, 24, 621–629. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Statistics 2011. Available online: https://www.who.int/whosis/whostat/2011/en/ (accessed on 27 August 2020).
- Gelderblom, W.C.; Marasas, W.F.; Lebepe-Mazur, S.; Swanevelder, S.; Abel, S. Cancer initiating properties of fumonisin B1 in a short-term rat liver carcinogenesis assay. Toxicology 2008, 250, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Dekant, W.; Mally, A. Fumonisin B1 and the kidney: Modes of action for renal tumor formation by fumonisin B1 in rodents. Food Chem. Toxicol. 2012, 50, 3833–3846. [Google Scholar] [CrossRef]
- Stockmann-Juvala, H.; Savolainen, K. A review of the toxic effects and mechanisms of action of fumonisin B1. Hum. Exp. Toxicol. 2008, 27, 799–809. [Google Scholar] [CrossRef]
- Abdel Nour, A.M.; Ringot, D.; Guéant, J.L.; Chango, A. Folate receptor and human reduced folate carrier expression in HepG2 cell line exposed to fumonisin B1 and folate deficiency. Carcinogenesis 2007, 28, 2291–2297. [Google Scholar] [CrossRef]
- Gelineau-van Waes, J.; Starr, L.; Maddox, J.; Aleman, F.; Voss, K.A.; Wilberding, J.; Riley, R.T. Maternal fumonisin exposure and risk for neural tube defects: Mechanisms in an in vivo mouse model. Birth Defects Res. A Clin. Mol. Teratol. 2005, 73, 487–497. [Google Scholar] [CrossRef]
- Stevens, V.L.; Tang, J. Fumonisin B1-induced sphingolipid depletion inhibits vitamin uptake via the glycosylphosphatidylinositol-anchored folate receptor. J. Biol. Chem. 1997, 272, 18020–18025. [Google Scholar] [CrossRef] [PubMed]
- Mobio, T.A.; Anane, R.; Baudrimont, I.; Carratú, M.-R.; Shier, T.W.; Dano, S.D.; Ueno, Y.; Creppy, E.E. Epigenetic Properties of Fumonisin B1: Cell Cycle Arrest and DNA Base Modification in C6 Glioma Cells. Toxicol. Appl. Pharmacol. 2000, 164, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Demirel, G.; Alpertunga, B.; Ozden, S. Role of fumonisin B1 on DNA methylation changes in rat kidney and liver cells. Pharm. Biol. 2015, 53, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.I. Penicillium viridicatum, Penicillium verrucosum, and production of ochratoxin A. Appl. Environ. Microbiol. 1987, 53, 266. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar] [CrossRef]
- Stojković, R.; Hult, K.; Gamulin, S.; Plestina, R. High affinity binding of ochratoxin A to plasma constituents. Biochem. Int. 1984, 9, 33–38. [Google Scholar]
- Hagelberg, S.; Hult, K.; Fuchs, R. Toxicokinetics of ochratoxin A in several species and its plasma-binding properties. J. Appl. Toxicol. 1989, 9, 91–96. [Google Scholar] [CrossRef]
- Palli, D.; Miraglia, M.; Saieva, C.; Masala, G.; Cava, E.; Colatosti, M.; Corsi, A.M.; Russo, A.; Brera, C. Serum levels of ochratoxin A in healthy adults in Tuscany: Correlation with individual characteristics and between repeat measurements. Cancer Epidemiol. Biomark. Prev. 1999, 8, 265–269. [Google Scholar]
- Boorman, G.A.; McDonald, M.R.; Imoto, S.; Persing, R. Renal lesions induced by ochratoxin A exposure in the F344 rat. Toxicol. Pathol. 1992, 20, 236–245. [Google Scholar] [CrossRef]
- Gagliano, N.; Donne, I.D.; Torri, C.; Migliori, M.; Grizzi, F.; Milzani, A.; Filippi, C.; Annoni, G.; Colombo, P.; Costa, F.; et al. Early cytotoxic effects of ochratoxin A in rat liver: A morphological, biochemical and molecular study. Toxicology 2006, 225, 214–224. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer; World Health Organization. Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins; World Health Organization: Geneva, Switzerland, 1993; Volume 56. [Google Scholar]
- Zhu, L.; Yu, T.; Qi, X.; Yang, B.; Shi, L.; Luo, H.; He, X.; Huang, K.; Xu, W. miR-122 plays an important role in ochratoxin A-induced hepatocyte apoptosis in vitro and in vivo. Toxicol Res. 2016, 5, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Giromini, C.; Rebucci, R.; Fusi, E.; Rossi, L.; Saccone, F.; Baldi, A. Cytotoxicity, apoptosis, DNA damage and methylation in mammary and kidney epithelial cell lines exposed to ochratoxin A. Cell Biol. Toxicol. 2016, 32, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Ozden, S.; Turgut Kara, N.; Sezerman, O.U.; Durasi, İ.M.; Chen, T.; Demirel, G.; Alpertunga, B.; Chipman, J.K.; Mally, A. Assessment of global and gene-specific DNA methylation in rat liver and kidney in response to non-genotoxic carcinogen exposure. Toxicol. Appl. Pharmacol. 2015, 289, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhang, Y.; Xu, W.; Luo, Y.; Hao, J.; Shen, X.L.; Yang, X.; Li, X.; Huang, K. Zinc protects HepG2 cells against the oxidative damage and DNA damage induced by ochratoxin A. Toxicol. Appl. Pharmacol. 2013, 268, 123–131. [Google Scholar] [CrossRef]
- Lanyasunya, T.; Wamae, L.; Musa, H.; Olowofeso, O.; Lokwaleput, I. The risk of mycotoxins contamination of dairy feed and milk on smallholder dairy farms in Kenya. Pak. J. Nutr. 2005, 4, 162–169. [Google Scholar]
- Strosnider, H.; Azziz-Baumgartner, E.; Banziger, M.; Bhat, R.V.; Breiman, R.; Brune, M.-N.; DeCock, K.; Dilley, A.; Groopman, J.; Hell, K. Workgroup report: Public health strategies for reducing aflatoxin exposure in developing countries. Environ. Health Perspect. 2006, 114, 1898–1903. [Google Scholar] [CrossRef]
- Torres, A.M.; Barros, G.G.; Palacios, S.A.; Chulze, S.N.; Battilani, P. Review on pre-and post-harvest management of peanuts to minimize aflatoxin contamination. Food Res. Int. 2014, 62, 11–19. [Google Scholar] [CrossRef]
- Qi, L.N.; Bai, T.; Chen, Z.S.; Wu, F.X.; Chen, Y.Y.; De Xiang, B.; Peng, T.; Han, Z.G.; Li, L.Q. The p53 mutation spectrum in hepatocellular carcinoma from Guangxi, China: Role of chronic hepatitis B virus infection and aflatoxin B1 exposure. Liver Int. 2015, 35, 999–1009. [Google Scholar] [CrossRef]
- Soini, Y.; Chia, S.; Bennett, W.; Groopman, J.D.; Wang, J.-S.; DeBenedetti, V.; Cawley, H.; Welsh, J.; Hansen, C.; Bergasa, N.V. An aflatoxin-associated mutational hotspot at codon 249 in the p53 tumor suppressor gene occurs in hepatocellular carcinomas from Mexico. Carcinogenesis 1996, 17, 1007–1012. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Ahsan, H.; Chen, Y.; Lunn, R.M.; Wang, L.Y.; Chen, S.Y.; Lee, P.H.; Chen, C.J.; Santella, R.M. High frequency of promoter hypermethylation of RASSF1A and p16 and its relationship to aflatoxin B1–DNA adduct levels in human hepatocellular carcinoma. Mol. Carcinog. 2002, 35, 85–92. [Google Scholar] [CrossRef]
- Wu, H.-C.; Wang, Q.; Yang, H.-I.; Tsai, W.-Y.; Chen, C.-J.; Santella, R.M. Global DNA methylation in a population with aflatoxin B1 exposure. Epigenetics 2013, 8, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Kriszt, R.; Krifaton, C.; Szoboszlay, S.; Cserháti, M.; Kriszt, B.; Kukolya, J.; Czéh, Á.; Fehér-Tóth, S.; Török, L.; Szőke, Z.; et al. A New Zearalenone Biodegradation Strategy Using Non-Pathogenic Rhodococcus pyridinivorans K408 Strain. PLoS ONE 2012, 7, e43608. [Google Scholar] [CrossRef] [PubMed]
- Frizzell, C.; Ndossi, D.; Verhaegen, S.; Dahl, E.; Eriksen, G.; Sørlie, M.; Ropstad, E.; Muller, M.; Elliott, C.T.; Connolly, L. Endocrine disrupting effects of zearalenone, alpha- and beta-zearalenol at the level of nuclear receptor binding and steroidogenesis. Toxicol. Lett. 2011, 206, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Abid-Essefi, S.; Ouanes, Z.; Hassen, W.; Baudrimont, I.; Creppy, E.; Bacha, H. Cytotoxicity, inhibition of DNA and protein syntheses and oxidative damage in cultured cells exposed to zearalenone. Toxicol. Vitr. 2004, 18, 467–474. [Google Scholar] [CrossRef]
- Wang, M.; Wu, W.; Li, L.; He, J.; Huang, S.; Chen, S.; Chen, J.; Long, M.; Yang, S.; Li, P. Analysis of the miRNA Expression Profiles in the Zearalenone-Exposed TM3 Leydig Cell Line. Int. J. Mol. Sci. 2019, 20, 635. [Google Scholar] [CrossRef]
- So, M.Y.; Tian, Z.; Phoon, Y.S.; Sha, S.; Antoniou, M.N.; Zhang, J.; Wu, R.S.; Tan-Un, K.C. Gene expression profile and toxic effects in human bronchial epithelial cells exposed to zearalenone. PLoS ONE 2014, 9, e96404. [Google Scholar] [CrossRef]
- Kouadio, J.H.; Dano, S.D.; Moukha, S.; Mobio, T.A.; Creppy, E.E. Effects of combinations of Fusarium mycotoxins on the inhibition of macromolecular synthesis, malondialdehyde levels, DNA methylation and fragmentation, and viability in Caco-2 cells. Toxicon 2007, 49, 306–317. [Google Scholar] [CrossRef]
- Karaman, E.F.; Zeybel, M.; Ozden, S. Evaluation of the epigenetic alterations and gene expression levels of HepG2 cells exposed to zearalenone and α-zearalenol. Toxicol. Lett. 2020, 325, 52–60. [Google Scholar] [CrossRef]
- Zhu, C.-C.; Hou, Y.-J.; Han, J.; Cui, X.-S.; Kim, N.-H.; Sun, S.-C. Zearalenone exposure affects epigenetic modifications of mouse eggs. Mutagenesis 2014, 29, 489–495. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, Y.; Zhang, H.; Zhang, P.; Liu, J.; Feng, Y.; Men, Y.; Li, L.; Shen, W.; Sun, Z. Pubertal exposure to low doses of zearalenone disrupting spermatogenesis through ERα related genetic and epigenetic pathways. Toxicol. Lett. 2019, 315, 31–38. [Google Scholar] [CrossRef]
- Pestka, J.J. Deoxynivalenol: Mechanisms of action, human exposure, and toxicological relevance. Arch. Toxicol. 2010, 84, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wang, Q.-C.; Zhu, C.-C.; Liu, J.; Zhang, Y.; Cui, X.-S.; Kim, N.-H.; Sun, S.-C. Deoxynivalenol exposure induces autophagy/apoptosis and epigenetic modification changes during porcine oocyte maturation. Toxicol. Appl. Pharmacol. 2016, 300, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Hu, S.; Zhu, X.; Ihsan, A.; Wu, Q.; Sun, L.; Wang, X. Dietary Exposure of Deoxynivalenol Affected Cytochrome P450 and Growth Related-Gene Expression via DNA Methylation in Piglet Liver. Available online: https://www.researchsquare.com/article/rs-34338/latest.pdf (accessed on 27 August 2020).
- Makowska, K.; Obremski, K.; Gonkowski, S. The impact of T-2 toxin on vasoactive intestinal polypeptide-like immunoreactive (VIP-LI) nerve structures in the wall of the porcine stomach and duodenum. Toxins 2018, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Tiemann, U.; Dänicke, S. In vivo and in vitro effects of the mycotoxins zearalenone and deoxynivalenol on different non-reproductive and reproductive organs in female pigs: A review. Food Addit. Contam. 2007, 24, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Pelyhe, C.; Kövesi, B.; Zándoki, E.; Kovács, B.; Erdélyi, M.; Kulcsár, S.; Mézes, M.; Balogh, K. Multi-trichothecene mycotoxin exposure activates glutathione-redox system in broiler chicken. Toxicon 2018, 153, 53–57. [Google Scholar] [CrossRef]
- Nakade, M.; Pelyhe, C.; Kövesi, B.; Balogh, K.; Kovács, B.; Szabó-Fodor, J.; Zándoki, E.; Mézes, M.; Erdélyi, M. Short-term effects of T-2 toxin or deoxynivalenol on glutathione status and expression of its regulatory genes in chicken. Acta Vet. Hung. 2018, 66, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, Y.; Yuan, Z.; Yi, J.; Chen, J.; Wang, N.; Tian, Y. Autophagy and apoptosis interact to modulate T-2 toxin-induced toxicity in liver cells. Toxins 2019, 11, 45. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef]
- Chow, L.S.-N.; Lo, K.-W.; Kwong, J.; Wong, A.Y.-H.; Huang, D.P. Aberrant methylation of RASSF4/AD037 in nasopharyngeal carcinoma. Oncol. Rep. 2004, 12, 781–787. [Google Scholar] [CrossRef]
- Han, Y.; Dong, Q.; Hao, J.; Fu, L.; Han, X.; Zheng, X.; Wang, E. RASSF4 is downregulated in nonsmall cell lung cancer and inhibits cancer cell proliferation and invasion. Tumor Biol. 2016, 37, 4865–4871. [Google Scholar] [CrossRef]
- Zheng, W.; Zhao, L.; Wang, G.; Kang, X.-C.; Qin, L.; Ji, J.-J.; Hao, S. Promoter methylation and expression of RASSF1A genes as predictors of disease progression in colorectal cancer. Int. J. Clin. Exp. Med. 2016, 9, 2027–2036. [Google Scholar]
- Steinmann, K.; Sandner, A.; Schagdarsurengin, U.; Dammann, R.H. Frequent promoter hypermethylation of tumor-related genes in head and neck squamous cell carcinoma. Oncol. Rep. 2009, 22, 1519–1526. [Google Scholar] [PubMed]
- Liu, A.; Xu, X.; Hou, R.; Badawy, S.; Tao, Y.; Chen, D.; Ihsan, A.; Wang, X.; Wu, Q.; Yuan, Z. DNA methylation and RASSF4 expression are involved in T-2 toxin-induced hepatotoxicity. Toxicology 2019, 425, 152246. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type and Model | Concentration of Folic Acid | Duration of Treatment | Effect on DNA Methylation and Cellular Outcome | Reference |
|---|---|---|---|---|
| Breast cancer: MCF-7 and MDA-MB-231 cells | 4–8 mg/L | 4 days | MCF-7 and MDA-MB-231 cells: ↑ DNMT1 expression; ↑ Promoter methylation of PTEN, APC, and RARβ2; ↑ caspase-3-dependent apoptosis | [33] |
| Colorectal cancer: HCT116, LS174T, and SW480 cells | 4–16 mg/L | 7 days | HCT116 cells: ↑ DNMT1 expression; ↓ DNMT3A and DNMT3B expression; ↓ Global DNA methylation; ↑ Cell proliferation; ↑ Colonosphere formation LS174T cells: ↑ DNMT1 expression; DNMT3A not expressed; No change in DNMT3B expression; ↓ Global DNA methylation; ↑ Cell proliferation; ↑ Colonosphere formation SW480 cells: ↓ DNMT1, DNMT3A, and DNMT3B expression; No change in global DNA methylation; ↑ Cell proliferation; ↑ Colonosphere formation | [34] |
| Colon cancer: HCT116 and Caco-2 cells | 0–2.3 µM | 20 days | Folic acid deficient (0 µM) HCT116 cells: ↓ DNMT1 and DNMT3A expression; No change in DNMT activity; No change in global DNA methylation; ↑ ER promoter methylation; No change in ER expression; ↓ Cell growth Folic acid deficient (0 µM) Caco-2 cells: ↓ DNMT1 and DNMT3A expression; ↓ DNMT activity; No change in global DNA methylation; ↑ ER promoter methylation; No change in ER expression; ↓ Cell growth | [46] |
| Colon cancer: SW620 cells | 0–3 µmol/L | 14 days | Folic acid deficiency (0 µmol/L): ↓ Global DNA methylation; ↓ p53 gene-specific DNA methylation. In both cases, the effects of folic acid depletion were reversed by folic acid (3 µmol/L) supplementation | [47] |
| Colon cancer: HCT116 and SW480 cells | Commercial folate-deficient RPMI 1640 medium | HCT116 cells: 24–48 h SW480 cells: 24–72 h | HCT116 and SW480 cells: ↓ Shh gene promoter methylation; ↑ Shh gene and protein expression; ↑ Activation of Shh signalling; ↑ Migration and invasiveness | [48,49] |
| Colon cancer: Caco-2 cells | 20 µM | 48 h | ↑ Promoter methylation of ESR1, p15INK4b, and p16INK4a; ↑ Cell proliferation | [50] |
| Nasopharyngeal cancer: KB cells | 2–10 nM | - | ↑ Promoter methylation of H-cadherin; ↓ H-cadherin expression; Promotes malignant phenotype | [51] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghazi, T.; Arumugam, T.; Foolchand, A.; Chuturgoon, A.A. The Impact of Natural Dietary Compounds and Food-Borne Mycotoxins on DNA Methylation and Cancer. Cells 2020, 9, 2004. https://doi.org/10.3390/cells9092004
Ghazi T, Arumugam T, Foolchand A, Chuturgoon AA. The Impact of Natural Dietary Compounds and Food-Borne Mycotoxins on DNA Methylation and Cancer. Cells. 2020; 9(9):2004. https://doi.org/10.3390/cells9092004
Chicago/Turabian StyleGhazi, Terisha, Thilona Arumugam, Ashmika Foolchand, and Anil A. Chuturgoon. 2020. "The Impact of Natural Dietary Compounds and Food-Borne Mycotoxins on DNA Methylation and Cancer" Cells 9, no. 9: 2004. https://doi.org/10.3390/cells9092004
APA StyleGhazi, T., Arumugam, T., Foolchand, A., & Chuturgoon, A. A. (2020). The Impact of Natural Dietary Compounds and Food-Borne Mycotoxins on DNA Methylation and Cancer. Cells, 9(9), 2004. https://doi.org/10.3390/cells9092004