Clinical Importance of the Human Umbilical Artery Potassium Channels




Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Importance of the K+ Channels in Physiological Regulation of HUA
3. Diversity of K+ Channels in HUA
3.1. Voltage-Dependent K+ (Kv) Channels
3.2. Calcium-Activated K+ (Kca) Channels
3.3. Inward Rectifier K+ (Kir) Channels
ATP-Sensitive K+ (KATP) Channels
3.4. The 2-Pore Domain K+ (K2P) Channels
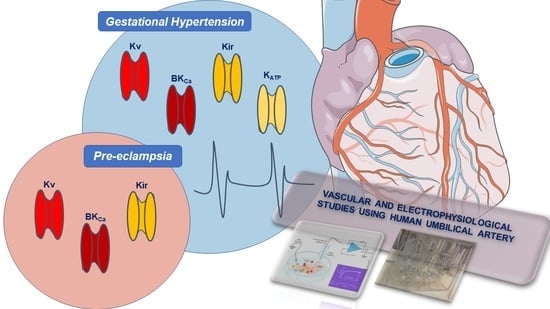
4. Clinical Importance and Medical Uses of K+-Channels
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| (Ca2+)i | intracellular Ca2+ concentration |
| 4-AP | 4-aminopyridine |
| ANP | atrial natriuretic peptide |
| Ba2+ | barium ion |
| BKCa | large-conductance Ca2+-activated K+ channels |
| Ca2+ | calcium |
| CaM | calmodulin |
| CaM-BD | calmodulin-binding domain |
| cGMP | cyclic guanosine monophosphate |
| Cl- | chloride |
| CPA | chorionic plaque arteries |
| Cs+ | caesium ion |
| CV | cardiovascular |
| CVD | cardiovascular diseases |
| DHS-1 | dedydrosoyasaponin-1 |
| EC | endothelial cells |
| EK | K+ equilibrium potential |
| eNOS | endothelial oxide nitric synthase |
| HUA | human umbilical artery |
| HUASMC | human umbilical artery smooth muscle cells |
| HUV | human umbilical vein |
| IK | K+ currents |
| IKCa | intermediate-conductance Ca2+-activated K+ channels |
| iNOS | inducible oxide nitric synthase |
| K+ | Potassium |
| K2P | 2-pore domains K+ channels |
| KATP | ATP-sensitive K+ channels |
| KCa | Ca2+-activated K+ channels |
| Kir | inward rectifier K+ channels |
| KV | voltage-dependent K+ channels |
| MP | resting membrane potential |
| Na2S | sodium sulfide |
| NO | nitric oxide |
| NP | natriuretic peptides |
| PDE | phosphodiesteras’s |
| pGC | particulate guanylyl cyclase |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PKG | protein kinase G |
| sGC | soluble guanylyl cyclase |
| SKCa | small-conductance Ca2+-activated K+ channels |
| SM | smooth muscle |
| SMC | smooth muscle cells |
| STOC | spontaneous transient outward currents |
| SUR | sulfonylurea receptor |
| TASK | acid-sensitive rectifiers K+ channels |
| TEA | tetraethylammonium |
| TWIK | lipid sensitive mechano-gated K+ channels |
| TREK | weak inward rectifiers K+ channels |
| UC | umbilical cord |
| VGCC | voltage-gated Ca2+-channels |
| Zn+2 | zinc ion |
References
- Dogan, M.F.; Yildiz, O.; Arslan, S.O.; Ulusoy, K.G. Potassium channels in vascular smooth muscle: A pathophysiological and pharmacological perspective. Fundam. Clin. Pharmacol. 2019, 33, 504–523. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.F. KV channels and the regulation of vascular smooth muscle tone. Microcirculation 2018, 25. [Google Scholar] [CrossRef]
- Hayabuchi, Y. The Action of Smooth Muscle Cell Potassium Channels in the Pathology of Pulmonary Arterial Hypertension. Pediatr. Cardiol. 2017, 38, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Burg, E.D.; Remillard, C.V.; Yuan, J.X. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: Pharmacotherapeutic implications. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S99–S111. [Google Scholar] [CrossRef]
- Nelson, M.T.; Quayle, J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol 1995, 268, C799–C822. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y. Electrical and Mechanical Properties of Vascular Smooth Muscle. In Biology of Vascular Smooth Muscle: Vasoconstriction and Dilatation; Springer: Berlin, Germany, 2017; pp. 41–55. [Google Scholar] [CrossRef]
- Tykocki, N.R.; Boerman, E.M.; Jackson, W.F. Smooth Muscle Ion Channels and Regulation of Vascular Tone in Resistance Arteries and Arterioles. Compr. Physiol. 2017, 7, 485–581. [Google Scholar] [CrossRef]
- Jackson, W.F. Potassium Channels in Regulation of Vascular Smooth Muscle Contraction and Growth. Adv. Pharmacol. 2017, 78, 89–144. [Google Scholar] [CrossRef]
- Lorigo, M.; Mariana, M.; Feiteiro, J.; Cairrao, E. How is the human umbilical artery regulated? J. Obs. Gynaecol Res. 2018. [Google Scholar] [CrossRef]
- Margarida Lorigo, M.; Melissa, M.; Joana, F.; Elisa, C. Human Umbilical Artery Smooth Muscle Cells: Vascular Function and Clinical Importance. In Horizons in World Cardiovascular Research; Bennington, E.H., Ed.; Nova Science Publisher: New York, NY, USA, 2019; Volume 16. [Google Scholar]
- Meyer, W.W.; Rumpelt, H.J.; Yao, A.C.; Lind, J. Structure and closure mechanism of the human umbilical artery. Eur J. Pediatr. 1978, 128, 247–259. [Google Scholar] [CrossRef]
- Ferguson, V.L.; Dodson, R.B. Bioengineering aspects of the umbilical cord. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 144 (Suppl. 1), S108–S113. [Google Scholar] [CrossRef]
- Chillakuru, S.; Velichety, S.D.; Rajagopalan, V. Human umbilical cord and its vessels: A histomorphometric study in difference severity of hypertensive disorders of pregnancy. Anat. Cell Biol. 2020, 53, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Cairrao, E.; Santos-Silva, A.J.; Alvarez, E.; Correia, I.; Verde, I. Isolation and culture of human umbilical artery smooth muscle cells expressing functional calcium channels. Vitr. Cell. Dev. Biol. Anim. 2009, 45, 175–184. [Google Scholar] [CrossRef]
- Martin, P.; Rebolledo, A.; Palomo, A.R.; Moncada, M.; Piccinini, L.; Milesi, V. Diversity of potassium channels in human umbilical artery smooth muscle cells: A review of their roles in human umbilical artery contraction. Reprod. Sci. 2014, 21, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.F. Ion channels and vascular tone. Hypertension 2000, 35, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Brayden, J.E. Potassium channels in vascular smooth muscle. Clin. Exp. Pharm. Physiol. 1996, 23, 1069–1076. [Google Scholar] [CrossRef]
- Milesi, V.; Raingo, J.; Rebolledo, A.; Grassi de Gende, A.O. Potassium channels in human umbilical artery cells. J. Soc. Gynecol. Investig. 2003, 10, 339–346. [Google Scholar] [CrossRef]
- Ko, E.A.; Park, W.S.; Firth, A.L.; Kim, N.; Yuan, J.X.; Han, J. Pathophysiology of voltage-gated K+ channels in vascular smooth muscle cells: Modulation by protein kinases. Prog. Biophys. Mol. Biol. 2010, 103, 95–101. [Google Scholar] [CrossRef]
- Ko, E.A.; Han, J.; Jung, I.D.; Park, W.S. Physiological roles of K+ channels in vascular smooth muscle cells. J. Smooth Muscle Res. 2008, 44, 65–81. [Google Scholar] [CrossRef]
- Cairrao, E.; Alvarez, E.; Santos-Silva, A.J.; Verde, I. Potassium channels are involved in testosterone-induced vasorelaxation of human umbilical artery. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2008, 376, 375–383. [Google Scholar] [CrossRef]
- Santos-Silva, A.J.; Cairrao, E.; Verde, I. Study of the mechanisms regulating human umbilical artery contractility. Health 2010, 2, 321–331. [Google Scholar] [CrossRef]
- Cairrao, E.; Santos-Silva, A.J.; Verde, I. PKG is involved in testosterone-induced vasorelaxation of human umbilical artery. Eur. J. Pharmacol. 2010, 640, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Feiteiro, J.; Santos-Silva, A.J.; Verde, I.; Cairrao, E. Testosterone and atrial natriuretic Peptide share the same pathway to induce vasorelaxation of human umbilical artery. J. Cardiovasc. Pharmacol. 2014, 63, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Feiteiro, J.; Verde, I.; Cairrao, E. Cyclic guanosine monophosphate compartmentation in human vascular smooth muscle cells. Cell. Signal. 2016, 28, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Firth, A.L.; Han, J.; Ko, E.A. Patho-, physiological roles of voltage-dependent K+ channels in pulmonary arterial smooth muscle cells. J. Smooth Muscle Res. 2010, 46, 89–105. [Google Scholar] [CrossRef]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov 2009, 8, 982–1001. [Google Scholar] [CrossRef]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [Google Scholar] [CrossRef]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science 2005, 309, 903–908. [Google Scholar] [CrossRef]
- Hasan, R.; Jaggar, J.H. KV channel trafficking and control of vascular tone. Microcirculation 2018, 25. [Google Scholar] [CrossRef]
- Lovren, F.; Triggle, C. Nitric oxide and sodium nitroprusside-induced relaxation of the human umbilical artery. Br. J. Pharmacol. 2000, 131, 521–529. [Google Scholar] [CrossRef]
- Yildiz, O.; Nacitarhan, C.; Seyrek, M. Potassium channels in the vasodilating action of levosimendan on the human umbilical artery. J. Soc. Gynecol. Investig. 2006, 13, 312–315. [Google Scholar] [CrossRef]
- Mathew John, C.; Khaddaj Mallat, R.; George, G.; Kim, T.; Mishra, R.C.; Braun, A.P. Pharmacologic targeting of endothelial Ca(2+)-activated K(+) channels: A strategy to improve cardiovascular function. Channels 2018, 12, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.M.; Shim, H.; Christophersen, P.; Wulff, H. Pharmacology of Small- and Intermediate-Conductance Calcium-Activated Potassium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 219–240. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.L.; Bai, Y.L.; Cai, B.Z. Calcium-Activated Potassium Channels: Potential Target for Cardiovascular Diseases. Adv. Protein Chem. Struct. Biol. 2016, 104, 233–261. [Google Scholar] [CrossRef] [PubMed]
- Marty, A. Ca-dependent K channels with large unitary conductance in chromaffin cell membranes. Nature 1981, 291, 497–500. [Google Scholar] [CrossRef]
- Gardos, G. The function of calcium in the potassium permeability of human erythrocytes. Biochim. Biophys. Acta 1958, 30, 653–654. [Google Scholar] [CrossRef]
- Blatz, A.L.; Magleby, K.L. Single apamin-blocked Ca-activated K+ channels of small conductance in cultured rat skeletal muscle. Nature 1986, 323, 718–720. [Google Scholar] [CrossRef]
- Ghatta, S.; Nimmagadda, D.; Xu, X.; O’Rourke, S.T. Large-conductance, calcium-activated potassium channels: Structural and functional implications. Pharmacology 2006, 110, 103–116. [Google Scholar] [CrossRef]
- Brenner, R.; Perez, G.J.; Bonev, A.D.; Eckman, D.M.; Kosek, J.C.; Wiler, S.W.; Patterson, A.J.; Nelson, M.T.; Aldrich, R.W. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 2000, 407, 870–876. [Google Scholar] [CrossRef]
- Eichhorn, B.; Dobrev, D. Vascular large conductance calcium-activated potassium channels: Functional role and therapeutic potential. Naunyn Schmiedebergs Arch. Pharm. 2007, 376, 145–155. [Google Scholar] [CrossRef]
- Sobey, C.G. Potassium channel function in vascular disease. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 28–38. [Google Scholar] [CrossRef]
- Lee, U.S.; Cui, J. BK channel activation: Structural and functional insights. Trends Neurosci. 2010, 33, 415–423. [Google Scholar] [CrossRef]
- Kohler, R. Single-nucleotide polymorphisms in vascular Ca2+-activated K+-channel genes and cardiovascular disease. Pflug. Arch. Eur. J. Physiol. 2010, 460, 343–351. [Google Scholar] [CrossRef]
- Gollasch, M.; Lohn, M.; Furstenau, M.; Nelson, M.T.; Luft, F.C.; Haller, H. Ca2+ channels, ‘quantized’ Ca2+ release, and differentiation of myocytes in the cardiovascular system. J. Hypertens. 2000, 18, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Cheng, H.; Rubart, M.; Santana, L.F.; Bonev, A.D.; Knot, H.J.; Lederer, W.J. Relaxation of arterial smooth muscle by calcium sparks. Science 1995, 270, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Perez, G.J.; Bonev, A.D.; Patlak, J.B.; Nelson, M.T. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J. Gen. Physiol. 1999, 113, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.Q.; Zhang, L. Function and regulation of large conductance Ca(2+)-activated K+ channel in vascular smooth muscle cells. Drug Discov. Today 2012, 17, 974–987. [Google Scholar] [CrossRef]
- Lu, R.; Alioua, A.; Kumar, Y.; Eghbali, M.; Stefani, E.; Toro, L. MaxiK channel partners: Physiological impact. J. Physiol. 2006, 570, 65–72. [Google Scholar] [CrossRef]
- Alioua, A.; Mahajan, A.; Nishimaru, K.; Zarei, M.M.; Stefani, E.; Toro, L. Coupling of c-Src to large conductance voltage- and Ca2+-activated K+ channels as a new mechanism of agonist-induced vasoconstriction. Proc. Natl. Acad. Sci. USA 2002, 99, 14560–14565. [Google Scholar] [CrossRef]
- Hill, M.A.; Yang, Y.; Ella, S.R.; Davis, M.J.; Braun, A.P. Large conductance, Ca2+-activated K+ channels (BKCa) and arteriolar myogenic signaling. FEBS Lett. 2010, 584, 2033–2042. [Google Scholar] [CrossRef]
- Kaczmarek, L.K.; Aldrich, R.W.; Chandy, K.G.; Grissmer, S.; Wei, A.D.; Wulff, H. International Union of Basic and Clinical Pharmacology. C. Nomenclature and Properties of Calcium-Activated and Sodium-Activated Potassium Channels. Pharmacol. Rev. 2017, 69, 1–11. [Google Scholar] [CrossRef]
- Clements, R.T.; Terentyev, D.; Sellke, F.W. Ca(2+)-activated K(+) channels as therapeutic targets for myocardial and vascular protection. Circ. J. Off. J. Jpn. Circ. Soc. 2015, 79, 455–462. [Google Scholar] [CrossRef]
- Wang, L.; Sigworth, F.J. Structure of the BK potassium channel in a lipid membrane from electron cryomicroscopy. Nature 2009, 461, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Leonetti, M.D.; Hsiung, Y.; MacKinnon, R. Open structure of the Ca2+ gating ring in the high-conductance Ca2+-activated K+ channel. Nature 2011, 481, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, D.C.; Muench, S.P.; Lippiat, J.D. Mechanism of inhibition of mouse Slo3 (KCa 5.1) potassium channels by quinine, quinidine and barium. Br. J. Pharmacol. 2015, 172, 4355–4363. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, J.; Werner, M.E.; Brayden, J.E.; Nelson, M.T. Calcium-activated potassium channels and the regulation of vascular tone. Physiol. (Bethesda) 2006, 21, 69–78. [Google Scholar] [CrossRef]
- Cui, J.; Yang, H.; Lee, U.S. Molecular mechanisms of BK channel activation. Cell. Mol. Life Sci. CMLS 2009, 66, 852–875. [Google Scholar] [CrossRef]
- Standen, N.B.; Quayle, J.M. K+ channel modulation in arterial smooth muscle. Acta Physiol. Scand. 1998, 164, 549–557. [Google Scholar] [CrossRef]
- Waldron, G.J.; Cole, W.C. Activation of vascular smooth muscle K+ channels by endothelium-derived relaxing factors. Clin. Exp. Pharm. Physiol. 1999, 26, 180–184. [Google Scholar] [CrossRef]
- Wallner, M.; Meera, P.; Ottolia, M.; Kaczorowski, G.J.; Latorre, R.; Garcia, M.L.; Stefani, E.; Toro, L. Characterization of and modulation by a beta-subunit of a human maxi KCa channel cloned from myometrium. Recept. Channels 1995, 3, 185–199. [Google Scholar]
- Trombetta-Lima, M.; Krabbendam, I.E.; Dolga, A.M. Calcium-activated potassium channels: Implications for aging and age-related neurodegeneration. Int. J. Biochem. Cell Biol. 2020, 123, 105748. [Google Scholar] [CrossRef]
- Tanaka, Y.; Koike, K.; Toro, L. MaxiK channel roles in blood vessel relaxations induced by endothelium-derived relaxing factors and their molecular mechanisms. J. Smooth Muscle Res. 2004, 40, 125–153. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wallner, M.; Meera, P.; Toro, L. Human and rodent MaxiK channel beta-subunit genes: Cloning and characterization. Genomics 1999, 55, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Aldrich, R.W. BK potassium channel modulation by leucine-rich repeat-containing proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 7917–7922. [Google Scholar] [CrossRef] [PubMed]
- Radenkovic, M.; Grbovic, L.; Radunovic, N.; Momcilov, P. Pharmacological evaluation of bradykinin effect on human umbilical artery in normal, hypertensive and diabetic pregnancy. Pharm. Rep. 2007, 59, 64–73. [Google Scholar]
- Saldanha, P.A.; Cairrao, E.; Maia, C.J.; Verde, I. Long- and short-term effects of androgens in human umbilical artery smooth muscle. Clin. Exp. Pharmacol. Physiol. 2013, 40, 181–189. [Google Scholar] [CrossRef]
- Evaristo Rodrigues da Silva, R.; de Alencar Silva, A.; Pereira-de-Morais, L.; de Sousa Almeida, N.; Iriti, M.; Kerntopf, M.R.; Menezes, I.R.A.d.; Coutinho, H.D.M.; Barbosa, R. Relaxant Effect of Monoterpene (−)-Carveol on Isolated Human Umbilical Cord Arteries and the Involvement of Ion Channels. Molecules 2020, 25, 2681. [Google Scholar] [CrossRef]
- Rebolledo, A.; Raingo, J.; Rinaldi, G.; Grassi, A.; Milesi, V. 05 Cyclic GMP activates the big Ca-sensitive K channel in smooth muscle cells of human umbilical artery. J. Mol. Cell. Cardiol. 2002, 34, A3. [Google Scholar] [CrossRef]
- Martin, P.; Enrique, N.; Palomo, A.R.; Rebolledo, A.; Milesi, V. Bupivacaine inhibits large conductance, voltage- and Ca2+- activated K+ channels in human umbilical artery smooth muscle cells. Channels 2012, 6, 174–180. [Google Scholar] [CrossRef]
- Bernsteiner, H.; Zangerl-Plessl, E.M.; Chen, X.; Stary-Weinzinger, A. Conduction through a narrow inward-rectifier K(+) channel pore. J. Gen. Physiol. 2019, 151, 1231–1246. [Google Scholar] [CrossRef]
- Chen, R.; Swale, D.R. Inwardly Rectifying Potassium (Kir) Channels Represent a Critical Ion Conductance Pathway in the Nervous Systems of Insects. Sci. Rep. 2018, 8, 1617. [Google Scholar] [CrossRef]
- Nichols, C.G.; Lopatin, A.N. Inward rectifier potassium channels. Annu. Rev. Physiol. 1997, 59, 171–191. [Google Scholar] [CrossRef]
- Lu, Z. Mechanism of rectification in inward-rectifier K+ channels. Annu. Rev. Physiol. 2004, 66, 103–129. [Google Scholar] [CrossRef] [PubMed]
- Logothetis, D.E.; Jin, T.; Lupyan, D.; Rosenhouse-Dantsker, A. Phosphoinositide-mediated gating of inwardly rectifying K(+) channels. Pflug. Arch. Eur. J. Physiol. 2007, 455, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Park, W.S.; Han, J.; Earm, Y.E. Physiological role of inward rectifier K(+) channels in vascular smooth muscle cells. Pflug. Arch. Eur. J. Physiol. 2008, 457, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Doyle, D.A.; Morais Cabral, J.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef] [PubMed]
- Black, K.A.; He, S.; Jin, R.; Miller, D.M.; Bolla, J.R.; Clarke, O.B.; Johnson, P.; Windley, M.; Burns, C.J.; Hill, A.P.; et al. A constricted opening in Kir channels does not impede potassium conduction. Nat. Commun. 2020, 11, 3024. [Google Scholar] [CrossRef]
- Wu, B.N.; Luykenaar, K.D.; Brayden, J.E.; Giles, W.R.; Corteling, R.L.; Wiehler, W.B.; Welsh, D.G. Hyposmotic challenge inhibits inward rectifying K+ channels in cerebral arterial smooth muscle cells. Am. J. Physiology. Heart Circ. Physiol. 2007, 292, H1085–H1094. [Google Scholar] [CrossRef]
- Tennant, B.P.; Cui, Y.; Tinker, A.; Clapp, L.H. Functional expression of inward rectifier potassium channels in cultured human pulmonary smooth muscle cells: Evidence for a major role of Kir2.4 subunits. J. Membr. Biol. 2006, 213, 19–29. [Google Scholar] [CrossRef][Green Version]
- Longden, T.A.; Nelson, M.T. Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirculation 2015, 22, 183–196. [Google Scholar] [CrossRef]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. CMLS 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Tang, J.; Zhou, X.; Xiang, S.; Zhu, X.; Li, N.; Shi, R.; Zhong, Y.; Zhang, L.; Sun, M.; et al. Roles of ion channels in regulation of acetylcholine-mediated vasoconstrictions in umbilical cords of rabbit/rats. Reprod. Toxicol. 2016, 65, 95–103. [Google Scholar] [CrossRef]
- Noma, A. ATP-regulated K+ channels in cardiac muscle. Nature 1983, 305, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hu, D.; Huang, C.; Nichols, C.G. Genetic Discovery of ATP-Sensitive K(+) Channels in Cardiovascular Diseases. Circulation. Arrhythmia Electrophysiol. 2019, 12, e007322. [Google Scholar] [CrossRef] [PubMed]
- Cole, W.C.; Clement-Chomienne, O. ATP-sensitive K+ channels of vascular smooth muscle cells. J. Cardiovasc. Electrophysiol. 2003, 14, 94–103. [Google Scholar] [CrossRef]
- Li, H.; Shin, S.E.; Seo, M.S.; An, J.R.; Ha, K.S.; Han, E.T.; Hong, S.H.; Kim, J.; Yim, M.J.; Lee, J.M.; et al. Alterations of ATP-sensitive K(+) channels in human umbilical arterial smooth muscle during gestational diabetes mellitus. Pflug. Arch. Eur. J. Physiol. 2018, 470, 1325–1333. [Google Scholar] [CrossRef]
- Shi, X.; Zhen, L.; Ding, H.; Chen, J.; Zhang, S.; Fu, Y. Role of ATP-sensitive potassium channels and inflammatory response of basilar artery smooth muscle cells in subarachnoid hemorrhage of rabbit and immune-modulation by shikonin. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2019, 134, 110804. [Google Scholar] [CrossRef]
- Szeto, V.; Chen, N.H.; Sun, H.S.; Feng, Z.P. The role of KATP channels in cerebral ischemic stroke and diabetes. Acta Pharmacol. Sin. 2018, 39, 683–694. [Google Scholar] [CrossRef]
- Miki, T.; Seino, S. Roles of KATP channels as metabolic sensors in acute metabolic changes. J. Mol. Cell Cardiol. 2005, 38, 917–925. [Google Scholar] [CrossRef]
- Seino, S. Physiology and pathophysiology of K(ATP) channels in the pancreas and cardiovascular system: A review. J. Diabetes Complic. 2003, 17, 2–5. [Google Scholar] [CrossRef]
- Brayden, J.E. Functional roles of KATP channels in vascular smooth muscle. Clin. Exp. Pharm. Physiol. 2002, 29, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Pipatpolkai, T.; Usher, S.; Stansfeld, P.J.; Ashcroft, F.M. New insights into KATP channel gene mutations and neonatal diabetes mellitus. Nat. Rev. Endocrinol. 2020, 16, 378–393. [Google Scholar] [CrossRef]
- Rubaiy, H.N. The therapeutic agents that target ATP-sensitive potassium channels. Acta Pharm. 2016, 66, 23–34. [Google Scholar] [CrossRef]
- Mohammed, R.; Provitera, L.; Cavallaro, G.; Lattuada, D.; Ercoli, G.; Mosca, F.; Villamor, E. Vasomotor effects of hydrogen sulfide in human umbilical vessels. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2017, 68, 737–747. [Google Scholar]
- Bai, X.J.; Tian, H.Y.; Wang, T.Z.; Du, Y.; Xi, Y.T.; Wu, Y.; Gao, J.; Ma, A.Q. Oleic acid inhibits the K(ATP) channel subunit Kir6.1 and the K(ATP) current in human umbilical artery smooth muscle cells. Am. J. Med. Sci. 2013, 346, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Honore, E.; Maingret, F.; Lesage, F.; Fink, M.; Duprat, F.; Lazdunski, M. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 1998, 17, 4283–4290. [Google Scholar] [CrossRef] [PubMed]
- Ketchum, K.A.; Joiner, W.J.; Sellers, A.J.; Kaczmarek, L.K.; Goldstein, S.A. A new family of outwardly rectifying potassium channel proteins with two pore domains in tandem. Nature 1995, 376, 690–695. [Google Scholar] [CrossRef]
- Hughes, S.; Foster, R.G.; Peirson, S.N.; Hankins, M.W. Expression and localisation of two-pore domain (K2P) background leak potassium ion channels in the mouse retina. Sci. Rep. 2017, 7, 46085. [Google Scholar] [CrossRef]
- Ben Soussia, I.; El Mouridi, S.; Kang, D.; Leclercq-Blondel, A.; Khoubza, L.; Tardy, P.; Zariohi, N.; Gendrel, M.; Lesage, F.; Kim, E.J.; et al. Mutation of a single residue promotes gating of vertebrate and invertebrate two-pore domain potassium channels. Nat. Commun. 2019, 10, 787. [Google Scholar] [CrossRef]
- Wiedmann, F.; Rinne, S.; Donner, B.; Decher, N.; Katus, H.A.; Schmidt, C. Mechanosensitive TREK-1 two-pore-domain potassium (K2P) channels in the cardiovascular system. Prog. Biophys. Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Lesage, F.; Lazdunski, M. Molecular and functional properties of two-pore-domain potassium channels. Am. J. Physiology. Ren. Physiol. 2000, 279, F793–F801. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.J.; Honoré, E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci. 2001, 24, 339–346. [Google Scholar] [CrossRef]
- Renigunta, V.; Schlichthorl, G.; Daut, J. Much more than a leak: Structure and function of K(2)p-channels. Pflug. Arch. Eur. J. Physiol. 2015, 467, 867–894. [Google Scholar] [CrossRef]
- Lotshaw, D.P. Biophysical, pharmacological, and functional characteristics of cloned and native mammalian two-pore domain K+ channels. Cell Biochem. Biophys. 2007, 47, 209–256. [Google Scholar] [CrossRef]
- Kim, D. Physiology and pharmacology of two-pore domain potassium channels. Curr. Pharm. Des. 2005, 11, 2717–2736. [Google Scholar] [CrossRef]
- Lesage, F. Pharmacology of neuronal background potassium channels. Neuropharmacology 2003, 44, 1–7. [Google Scholar] [CrossRef]
- Goldstein, S.A.; Bockenhauer, D.; O’Kelly, I.; Zilberberg, N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat. Rev. Neurosci. 2001, 2, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Pathan, A.R.; Rusch, N.J. Two-pore domain K(+) channels: Evidence for TWIK-2 in blood pressure regulation. Hypertension 2011, 58, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Ledoux, J. K+ channels in biological processes: Vascular K+ channels in the regulation of blood pressure. J. Recept. Ligand Channel Res. 2014, 51. [Google Scholar] [CrossRef]
- Pandit, L.M.; Lloyd, E.E.; Reynolds, J.O.; Lawrence, W.S.; Reynolds, C.; Wehrens, X.H.; Bryan, R.M. TWIK-2 channel deficiency leads to pulmonary hypertension through a rho-kinase-mediated process. Hypertension 2014, 64, 1260–1265. [Google Scholar] [CrossRef]
- Lloyd, E.E.; Crossland, R.F.; Phillips, S.C.; Marrelli, S.P.; Reddy, A.K.; Taffet, G.E.; Hartley, C.J.; Bryan, R.M., Jr. Disruption of K(2P)6.1 produces vascular dysfunction and hypertension in mice. Hypertension 2011, 58, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Gurney, A.M.; Osipenko, O.N.; MacMillan, D.; McFarlane, K.M.; Tate, R.J.; Kempsill, F.E. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ. Res. 2003, 93, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Gardener, M.J.; Johnson, I.T.; Burnham, M.P.; Edwards, G.; Heagerty, A.M.; Weston, A.H. Functional evidence of a role for two-pore domain potassium channels in rat mesenteric and pulmonary arteries. Br. J. Pharmacol. 2004, 142, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, A.; Veale, E.L.; Nagy, B.M.; Nagaraj, C.; Kwapiszewska, G.; Antigny, F.; Lambert, M.; Humbert, M.; Czirjak, G.; Enyedi, P. TASK-1 (KCNK3) channels in the lung: From cell biology to clinical implications. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- Antigny, F.; Hautefort, A.; Meloche, J.; Belacel-Ouari, M.; Manoury, B.; Rucker-Martin, C.; Pechoux, C.; Potus, F.; Nadeau, V.; Tremblay, E.; et al. Potassium Channel Subfamily K Member 3 (KCNK3) Contributes to the Development of Pulmonary Arterial Hypertension. Circulation 2016, 133, 1371–1385. [Google Scholar] [CrossRef]
- Feliciangeli, S.; Chatelain, F.C.; Bichet, D.; Lesage, F. The family of K2P channels: Salient structural and functional properties. J. Physiol. 2015, 593, 2587–2603. [Google Scholar] [CrossRef] [PubMed]
- Chemin, J.; Patel, A.; Duprat, F.; Zanzouri, M.; Lazdunski, M.; Honore, E. Lysophosphatidic acid-operated K+ channels. J. Biol. Chem. 2005, 280, 4415–4421. [Google Scholar] [CrossRef] [PubMed]
- Mathie, A. Neuronal two-pore-domain potassium channels and their regulation by G protein-coupled receptors. J. Physiol. 2007, 578, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Rinne, S.; Renigunta, V.; Schlichthorl, G.; Zuzarte, M.; Bittner, S.; Meuth, S.G.; Decher, N.; Daut, J.; Preisig-Muller, R. A splice variant of the two-pore domain potassium channel TREK-1 with only one pore domain reduces the surface expression of full-length TREK-1 channels. Pflug. Arch. Eur. J. Physiol. 2014, 466, 1559–1570. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kang, D.; Kim, D. Functional properties of four splice variants of a human pancreatic tandem-pore K+ channel, TALK-1. Am. J. Physiology. Cell Physiol. 2003, 285, C529–C538. [Google Scholar] [CrossRef]
- Gu, W.; Schlichthorl, G.; Hirsch, J.R.; Engels, H.; Karschin, C.; Karschin, A.; Derst, C.; Steinlein, O.K.; Daut, J. Expression pattern and functional characteristics of two novel splice variants of the two-pore-domain potassium channel TREK-2. J. Physiol. 2002, 539, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Xian Tao, L.; Dyachenko, V.; Zuzarte, M.; Putzke, C.; Preisig-Muller, R.; Isenberg, G.; Daut, J. The stretch-activated potassium channel TREK-1 in rat cardiac ventricular muscle. Cardiovasc Res. 2006, 69, 86–97. [Google Scholar] [CrossRef]
- Thomas, D.; Plant, L.D.; Wilkens, C.M.; McCrossan, Z.A.; Goldstein, S.A. Alternative translation initiation in rat brain yields K2P2.1 potassium channels permeable to sodium. Neuron 2008, 58, 859–870. [Google Scholar] [CrossRef]
- Levitz, J.; Royal, P.; Comoglio, Y.; Wdziekonski, B.; Schaub, S.; Clemens, D.M.; Isacoff, E.Y.; Sandoz, G. Heterodimerization within the TREK channel subfamily produces a diverse family of highly regulated potassium channels. Proc. Natl. Acad. Sci. USA 2016, 113, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Czirjak, G.; Enyedi, P. Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J. Biol. Chem. 2002, 277, 5426–5432. [Google Scholar] [CrossRef]
- Hwang, E.M.; Kim, E.; Yarishkin, O.; Woo, D.H.; Han, K.S.; Park, N.; Bae, Y.; Woo, J.; Kim, D.; Park, M.; et al. A disulphide-linked heterodimer of TWIK-1 and TREK-1 mediates passive conductance in astrocytes. Nat. Commun. 2014, 5, 3227. [Google Scholar] [CrossRef]
- Wareing, M.; Bai, X.; Seghier, F.; Turner, C.M.; Greenwood, S.L.; Baker, P.N.; Taggart, M.J.; Fyfe, G.K. Expression and function of potassium channels in the human placental vasculature. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R437–R446. [Google Scholar] [CrossRef]
- Naderi, S.; Tsai, S.A.; Khandelwal, A. Hypertensive Disorders of Pregnancy. Curr. Atheroscler Rep. 2017, 19, 15. [Google Scholar] [CrossRef]
- Folk, D.M. Hypertensive Disorders of Pregnancy: Overview and Current Recommendations. J. Midwifery Womens Health 2018, 63, 289–300. [Google Scholar] [CrossRef]
- Fox, R.; Kitt, J.; Leeson, P.; Aye, C.Y.L.; Lewandowski, A.J. Preeclampsia: Risk Factors, Diagnosis, Management, and the Cardiovascular Impact on the Offspring. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef]
- Peres, G.M.; Mariana, M.; Cairrao, E. Pre-Eclampsia and Eclampsia: An Update on the Pharmacological Treatment Applied in Portugal. J. Cardiovasc. Dev. Dis. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.H. Molecular determinants of voltage-gated potassium currents in vascular smooth muscle. Cell Biochem. Biophys. 2005, 42, 167–195. [Google Scholar] [CrossRef]
- Cox, R.H.; Folander, K.; Swanson, R. Differential Expression of Voltage-Gated K+ Channel Genes in Arteries From Spontaneously Hypertensive and Wistar-Kyoto Rats. Hypertension 2001, 37, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.H. Changes in the expression and function of arterial potassium channels during hypertension. Vasc. Pharm. 2002, 38, 13–23. [Google Scholar] [CrossRef]
- Pinterova, M.; Kunes, J.; Zicha, J. Altered neural and vascular mechanisms in hypertension. Physiol Res. 2011, 60, 381–402. [Google Scholar] [CrossRef]
- Djokic, V.; Jankovic-Raznatovic, S.; Novakovic, R.; Kostic, M.; Rajkovic, J.; Labudovic-Borovic, M.; Rakocevic, J.; Stanisic, J.; Djuric, M.; Gojkovic-Bukarica, L. Effect of gestational diabetes mellitus and pregnancy-induced hypertension on human umbilical vein smooth muscle KATP channels. Exp. Mol. Pathol. 2019, 111, 104323. [Google Scholar] [CrossRef]
- Djokic, V.; Jankovic, S.; Labudovic-Borovic, M.; Rakocevic, J.; Stanisic, J.; Rajkovic, J.; Novakovic, R.; Kostic, M.; Djuric, M.; Gostimirovic, M.; et al. Pregnancy-induced hypertension decreases Kv1.3 potassium channel expression and function in human umbilical vein smooth muscle. Eur. J. Pharm. 2020, 173281. [Google Scholar] [CrossRef]
- Wareing, M.; Greenwood, S.L.; Fyfe, G.K.; Baker, P.N. Reactivity of human placental chorionic plate vessels from pregnancies complicated by intrauterine growth restriction (IUGR). Biol. Reprod. 2006, 75, 518–523. [Google Scholar] [CrossRef]
- Wareing, M. Oxygen sensitivity, potassium channels, and regulation of placental vascular tone. Microcirculation 2014, 21, 58–66. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, Y.; Yin, B.; Wen, J.; Cheng, J.; Fu, X. The expression and function of KCNQ potassium channels in human chorionic plate arteries from women with normal pregnancies and pre-eclampsia. PLoS ONE 2018, 13, e0192122. [Google Scholar] [CrossRef]
- Mills, T.A.; Greenwood, S.L.; Devlin, G.; Shweikh, Y.; Robinson, M.; Cowley, E.; Hayward, C.E.; Cottrell, E.C.; Tropea, T.; Brereton, M.F. Activation of KV7 channels stimulates vasodilatation of human placental chorionic plate arteries. Placenta 2015, 36, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.D.; McCallum, L.A.; Kurlak, L.O.; Greenwood, I.A.; Broughton Pipkin, F.; Tribe, R.M. Novel expression and regulation of voltage-dependent potassium channels in placentas from women with preeclampsia. Hypertension 2011, 58, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Manville, R.W.; Abbott, G.W. Isoform-Selective KCNA1 Potassium Channel Openers Built from Glycine. J. Pharm. Exp. 2020, 373, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Jepps, T.A.; Olesen, S.P.; Greenwood, I.A. One man’s side effect is another man’s therapeutic opportunity: Targeting Kv7 channels in smooth muscle disorders. Br. J. Pharmacol. 2013, 168, 19–27. [Google Scholar] [CrossRef]
- Lawson, K.; McKay, N.G. Modulation of potassium channels as a therapeutic approach. Curr. Pharm. Des. 2006, 12, 459–470. [Google Scholar] [CrossRef]
- Bartok, A.; Toth, A.; Somodi, S.; Szanto, T.G.; Hajdu, P.; Panyi, G.; Varga, Z. Margatoxin is a non-selective inhibitor of human Kv1.3 K+ channels. Toxicon Off. J. Int. Soc. Toxinol. 2014, 87, 6–16. [Google Scholar] [CrossRef]
- Khammy, M.M.; Kim, S.; Bentzen, B.H.; Lee, S.; Choi, I.; Aalkjaer, C.; Jepps, T.A. 4-Aminopyridine: A pan voltage-gated potassium channel inhibitor that enhances Kv 7.4 currents and inhibits noradrenaline-mediated contraction of rat mesenteric small arteries. Br. J. Pharmacol. 2018, 175, 501–516. [Google Scholar] [CrossRef]
- Lindquist, S.; Stangel, M. Update on treatment options for Lambert-Eaton myasthenic syndrome: Focus on use of amifampridine. Neuropsychiatr. Dis. Treat. 2011, 7, 341–349. [Google Scholar] [CrossRef]
- Moore, J.C.; Trager, L.; Anzia, L.E.; Saliba, W.; Bassiouny, M.; Bhargava, M.; Chung, M.; Desai, M.; Garberich, R.; Lever, H.; et al. Dofetilide for suppression of atrial fibrillation in hypertrophic cardiomyopathy: A case series and literature review. Pacing Clin. Electrophysiol. Pace 2018, 41, 396–401. [Google Scholar] [CrossRef]
- Colunga Biancatelli, R.M.; Congedo, V.; Calvosa, L.; Ciacciarelli, M.; Polidoro, A.; Iuliano, L. Adverse reactions of Amiodarone. J. Geriatr. Cardiol. JGC 2019, 16, 552–566. [Google Scholar] [CrossRef]
- Faraci, F.M.; Heistad, D.D. Regulation of the cerebral circulation: Role of endothelium and potassium channels. Physiol. Rev. 1998, 78, 53–97. [Google Scholar] [CrossRef] [PubMed]
- Rusch, N.J.; Liu, Y. Potassium channels in hypertension: Homeostatic pathways to buffer arterial contraction. J. Lab. Clin. Med. 1997, 130, 245–251. [Google Scholar] [CrossRef]
- Wang, R.; Wu, L. The chemical modification of KCa channels by carbon monoxide in vascular smooth muscle cells. J. Biol. Chem. 1997, 272, 8222–8226. [Google Scholar] [CrossRef] [PubMed]
- Amberg, G.C.; Bonev, A.D.; Rossow, C.F.; Nelson, M.T.; Santana, L.F. Modulation of the molecular composition of large conductance, Ca2+ activated K+ channels in vascular smooth muscle during hypertension. J. Clin. Investig. 2003, 112, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Amberg, G.C.; Santana, L.F. Downregulation of the BK channel beta1 subunit in genetic hypertension. Circ. Res. 2003, 93, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Leo, M.D.; Jaggar, J.H. Endothelin-1 Stimulates Vasoconstriction Through Rab11A Serine 177 Phosphorylation. Circ. Res. 2017, 121, 650–661. [Google Scholar] [CrossRef]
- Leo, M.D.; Zhai, X.; Yin, W.; Jaggar, J.H. Impaired Trafficking of beta1 Subunits Inhibits BK Channels in Cerebral Arteries of Hypertensive Rats. Hypertension 2018, 72, 765–775. [Google Scholar] [CrossRef]
- Joseph, B.K.; Thakali, K.M.; Moore, C.L.; Rhee, S.W. Ion channel remodeling in vascular smooth muscle during hypertension: Implications for novel therapeutic approaches. Pharmacol. Res. 2013, 70, 126–138. [Google Scholar] [CrossRef]
- He, M.; Li, F.; Yang, M.; Fan, Y.; Beejadhursing, R.; Xie, Y.; Zhou, Y.; Deng, D. Impairment of BKca channels in human placental chorionic plate arteries is potentially relevant to the development of preeclampsia. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2018, 41, 126–134. [Google Scholar] [CrossRef]
- Li, F.F.; He, M.Z.; Xie, Y.; Wu, Y.Y.; Yang, M.T.; Fan, Y.; Qiao, F.Y.; Deng, D.R. Involvement of dysregulated IKCa and SKCa channels in preeclampsia. Placenta 2017, 58, 9–16. [Google Scholar] [CrossRef]
- Bentzen, B.H.; Olesen, S.P.; Ronn, L.C.; Grunnet, M. BK channel activators and their therapeutic perspectives. Front. Physiol. 2014, 5, 389. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Wang, M.; Patel, P.N.; Kalogeris, T.; Liu, Y.; Durante, W.; Korthuis, R.J. Preconditioning with the BKCa channel activator NS-1619 prevents ischemia-reperfusion-induced inflammation and mucosal barrier dysfunction: Roles for ROS and heme oxygenase-1. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H988–H999. [Google Scholar] [CrossRef] [PubMed]
- Olver, T.D.; Edwards, J.C.; Ferguson, B.S.; Hiemstra, J.A.; Thorne, P.K.; Hill, M.A.; Laughlin, M.H.; Emter, C.A. Chronic interval exercise training prevents BKCa channel-mediated coronary vascular dysfunction in aortic-banded miniswine. J. Appl. Physiol. 2018, 125, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Tajada, S.; Cidad, P.; Moreno-Dominguez, A.; Perez-Garcia, M.T.; Lopez-Lopez, J.R. High blood pressure associates with the remodelling of inward rectifier K+ channels in mice mesenteric vascular smooth muscle cells. J. Physiol. 2012, 590, 6075–6091. [Google Scholar] [CrossRef]
- Caballero, R.; Dolz-Gaiton, P.; Gomez, R.; Amoros, I.; Barana, A.; Gonzalez de la Fuente, M.; Osuna, L.; Duarte, J.; Lopez-Izquierdo, A.; Moraleda, I.; et al. Flecainide increases Kir2.1 currents by interacting with cysteine 311, decreasing the polyamine-induced rectification. Proc. Natl. Acad. Sci. USA 2010, 107, 15631–15636. [Google Scholar] [CrossRef]
- Walsh, K.B. Screening Technologies for Inward Rectifier Potassium Channels: Discovery of New Blockers and Activators. Slas Discov. Adv. Life Sci. R D 2020, 25, 420–433. [Google Scholar] [CrossRef]
- Rodriguez-Menchaca, A.A.; Navarro-Polanco, R.A.; Ferrer-Villada, T.; Rupp, J.; Sachse, F.B.; Tristani-Firouzi, M.; Sanchez-Chapula, J.A. The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc. Natl. Acad. Sci. USA 2008, 105, 1364–1368. [Google Scholar] [CrossRef]
- Sánchez-Chapula, J.; Salinas-Stefanon, E.; Torres-Jácome, J.; Benavides-Haro, D.; Navarro-Polanco, R. Blockade of currents by the antimalarial drug chloroquine in feline ventricular myocytes. J. Pharmacol. Exp. Ther. 2001, 297, 437–445. [Google Scholar]
- Takanari, H.; Nalos, L.; Stary-Weinzinger, A.; de Git, K.C.; Varkevisser, R.; Linder, T.; Houtman, M.J.; Peschar, M.; de Boer, T.P.; Tidwell, R.R.; et al. Efficient and specific cardiac IK(1) inhibition by a new pentamidine analogue. Cardiovasc Res. 2013, 99, 203–214. [Google Scholar] [CrossRef]
- Wang, H.R.; Wu, M.; Yu, H.; Long, S.; Stevens, A.; Engers, D.W.; Sackin, H.; Daniels, J.S.; Dawson, E.S.; Hopkins, C.R.; et al. Selective inhibition of the K(ir)2 family of inward rectifier potassium channels by a small molecule probe: The discovery, SAR, and pharmacological characterization of ML133. ACS Chem. Biol. 2011, 6, 845–856. [Google Scholar] [CrossRef]
- Van de Voorde, J.; Vanheel, B.; Leusen, I. Endothelium-dependent relaxation and hyperpolarization in aorta from control and renal hypertensive rats. Circ. Res. 1992, 70, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kitazono, T.; Heistad, D.D.; Faraci, F.M. ATP-sensitive potassium channels in the basilar artery during chronic hypertension. Hypertension 1993, 22, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Kam, K.L.; Pfaffendorf, M.; van Zwieten, P.A. Drug-induced endothelium-dependent and -independent relaxations in isolated resistance vessels taken from simultaneously hypertensive and streptozotocin-diabetic rats. Blood Press. 1994, 3, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Hanna, S.T.; Wang, R.; McNeill, J.R. Altered vascular reactivity and KATP channel currents in vascular smooth muscle cells from deoxycorticosterone acetate (DOCA)-salt hypertensive rats. J. Cardiovasc. Pharmacol. 2004, 44, 525–531. [Google Scholar] [CrossRef]
- Kalliovalkama, J. Arterial function in nitric oxide-deficient hypertension: Influence of long-term angiotensin II receptor antagonism. Cardiovasc. Res. 1999, 42, 773–782. [Google Scholar] [CrossRef]
- Takaba, H.; Nagao, T.; Ibayashi, S.; Kitazono, T.; Fujii, K.; Fujishima, M. Altered cerebrovascular response to a potassium channel opener in hypertensive rats. Hypertension 1996, 28, 143–146. [Google Scholar] [CrossRef]
- Kawata, T.; Mimuro, T.; Onuki, T.; Tsuchiya, K.; Nihei, H.; Koike, T. The K(ATP) channel opener nicorandil: Effect on renal hemodynamics in spontaneously hypertensive and Wistar Kyoto rats. Kidney Int. Suppl. 1998, 67, S231–S233. [Google Scholar] [CrossRef]
- Gutterman, D.D.; Miura, H.; Liu, Y. Redox modulation of vascular tone: Focus of potassium channel mechanisms of dilation. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 671–678. [Google Scholar] [CrossRef]
- Bisseling, T.M.; Versteegen, M.G.; van der Wal, S.; Copius Peereboom-Stegeman, J.J.; Borggreven, J.M.; Steegers, E.A.; van der Laak, J.A.; Russel, F.G.; Smits, P. Impaired KATP channel function in the fetoplacental circulation of patients with type 1 diabetes mellitus. Am. J. Obstet. Gynecol. 2005, 192, 973–979. [Google Scholar] [CrossRef]
- Taricco, E.; Radaelli, T.; Rossi, G.; Nobile de Santis, M.S.; Bulfamante, G.P.; Avagliano, L.; Cetin, I. Effects of gestational diabetes on fetal oxygen and glucose levels in vivo. BJOG Int. J. Obstet. Gynaecol. 2009, 116, 1729–1735. [Google Scholar] [CrossRef]
- Toljic, M.; Egic, A.; Munjas, J.; Karadzov Orlic, N.; Milovanovic, Z.; Radenkovic, A.; Vuceljic, J.; Joksic, I. Increased oxidative stress and cytokinesis-block micronucleus cytome assay parameters in pregnant women with gestational diabetes mellitus and gestational arterial hypertension. Reprod. Toxicol. 2017, 71, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Tang, P.L.; Liu, P.Y.; Huang, W.C.; Chen, Y.Y.; Wang, H.P.; Chang, J.T.; Lin, L.T. Maternal pregnancy-induced hypertension increases subsequent neonatal necrotizing enterocolitis risk: A nationwide population-based retrospective cohort study in Taiwan. Medicine 2018, 97, e11739. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.M.; Braunstein, T.H.; Holstein-Rathlou, N.H.; Salomonsson, M. Role of vascular potassium channels in the regulation of renal hemodynamics. Am. J. Physiol. Ren. Physiol. 2012, 302, F505–F518. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Xiao, D.; Zhang, L. Potassium channels and uterine vascular adaptation to pregnancy and chronic hypoxia. Curr. Vasc. Pharmacol. 2013, 11, 737–747. [Google Scholar] [CrossRef]
- Li, A.; Knutsen, R.H.; Zhang, H.; Osei-Owusu, P.; Moreno-Dominguez, A.; Harter, T.M.; Uchida, K.; Remedi, M.S.; Dietrich, H.H.; Bernal-Mizrachi, C.; et al. Hypotension due to Kir6.1 gain-of-function in vascular smooth muscle. J. Am. Heart Assoc. 2013, 2, e000365. [Google Scholar] [CrossRef]
- Liu, X.; Duan, P.; Hu, X.; Li, R.; Zhu, Q. Altered KATP Channel Subunits Expression and Vascular Reactivity in Spontaneously Hypertensive Rats With Age. J. Cardiovasc. Pharmacol. 2016, 68, 143–149. [Google Scholar] [CrossRef]
- Ohya, Y.; Setoguchi, M.; Fujii, K.; Nagao, T.; Abe, I.; Fujishima, M. Impaired action of levcromakalim on ATP-sensitive K+ channels in mesenteric artery cells from spontaneously hypertensive rats. Hypertension 1996, 27, 1234–1239. [Google Scholar] [CrossRef]
- Du, Q.; Jovanovic, S.; Tulic, L.; Tulic, I.; Jovanovic, A. Pregnancy-induced hypertension is associated with down-regulation of Kir6.1 in human myometrium. Pregnancy Hypertens. 2019, 18, 96–98. [Google Scholar] [CrossRef]
- Blanco-Rivero, J.; Gamallo, C.; Aras-Lopez, R.; Cobeno, L.; Cogolludo, A.; Perez-Vizcaino, F.; Ferrer, M.; Balfagon, G. Decreased expression of aortic KIR6.1 and SUR2B in hypertension does not correlate with changes in the functional role of K(ATP) channels. Eur J. Pharm. 2008, 587, 204–208. [Google Scholar] [CrossRef]
- Thorne, G.D.; Conforti, L.; Paul, R.J. Hypoxic vasorelaxation inhibition by organ culture correlates with loss of Kv channels but not Ca(2+) channels. Am. J. Physiology. Heart Circ. Physiol. 2002, 283, H247–H253. [Google Scholar] [CrossRef]
- Khan, R.N.; Morrison, J.J.; Smith, S.K.; Ashford, M.L.J. Activation of large-conductance potassium channels in pregnant human myometrium by pinacidil. Am. J. Obstet. Gynecol. 1998, 178, 1027–1034. [Google Scholar] [CrossRef]
- Novakovic, A.; Pavlovic, M.; Milojevic, P.; Stojanovic, I.; Nenezic, D.; Jovic, M.; Ugresic, N.; Kanjuh, V.; Yang, Q.; He, G.W. Different potassium channels are involved in relaxation of rat renal artery induced by P1075. Basic Clin. Pharmacol. Toxicol. 2012, 111, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Stockbridge, N.; Zhang, H.; Weir, B. Effects of K+ channel agonists cromakalim and pinacidil on rat basilar artery smooth muscle cells are mediated by Ca++ activated K+ channels. Biochem. Biophys. Res. Commun. 1991, 181, 172–178. [Google Scholar] [CrossRef]
- Kolias, T.J.; Chai, S.; Webb, R.C. Potassium channel antagonists and vascular reactivity in stroke-prone spontaneously hypertensive rats. Am. J. Hypertens. 1993, 6, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Hutri-Kahonen, N.; Kahonen, M.; Wu, X.; Sand, J.; Nordback, I.; Taurio, J.; Porsti, I. Control of vascular tone in isolated mesenteric arterial segments from hypertensive patients. Br. J. Pharmacol. 1999, 127, 1735–1743. [Google Scholar] [CrossRef]
- Miyata, N.; Tsuchida, K.; Otomo, S. Functional changes in potassium channels in carotid arteries from stroke-prone spontaneously hypertensive rats. Eur. J. Pharmacol. 1990, 182, 209–210. [Google Scholar] [CrossRef]
- Furspan, P.B.; Webb, R.C. Decreased ATP sensitivity of a K+ channel and enhanced vascular smooth muscle relaxation in genetically hypertensive rats. J. Hypertens. 1993, 11, 1067–1072. [Google Scholar] [CrossRef]
- Jahangir, A.; Terzic, A. K(ATP) channel therapeutics at the bedside. J. Mol. Cell Cardiol. 2005, 39, 99–112. [Google Scholar] [CrossRef]
- Roy Chowdhury, U.; Dosa, P.I.; Fautsch, M.P. ATP sensitive potassium channel openers: A new class of ocular hypotensive agents. Exp. Eye Res. 2017, 158, 85–93. [Google Scholar] [CrossRef]
- Wiedmann, F.; Schmidt, C.; Lugenbiel, P.; Staudacher, I.; Rahm, A.K.; Seyler, C.; Schweizer, P.A.; Katus, H.A.; Thomas, D. Therapeutic targeting of two-pore-domain potassium (K(2P)) channels in the cardiovascular system. Clin. Sci. 2016, 130, 643–650. [Google Scholar] [CrossRef]
- Abraham, D.M.; Lee, T.E.; Watson, L.J.; Mao, L.; Chandok, G.; Wang, H.G.; Frangakis, S.; Pitt, G.S.; Shah, S.H.; Wolf, M.J. The two-pore domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction. J. Clin. Investig. 2018, 128, 4843–4855. [Google Scholar] [CrossRef] [PubMed]
- Lamas, J.A.; Fernandez-Fernandez, D. Tandem pore TWIK-related potassium channels and neuroprotection. Neural Regen. Res. 2019, 14, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Loucif, A.J.C.; Saintot, P.P.; Liu, J.; Antonio, B.M.; Zellmer, S.G.; Yoger, K.; Veale, E.L.; Wilbrey, A.; Omoto, K.; Cao, L. GI-530159, a novel, selective, mechanosensitive two-pore-domain potassium (K2P ) channel opener, reduces rat dorsal root ganglion neuron excitability. Br. J. Pharmacol. 2018, 175, 2272–2283. [Google Scholar] [CrossRef] [PubMed]
- Veale, E.L.; Al-Moubarak, E.; Bajaria, N.; Omoto, K.; Cao, L.; Tucker, S.J.; Stevens, E.B.; Mathie, A. Influence of the N terminus on the biophysical properties and pharmacology of TREK1 potassium channels. Mol. Pharmacol. 2014, 85, 671–681. [Google Scholar] [CrossRef]
- Bagriantsev, S.N.; Ang, K.H.; Gallardo-Godoy, A.; Clark, K.A.; Arkin, M.R.; Renslo, A.R.; Minor, D.L., Jr. A high-throughput functional screen identifies small molecule regulators of temperature- and mechano-sensitive K2P channels. ACS Chem. Biol. 2013, 8, 1841–1851. [Google Scholar] [CrossRef]
- Cunningham, K.; Veale, E.; Clapp, L.; Mathie, A. The Role of the K2P Channels TASK-1, TREK-1 and TREK-2 in the Use of Treprostinil Therapy in Pulmonary Arterial Hypertension. FASEB J. 2018, 32. [Google Scholar] [CrossRef]
- Mathie, A.; Veale, E.L.; Cunningham, K.P.; Holden, R.G.; Wright, P.D. Two-Pore Domain Potassium Channels as Drug Targets: Anesthesia and Beyond. Annu. Rev. Pharmacol. Toxicol. 2020, 61. [Google Scholar] [CrossRef]
- Mazella, J.; Petrault, O.; Lucas, G.; Deval, E.; Beraud-Dufour, S.; Gandin, C.; El-Yacoubi, M.; Widmann, C.; Guyon, A.; Chevet, E. Spadin, a sortilin-derived peptide, targeting rodent TREK-1 channels: A new concept in the antidepressant drug design. Plos Biol. 2010, 8, e1000355. [Google Scholar] [CrossRef]
- Takahira, M.; Sakurai, M.; Sakurada, N.; Sugiyama, K. Fenamates and diltiazem modulate lipid-sensitive mechano-gated 2P domain K(+) channels. Pflug. Arch. Eur. J. Physiol. 2005, 451, 474–478. [Google Scholar] [CrossRef]
- Bodnar, M.; Schlichthorl, G.; Daut, J. The potassium current carried by TREK-1 channels in rat cardiac ventricular muscle. Pflug. Arch. Eur. J. Physiol. 2015, 467, 1069–1079. [Google Scholar] [CrossRef]
- Fink, M.; Lesage, F.; Duprat, F.; Heurteaux, C.; Reyes, R.; Fosset, M.; Lazdunski, M. A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J. 1998, 17, 3297–3308. [Google Scholar] [CrossRef] [PubMed]
- Meadows, H.J.; Chapman, C.G.; Duckworth, D.M.; Kelsell, R.E.; Murdock, P.R.; Nasir, S.; Rennie, G.; Randall, A.D. The neuroprotective agent sipatrigine (BW619C89) potently inhibits the human tandem pore-domain K+ channels TREK-1 and TRAAK. Brain Res. 2001, 892, 94–101. [Google Scholar] [CrossRef]
- Thummler, S.; Duprat, F.; Lazdunski, M. Antipsychotics inhibit TREK but not TRAAK channels. Biochem. Biophys. Res. Commun. 2007, 354, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Lee, D.K.; Hong, S.G.; Han, J.; Kang, D. Activation of TREK-1, but Not TREK-2, Channel by Mood Stabilizers. Int. J. Mol. Sci. 2017, 18, 2460. [Google Scholar] [CrossRef]
- Maingret, F.; Patel, A.J.; Lesage, F.; Lazdunski, M.; Honore, E. Lysophospholipids open the two-pore domain mechano-gated K(+) channels TREK-1 and TRAAK. J. Biol. Chem. 2000, 275, 10128–10133. [Google Scholar] [CrossRef]

 —G-protein coupled receptor;
—G-protein coupled receptor;  —peptide receptor;
—peptide receptor;  /green arrows—stimulation;
/green arrows—stimulation;  /red arrows—inhibition; ?—unknown mechanism; [Ca2+]I—intracellular Ca2+ concentration; BKCa—large-conductance Ca2+-activated K+ channels; Ca2+—calcium; cAMP—cyclic adenosine monophosphate; cGMP—cyclic guanosine monophosphate; eNOS—endothelial nitric oxide synthase; K+—potassium; Kv—voltage-gated K+ channels; NO—nitric oxide; PDE—phosphodiesterase; PKA—protein kinase A; PKG—protein kinase G; SKCa—small-conductance Ca2+-activated K+ channels; VGCC—voltage-gated Ca2+ channels.
—G-protein coupled receptor; —peptide receptor; /green arrows—stimulation; /red arrows—inhibition; ?—unknown mechanism; [Ca2+]I—intracellular Ca2+ concentration; BKCa—large-conductance Ca2+-activated K+ channels; Ca2+—calcium; cAMP—cyclic adenosine monophosphate; cGMP—cyclic guanosine monophosphate; eNOS—endothelial nitric oxide synthase; K+—potassium; Kv—voltage-gated K+ channels; NO—nitric oxide; PDE—phosphodiesterase; PKA—protein kinase A; PKG—protein kinase G; SKCa—small-conductance Ca2+-activated K+ channels; VGCC—voltage-gated Ca2+ channels.
/red arrows—inhibition; ?—unknown mechanism; [Ca2+]I—intracellular Ca2+ concentration; BKCa—large-conductance Ca2+-activated K+ channels; Ca2+—calcium; cAMP—cyclic adenosine monophosphate; cGMP—cyclic guanosine monophosphate; eNOS—endothelial nitric oxide synthase; K+—potassium; Kv—voltage-gated K+ channels; NO—nitric oxide; PDE—phosphodiesterase; PKA—protein kinase A; PKG—protein kinase G; SKCa—small-conductance Ca2+-activated K+ channels; VGCC—voltage-gated Ca2+ channels.
—G-protein coupled receptor; —peptide receptor; /green arrows—stimulation; /red arrows—inhibition; ?—unknown mechanism; [Ca2+]I—intracellular Ca2+ concentration; BKCa—large-conductance Ca2+-activated K+ channels; Ca2+—calcium; cAMP—cyclic adenosine monophosphate; cGMP—cyclic guanosine monophosphate; eNOS—endothelial nitric oxide synthase; K+—potassium; Kv—voltage-gated K+ channels; NO—nitric oxide; PDE—phosphodiesterase; PKA—protein kinase A; PKG—protein kinase G; SKCa—small-conductance Ca2+-activated K+ channels; VGCC—voltage-gated Ca2+ channels.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorigo, M.; Oliveira, N.; Cairrao, E. Clinical Importance of the Human Umbilical Artery Potassium Channels. Cells 2020, 9, 1956. https://doi.org/10.3390/cells9091956
Lorigo M, Oliveira N, Cairrao E. Clinical Importance of the Human Umbilical Artery Potassium Channels. Cells. 2020; 9(9):1956. https://doi.org/10.3390/cells9091956
Chicago/Turabian StyleLorigo, Margarida, Nelson Oliveira, and Elisa Cairrao. 2020. "Clinical Importance of the Human Umbilical Artery Potassium Channels" Cells 9, no. 9: 1956. https://doi.org/10.3390/cells9091956
APA StyleLorigo, M., Oliveira, N., & Cairrao, E. (2020). Clinical Importance of the Human Umbilical Artery Potassium Channels. Cells, 9(9), 1956. https://doi.org/10.3390/cells9091956