Detecting Interactive Gene Groups for Single-Cell RNA-Seq Data Based on Co-Expression Network Analysis and Subgraph Learning



Abstract
1. Introduction
2. Materials and Methods
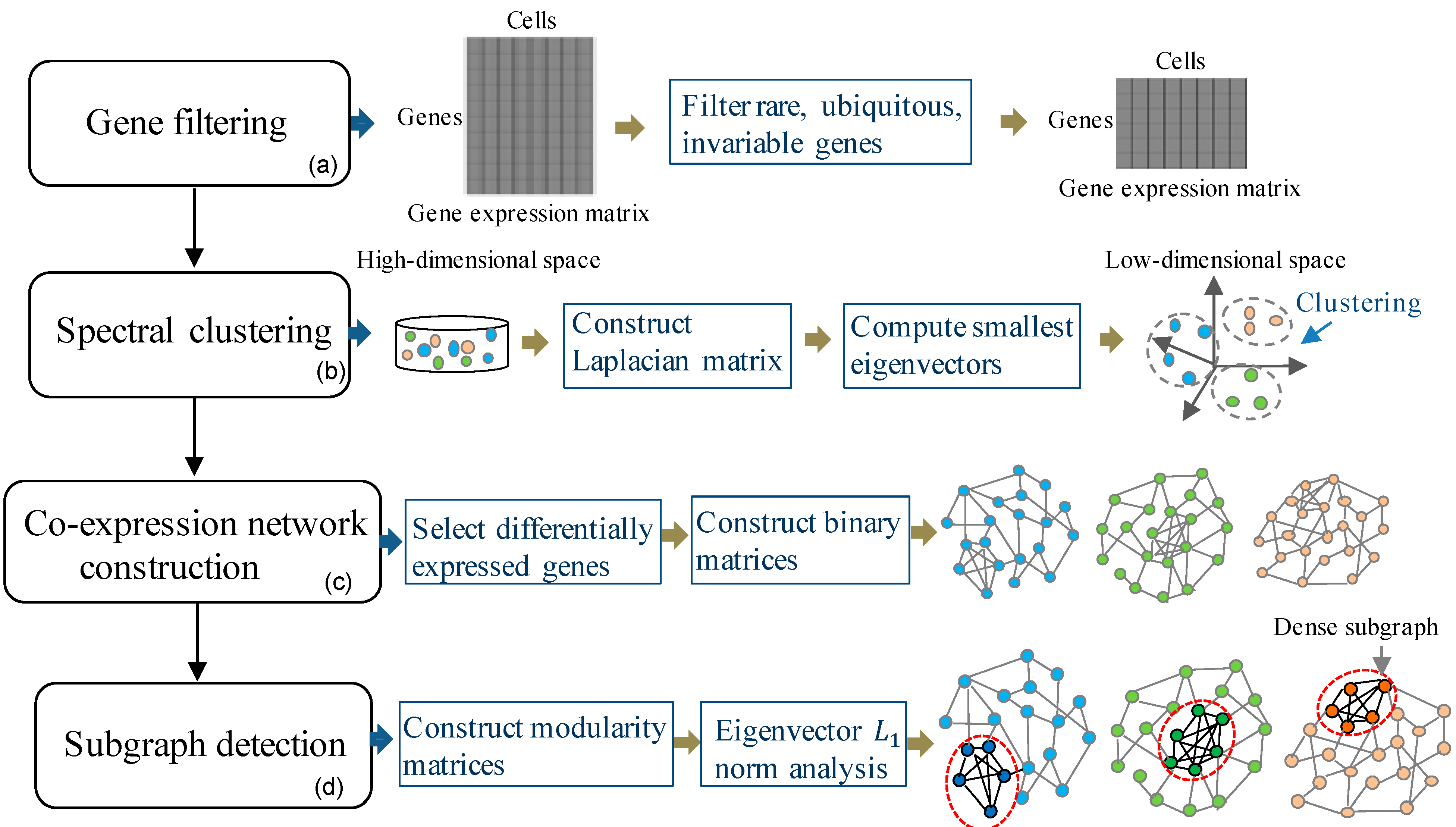
2.1. Methods: The Proposed Learning Framework
2.1.1. Gene Filtering
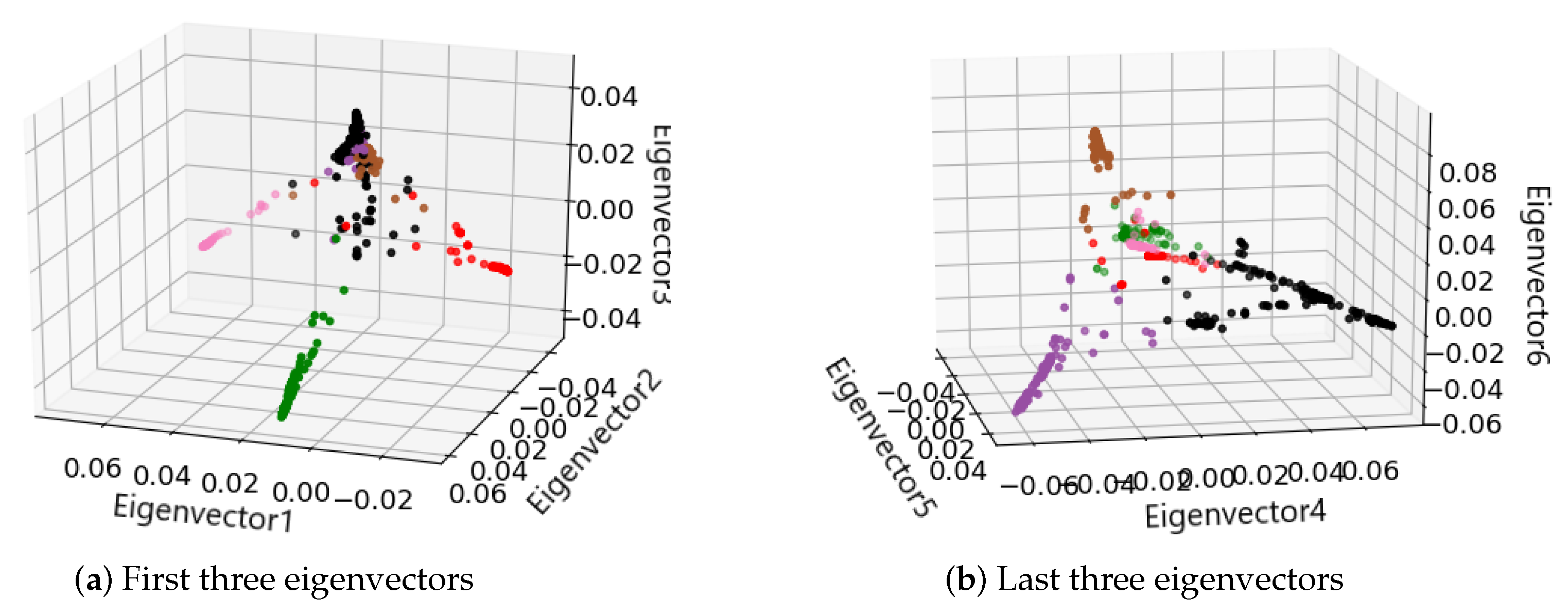
2.1.2. Spectral Clustering to Identify Cell Subpopulations
2.1.3. Differentially Expressed Gene Selection
2.1.4. Gene Co-Expression Network Construction
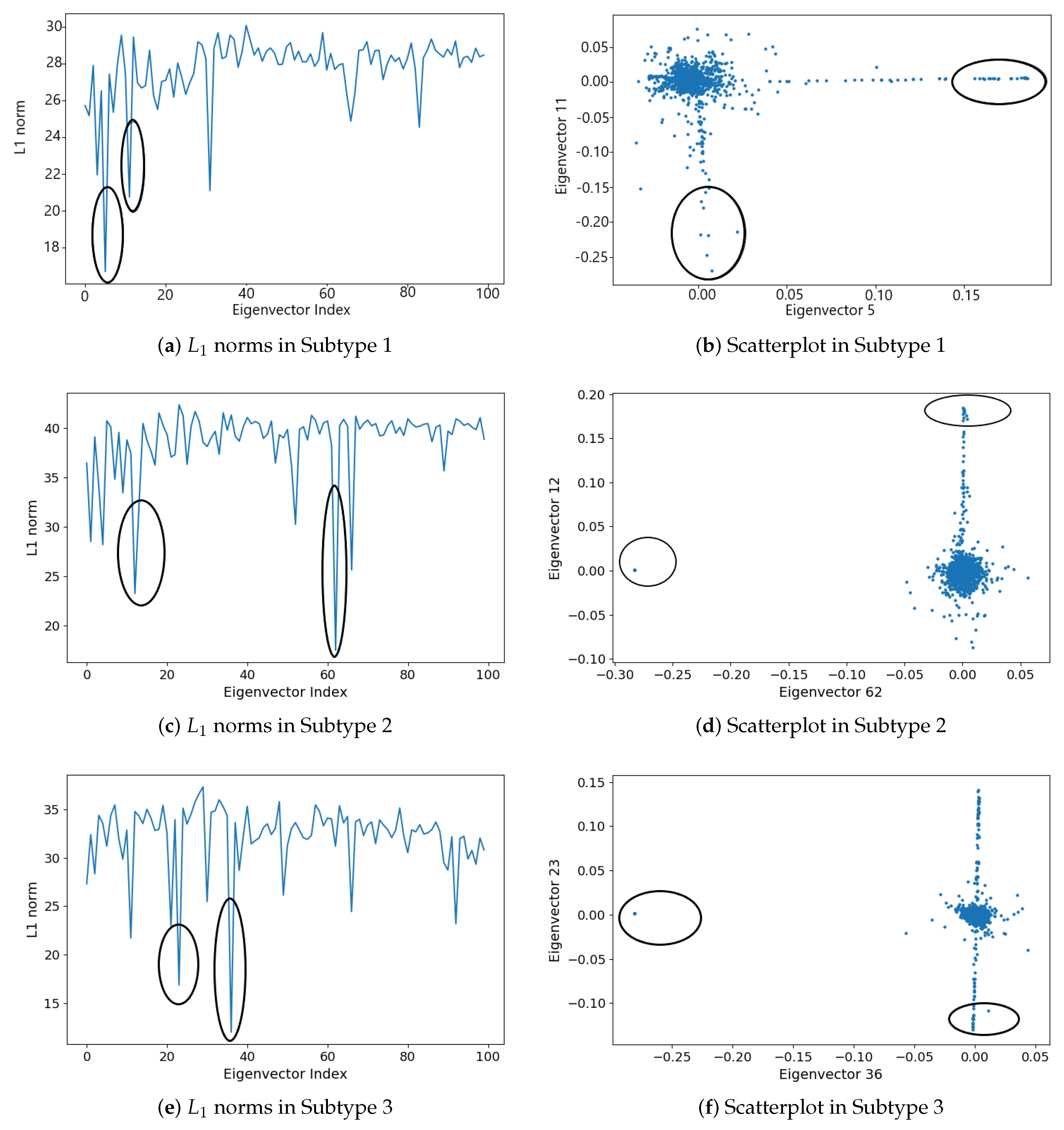
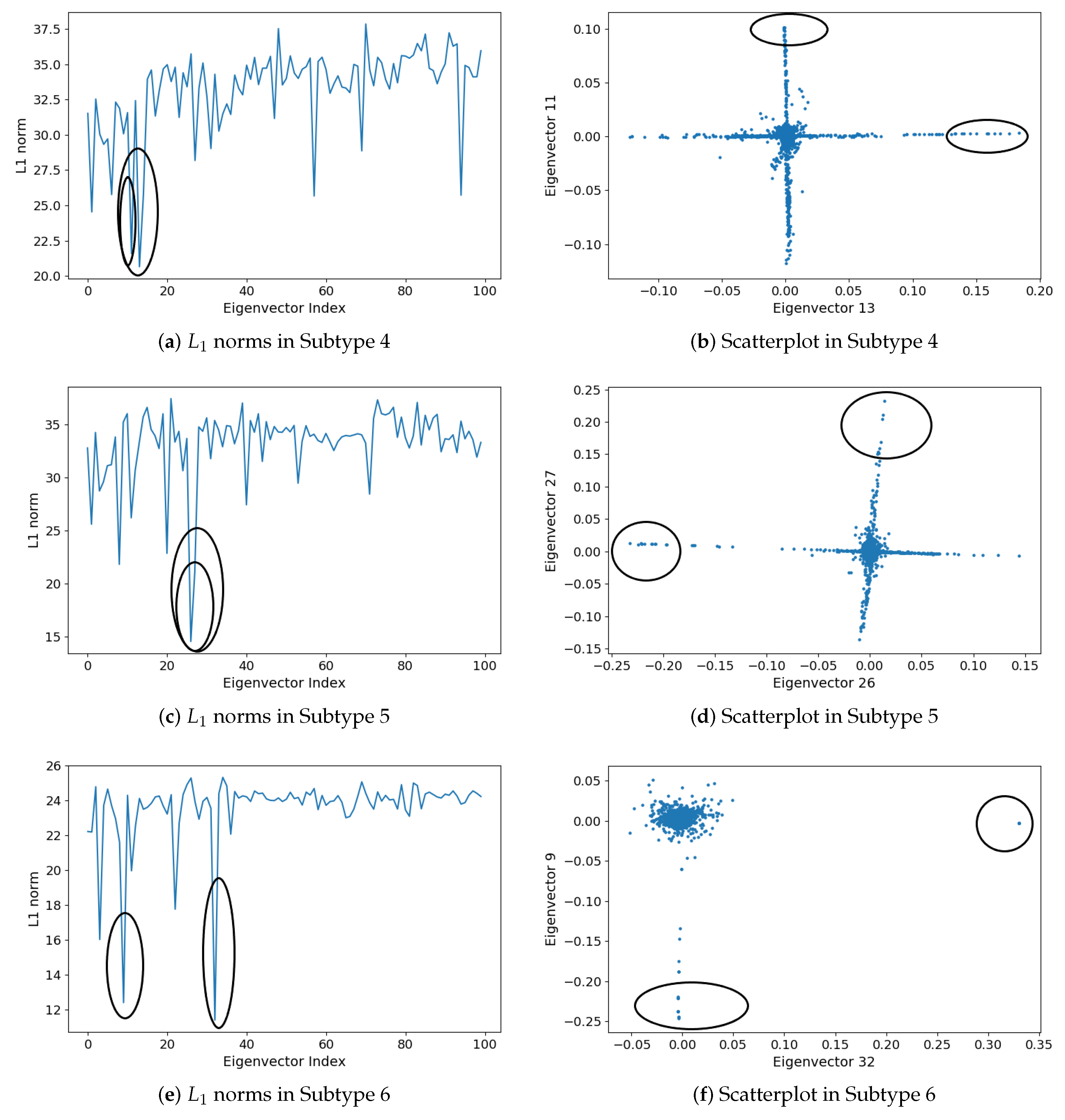
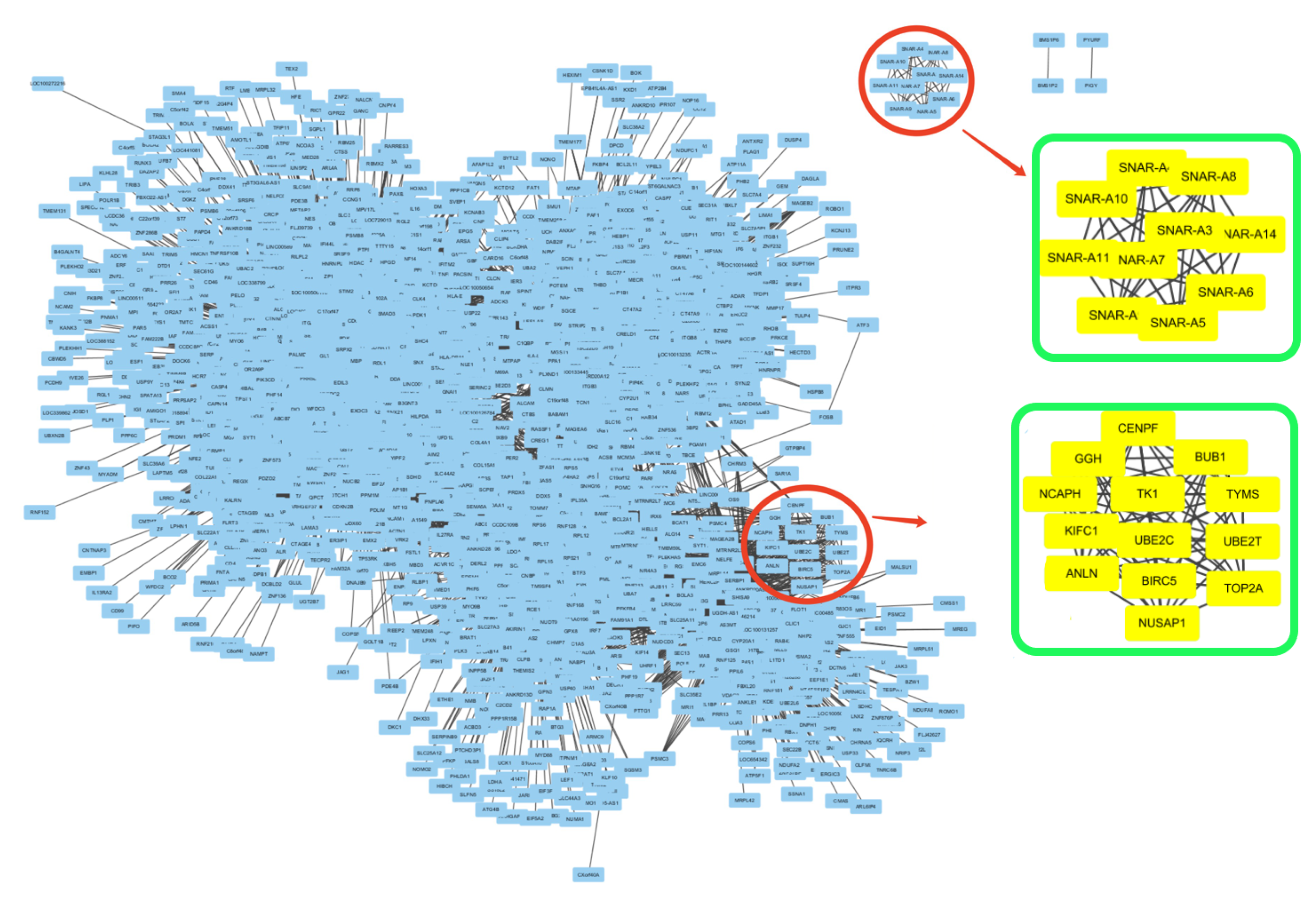
2.1.5. Subgraph Detection
2.2. Materials
3. Results
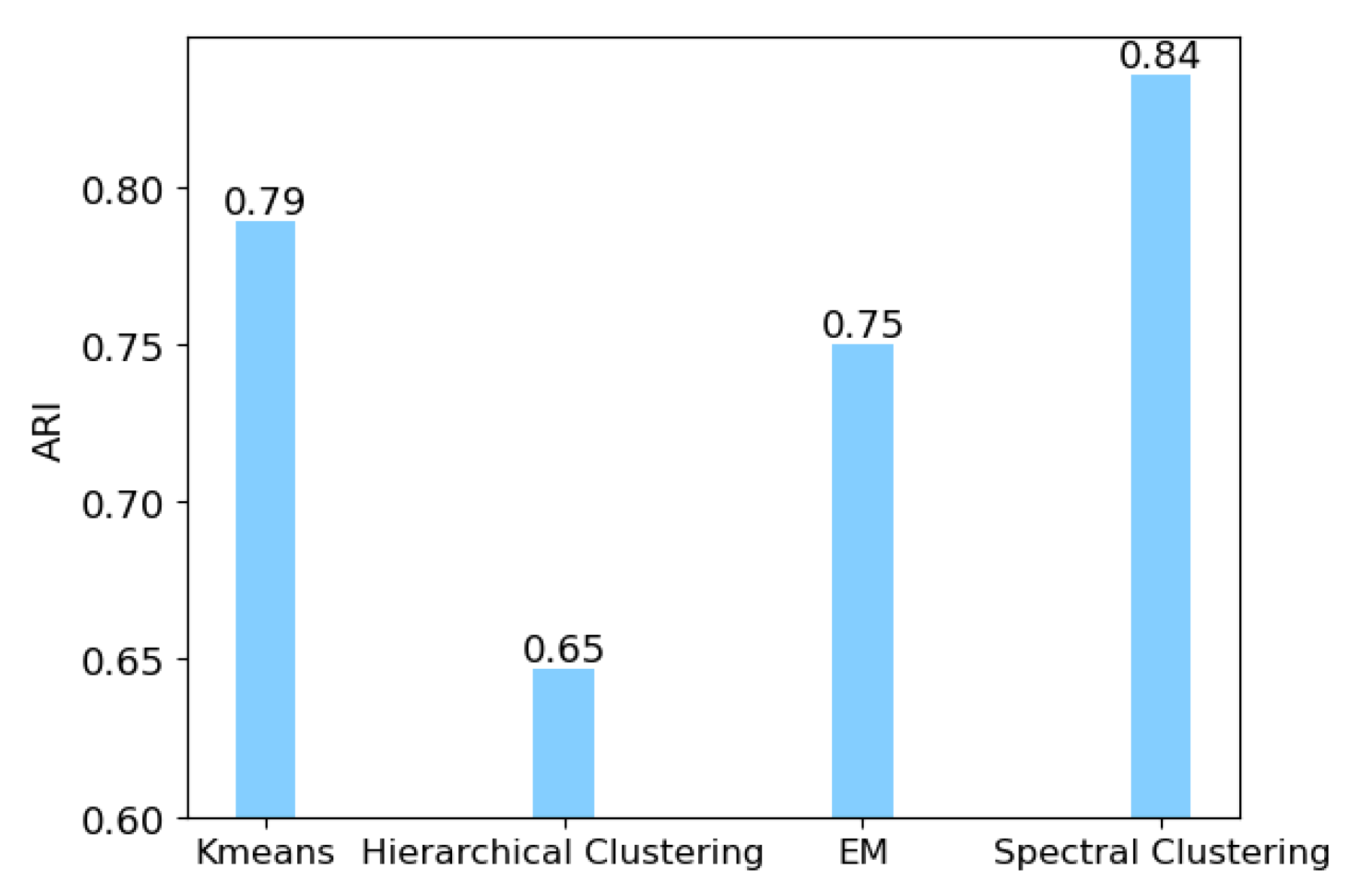
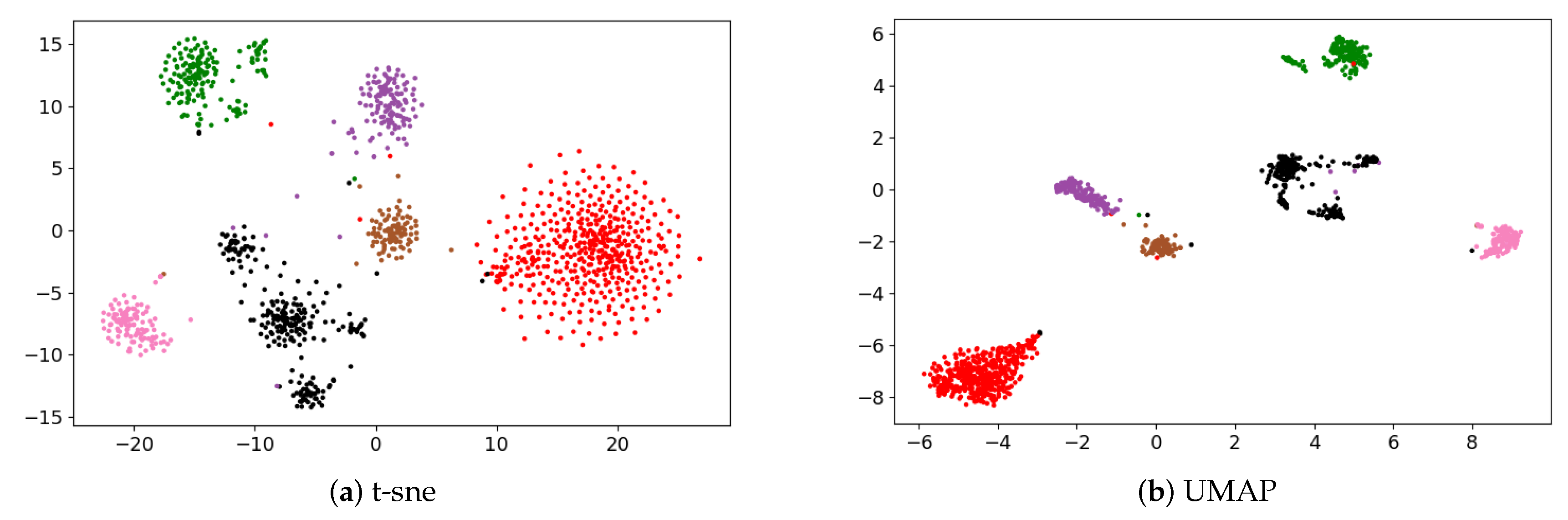
3.1. Identification of Cancer Subtypes
3.2. Detecting Interactive Gene Groups
4. Conclusions and Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Villani, A.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef]
- Wang, Z.; Ding, H.; Zou, Q. Identifying cell types to interpret scRNA-seq data: How, why and more possibilities. Brief. Funct. Genom. 2020, 19, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Ma, A.; Ma, Q.; Zou, Q. Clustering and classification methods for single-cell RNA-sequencing data. Brief. Bioinform. 2019, 7, 1–13. [Google Scholar]
- Hwang, B.; Lee, J.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Kanter, I.; Dalerba, P.; Kalisky, T. A cluster robustness score for identifying cell subpopulations in single cell gene expression datasets from heterogeneous tissues and tumors. Bioinformatics 2019, 35, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Davis-Marcisak, E.F.; Sherman, T.D.; Orugunta, P.; Stein-O’Brien, G.L.; Puram, S.V.; Torres, E.T.R.; Hopkins, A.C.; Jaffee, E.M.; Favorov, A.V.; Afsari, B.; et al. Differential variation analysis enables detection of tumor heterogeneity using single-cell RNA-sequencing data. Cancer Res. 2019, 79, 5102–5112. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Yu, K.; Lee, P.C. Cancer classification of single-cell gene expression data by neural network. Bioinformatics 2020, 36, 1360–1366. [Google Scholar] [CrossRef]
- Ye, X.; Sakurai, T. Unsupervised Feature Selection for Microarray Gene Expression Data Based on Discriminative Structure Learning. J. Univers. Comput. Sci. 2018, 24, 725–741. [Google Scholar]
- Wold, S.; Esbensen, K.; Geladi, P. Principal component analysis. Chemom. Intell. Lab. Syst. 1987, 2, 37–52. [Google Scholar] [CrossRef]
- Maaten, L.V.D.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019, 37, 38. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Li, H.; Imakura, A.; Sakurai, T. Distributed Collaborative Feature Selection Based on Intermediate Representation. In Proceedings of the 28th International Joint Conference on Artificial Intelligence, Macao, China, 10–16 August 2019; pp. 563–568. [Google Scholar]
- Ye, X.; Li, H.; Sakurai, T.; Shueng, P. Ensemble Feature Learning to Identify Risk Factors for Predicting Secondary Cancer. Int. J. Med. Sci. 2019, 16, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Liu, B. BioSeq-Analysis: A platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 2019, 20, 1280–1294. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gao, X.; Zhang, H. BioSeq-Analysis2.0: An updated platform for analyzing DNA, RNA and protein sequences at sequence level and residue level based on machine learning approaches. Nucleic Acids Res. 2019, 47, e127. [Google Scholar] [CrossRef]
- Menon, V. Clustering single cells: A review of approaches on high-and low-depth single-cell RNA-seq data. Brief. Funct. Genom. 2018, 17, 240–245. [Google Scholar] [CrossRef]
- Hartigan, J.A.; Wong, M.A. Algorithm AS 136: A k-means clustering algorithm. J. R. Stat. Soc. Ser. C 1979, 28, 100–108. [Google Scholar] [CrossRef]
- Yau, C. pcaReduce: Hierarchical clustering of single cell transcriptional profiles. BMC Bioinform. 2016, 17, 140. [Google Scholar]
- Dempster, A.P.; Laird, N.M.; Rubin, D.B. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Ser. B 1977, 39, 1–22. [Google Scholar]
- Ye, X.; Sakurai, T. Robust Similarity Measure for Spectral Clustering Based on Shared Neighbors. ETRI J. 2016, 38, 540–550. [Google Scholar] [CrossRef]
- Ye, X.; Sakurai, T. Spectral Clustering with Adaptive Similarity Measure in Kernel Space. Intell. Data Anal. 2018, 22, 751–765. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Wang, H.; Potter, S.S.; Whitsett, J.A.; Xu, Y. SINCERA: A pipeline for single-cell RNA-seq profiling analysis. PLoS Comput. Biol. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Troup, M.; Ho, J.W. CIDR: Ultrafast and accurate clustering through imputation for single-cell RNA-seq data. Genome Biol. 2017, 18, 59. [Google Scholar] [CrossRef]
- Kiselev, V.Y.; Kirschner, K.; Schaub, M.T.; Andrews, T.; Yiu, A.; Chandra, T.; Natarajan, K.N.; Reik, W.; Barahona, M.; Green, A.R.; et al. SC3: Consensus clustering of single-cell RNA-seq data. Nat. Methods 2017, 14, 483–486. [Google Scholar] [CrossRef]
- Xu, C.; Su, Z. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics 2015, 31, 1974–1980. [Google Scholar] [CrossRef]
- Kharchenko, P.V.; Silberstein, L.; Scadden, D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods 2014, 11, 740. [Google Scholar] [CrossRef]
- Ji, Z.; Ji, H. TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res. 2016, 44, e117. [Google Scholar] [CrossRef]
- Deng, Q.; Ramsköld, D.; Reinius, B.; Sandberg, R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 2014, 343, 193–196. [Google Scholar] [CrossRef]
- Tieri, P.; Farina, L.; Petti, M.; Astolfi, L.; Paci, P.; Castiglione, F. Network Inference and Reconstruction in Bioinformatics. Encycl. Bioinform. Comput. Biol. 2019, 2, 805–813. [Google Scholar]
- Gan, Y.; Li, N.; Zou, G.; Xin, Y.; Guan, J. Identification of cancer subtypes from single-cell RNA-seq data using a consensus clustering method. BMC Med. Genom. 2018, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Ralston, A. Gene Interaction and Disease. Nat. Educ. 2018, 1, 16. [Google Scholar]
- Gerring, Z.F.; Gamazon, E.R.; Derks, E.M. A gene co-expression network-based analysis of multiple brain tissues reveals novel genes and molecular pathways underlying major depression. PLoS Genet. 2019, 15, e1008245. [Google Scholar] [CrossRef]
- Caliński, T.; Harabasz, J. A dendrite method for cluster analysis. Commun. Stat. Theory Methods 1974, 3, 1–27. [Google Scholar] [CrossRef]
- Anjum, A.; Jaggi, S.; Varghese, E.; Lall, S.; Bhowmik, A.; Rai, A. Identification of differentially expressed genes in rna-seq data of arabidopsis thaliana: A compound distribution approach. J. Comput. Biol. 2016, 23, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Robinson, M.D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 2018, 15, 255. [Google Scholar] [CrossRef]
- Simes, R.J. An improved Bonferroni procedure for multiple tests of significance. Biometrika 1986, 73, 751–754. [Google Scholar] [CrossRef]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A gene-coexpression network for global discovery of conserved genetic modules. Science 2003, 302, 249–255. [Google Scholar] [CrossRef]
- Su, R.; Zhang, J.; Liu, X.; Wei, L. Identification of expression signatures for non-small-cell lung carcinoma subtype classification. Bioinformatics 2020, 36, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.; Bliss, N.; Wolfe, P.J. Subgraph detection using eigenvector L1 norms. In Proceedings of the 24th Annual Conference on Neural Information Processing Systems, Vancouver, BC, Canada, 6–9 December 2010; pp. 1633–1641. [Google Scholar]
- Futamura, Y.; Ye, X.; Imakura, A.; Sakurai, T. Spectral Anomaly Detection in Large Graphs Using a Complex Moment-Based Eigenvalue Solver. ASCE-ASME J. Risk Uncertain. Eng. Syst. Part A Civ. Eng. 2020, 6, 04020010. [Google Scholar] [CrossRef]
- Newman, M.E. Finding community structure in networks using the eigenvectors of matrices. Phys. Rev. E 2006, 74, 036104. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Hubert, L.; Arabie, P. Comparing partitions. J. Classif. 1985, 2, 193–218. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44. [Google Scholar]
- Li, W.; Sanki, A.; Karim, R.Z.; Thompson, J.F.; Lee, C.S.; Zhuang, L.; McCarthy, S.W.; Scolyer, R.A. The role of cell cycle regulatory proteins in the pathogenesis of melanoma. Pathology 2006, 38, 287–301. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Total Cells | Benign Cells (Percentage) | Malignant Cells (Percentage) |
|---|---|---|---|
| Melanoma_53 | 143 | 127 (88.8%) | 16 (11.2%) |
| Melanoma_58 | 142 | 142 (100%) | 0 |
| Melanoma_59 | 70 | 16 (22.9%) | 54 (77.1%) |
| Melanoma_60 | 226 | 217 (96.0%) | 9 (4.0%) |
| Melanoma_65 | 63 | 59 (93.7%) | 4 (6.3%) |
| Melanoma_67 | 95 | 95 (100%) | 0 |
| Melanoma_71 | 89 | 35 (39.3%) | 54 (60.7%) |
| Melanoma_72 | 181 | 181 (100%) | 0 |
| Melanoma_74 | 147 | 147 (100%) | 0 |
| Melanoma_75 | 344 | 341 (99.1%) | 3 (0.9%) |
| Melanoma_78 | 131 | 11 (8.4%) | 120 (91.6%) |
| Melanoma_79 | 896 | 428 (47.8%) | 468 (52.2%) |
| Melanoma_80 | 480 | 355 (74.0%) | 125 (26.0%) |
| Melanoma_81 | 205 | 72 (35.1%) | 133 (64.9%) |
| Melanoma_82 | 84 | 52 (61.9%) | 32 (38.1%) |
| Melanoma_84 | 159 | 145 (91.2%) | 14 (8.8%) |
| Melanoma_88 | 351 | 234 (66.7%) | 117 (33.3%) |
| Melanoma_89 | 475 | 377 (79.4%) | 98 (20.6%) |
| Melanoma_94 | 364 | 354 (97.3%) | 10 (2.7%) |
| Sample ID | Subtype 1 | Subtype 2 | Subtype 3 | Subtype 4 | Subtype 5 | Subtype 6 |
|---|---|---|---|---|---|---|
| Melanoma_53 | 0 | 0 | 16 (100%) | 0 | 0 | 0 |
| Melanoma_59 | 52 (96.3%) | 0 | 0 | 0 | 0 | 2 (3.7%) |
| Melanoma_60 | 1 (11.1%) | 0 | 0 | 6 (66.7%) | 1 (11.1%) | 1 (11.1%) |
| Melanoma_65 | 4 (100%) | 0 | 0 | 0 | 0 | 0 |
| Melanoma_71 | 50 (92.6%) | 1 (1.8%) | 0 | 0 | 0 | 3 (5.6%) |
| Melanoma_75 | 3 (100%) | 0 | 0 | 0 | 0 | 0 |
| Melanoma_78 | 6 (5.0%) | 0 | 0 | 114 (95.0%) | 0 | 0 |
| Melanoma_79 | 2 (0.4%) | 465 (99.4%) | 0 | 0 | 1 (0.2%) | 0 |
| Melanoma_80 | 0 | 0 | 0 | 0 | 0 | 125 (100%) |
| Melanoma_81 | 2 (1.5%) | 0 | 131 (98.5%) | 0 | 0 | 0 |
| Melanoma_82 | 0 | 0 | 32 (100%) | 0 | 0 | 0 |
| Melanoma_84 | 1 (7.1%) | 1 (7.1%) | 0 | 0 | 1 (7.1%) | 11 (68.7%) |
| Melanoma_88 | 116 (99.1%) | 0 | 0 | 0 | 0 | 1 (0.9%) |
| Melanoma_89 | 1 (1.0%) | 0 | 0 | 0 | 97 (99.0%) | 0 |
| Melanoma_94 | 6 (60.0%) | 1 (10.0%) | 2 (20.0%) | 0 | 1 (10.0%) | 0 |
| Subtype | Subgraph | Eigenvector | Subgraph Size | Subgraph Density |
|---|---|---|---|---|
| Subtype 1 | Subg 1 | 20 | ||
| Subg 2 | 9 | |||
| Subtype 2 | Subg 1 | 10 | ||
| Subg 2 | 13 | |||
| Subtype 3 | Subg 1 | 12 | ||
| Subg 2 | 15 | |||
| Subtype 4 | Subg 1 | 13 | ||
| Subg 2 | 13 | |||
| Subtype 5 | Subg 1 | 12 | ||
| Subg 2 | 9 | |||
| Subtype 6 | Subg 1 | 8 | ||
| Subg 2 | 13 |
| Subgraph | Gene List | Term Type & Name | p-Value |
|---|---|---|---|
| Subtype 1: Subg 1 | UHRF1, TK1, UBE2T, FANCI, | BP: cell cycle (18) | 1.4 × 10 |
| DTL, TYMS, CENPF, NUSAP1, | BP: nuclear division (13) | 1.2 × 10 | |
| BIRC5, TOP2A, UBE2C, CENPM, | CC: chromosome (11) | 6.4 × 10 | |
| TPX2, CDK1, ANLN, ASPM, | KEGG: Cell cycle (3) | 8.4 × 10 | |
| BUB1, MKI67, PKMYT1, AURKB | |||
| Subtype 2: Subg 2 | GGH, TK1, TYMS, | BP: sister chromatid segregation (8) | 5.4 × 10 |
| BUB1, UBE2C, BIRC5, CENPF, | BP: mitotic cell cycle process (10) | 6.5 × 10 | |
| ANLN, NUSAP1, UBE2T, | BP: chromosome organization (8) | 5.2 × 10 | |
| TOP2A, NCAPH, KIFC1 | KEGG: Pyrimidine metabolism (3) | 8.5 × 10 | |
| Subtype 3: Subg 2 | LMNB1, CKAP2, FOXM1, TTK, | BP: mitotic cell cycle process (13) | 6.2 × 10 |
| NDC80, DEPDC1B, KIF20A, | BP: cell division (10) | 4.2 × 10 | |
| KIF4A, CDKN3, FAM64A, KIF14, | CC: spindle (9) | 8.8 × 10 | |
| RACGAP1, CCNB1, SKA3, KIF20B | BP: microtubule-based process (9)) | 3.8 × 10 | |
| MF: microtubule binding (5) | 1.2 × 10 | ||
| KEGG:Cell cycle (2) | 1.8 × 10 | ||
| Subtype 4: Subg 2 | ORC6, KIF20B, RTKN2, EZH2, | BP: mitotic cell cycle (9) | 5.1 × 10 |
| CENPW, BRCA2, ARHGAP11B, | BP: organelle fission (6) | 4.6 × 10 | |
| KIAA1524, TIMELESS, | CC: centrosome (6) | 3.7 × 10 | |
| CEP55, PLK4, ESPL1, NEIL3 | BP: DNA metabolic process (5) | 4.3 × 10 | |
| Subtype 5: Subg 2 | CDCA7, MCM4, DSCC1, | BP: DNA replication (7) | 7.2 × 10 |
| CHAF1A, E2F7, HELLS, | CC: chromosomal part (7) | 8.5 × 10 | |
| GINS2, MCM5, MCM10 | MF: helicase activity (4) | 3.6 × 10 | |
| BP: cellular macromolecule (9) | 6.7 × 10 | ||
| KEGG: DNA replication (2) | 5.2 × 10 | ||
| Subtype 6: Subg 2 | FANCI, TYMS, BIRC5, ASPM, | BP: mitotic cell cycle (11) | 2.5 × 10 |
| PRC1, CENPF, TK1, | BP: organelle fission (9) | 1.6 × 10 | |
| TOP2A, KIF14, NDC80, | CC: condensed chromosome (6) | 4.4× 10 | |
| HMGB2, MKI67, CDC20 | KEGG: Pyrimidine metabolism (2) | 5.7 × 10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, X.; Zhang, W.; Futamura, Y.; Sakurai, T. Detecting Interactive Gene Groups for Single-Cell RNA-Seq Data Based on Co-Expression Network Analysis and Subgraph Learning. Cells 2020, 9, 1938. https://doi.org/10.3390/cells9091938
Ye X, Zhang W, Futamura Y, Sakurai T. Detecting Interactive Gene Groups for Single-Cell RNA-Seq Data Based on Co-Expression Network Analysis and Subgraph Learning. Cells. 2020; 9(9):1938. https://doi.org/10.3390/cells9091938
Chicago/Turabian StyleYe, Xiucai, Weihang Zhang, Yasunori Futamura, and Tetsuya Sakurai. 2020. "Detecting Interactive Gene Groups for Single-Cell RNA-Seq Data Based on Co-Expression Network Analysis and Subgraph Learning" Cells 9, no. 9: 1938. https://doi.org/10.3390/cells9091938
APA StyleYe, X., Zhang, W., Futamura, Y., & Sakurai, T. (2020). Detecting Interactive Gene Groups for Single-Cell RNA-Seq Data Based on Co-Expression Network Analysis and Subgraph Learning. Cells, 9(9), 1938. https://doi.org/10.3390/cells9091938