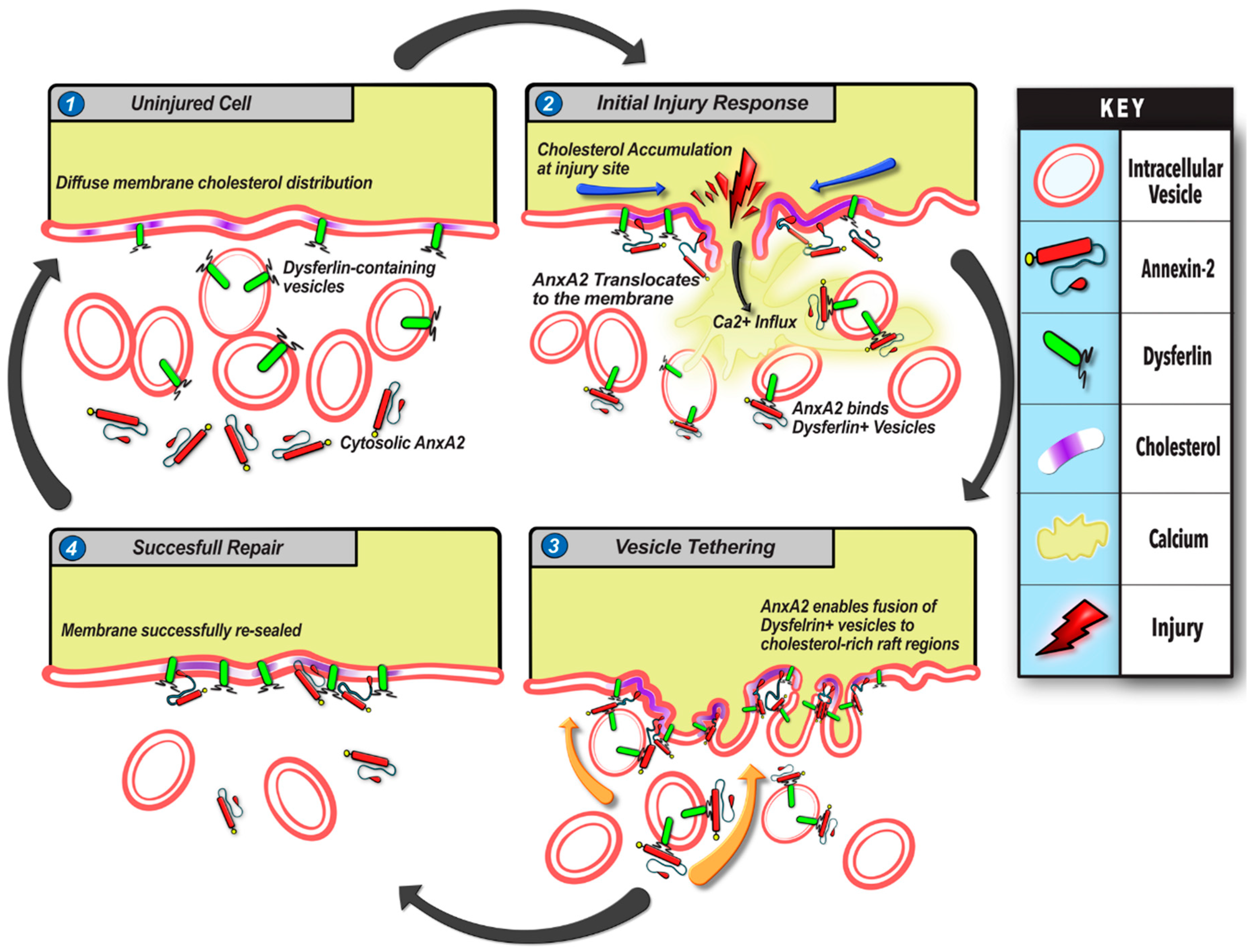
Annexin A2 Mediates Dysferlin Accumulation and Muscle Cell Membrane Repair

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Cell Culture and Treatment
Generating Immortalized Annexin A2 Knockout Myoblasts for Laser Injury Assays
2.2. Injury Assays
2.2.1. Laser Injury
2.2.2. Dysferlin Vesicle Fusion Assessment
2.2.3. Kymograph Analyses
2.2.4. Glass Bead Injury
2.3. Cell Membrane Cholesterol Response Assays
2.4. Cholesterol Depletion Assay
2.5. Western Blotting
2.6. Membrane Fraction Protein Analysis
2.7. Dysferlin Immunoprecipitation (IP)
2.8. Statistical Analysis
3. Results
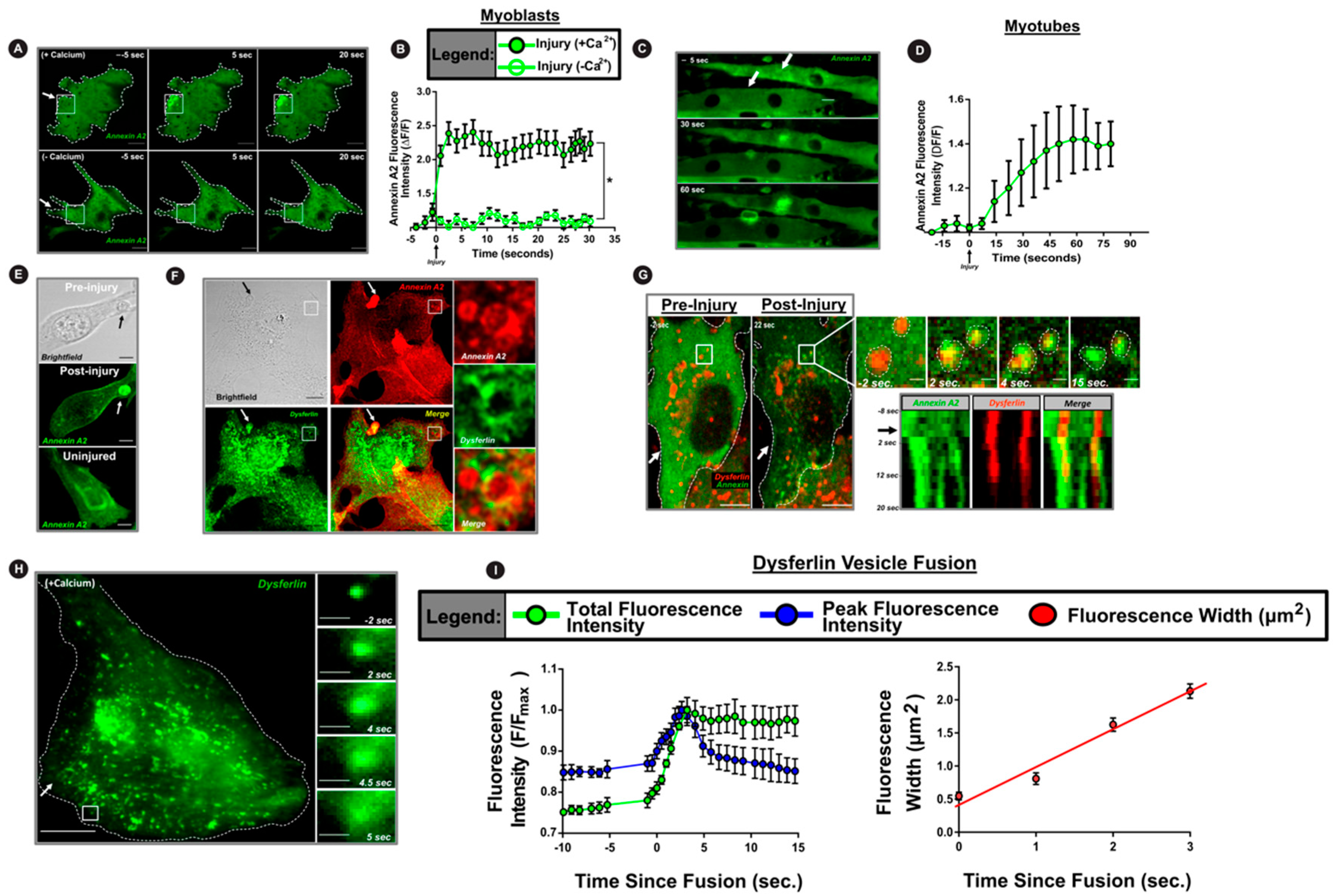
3.1. Cell Injury Triggers AnxA2 and Dysferlin Co-Accumulation on the Membrane
3.2. PM Translocation of AnxA2 Enables Ca2+-Triggered PM Accumulation of Dysferlin
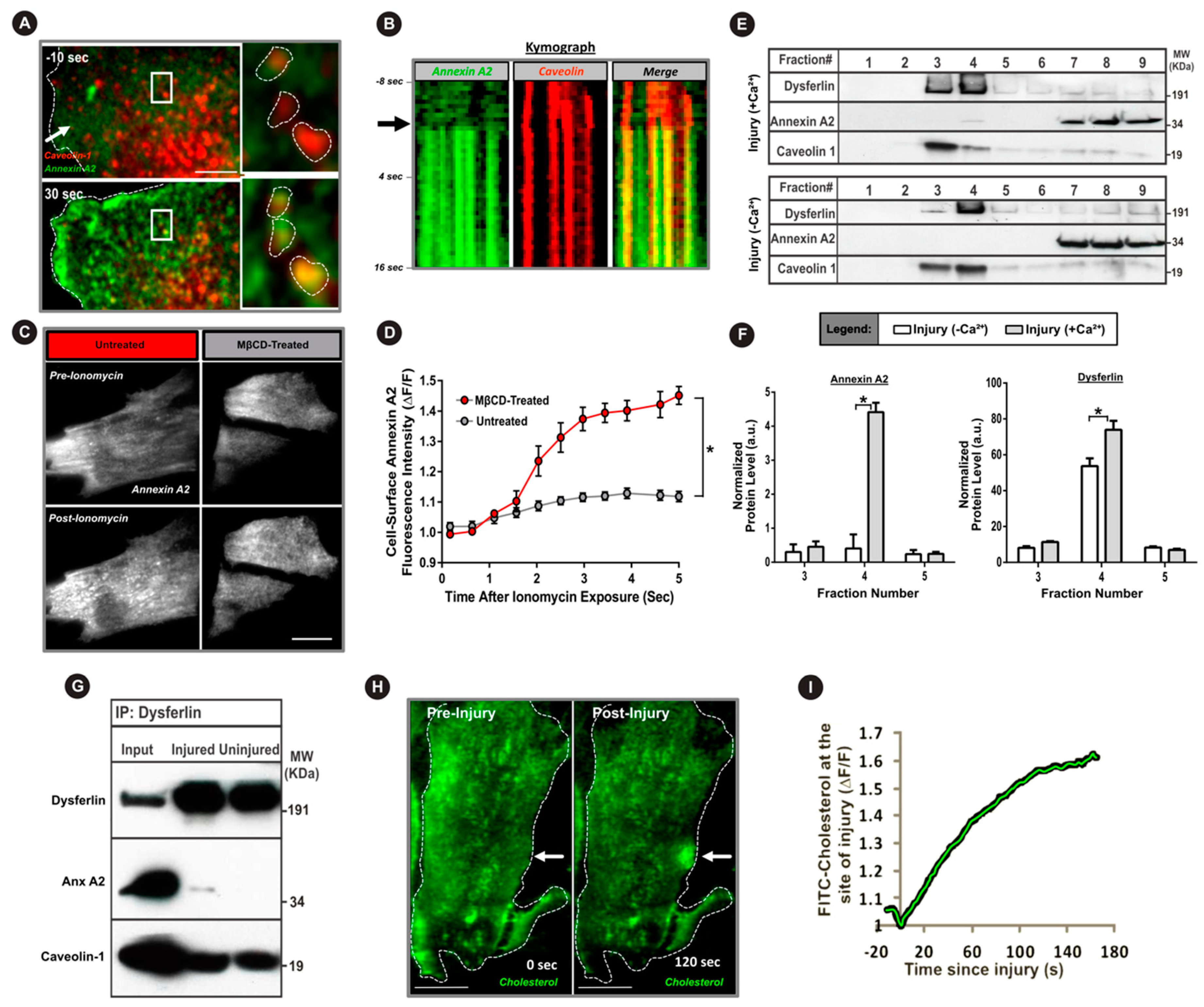
3.3. Cholesterol is Required for AnxA2 and Dysferlin Accumulation at the Injured PM
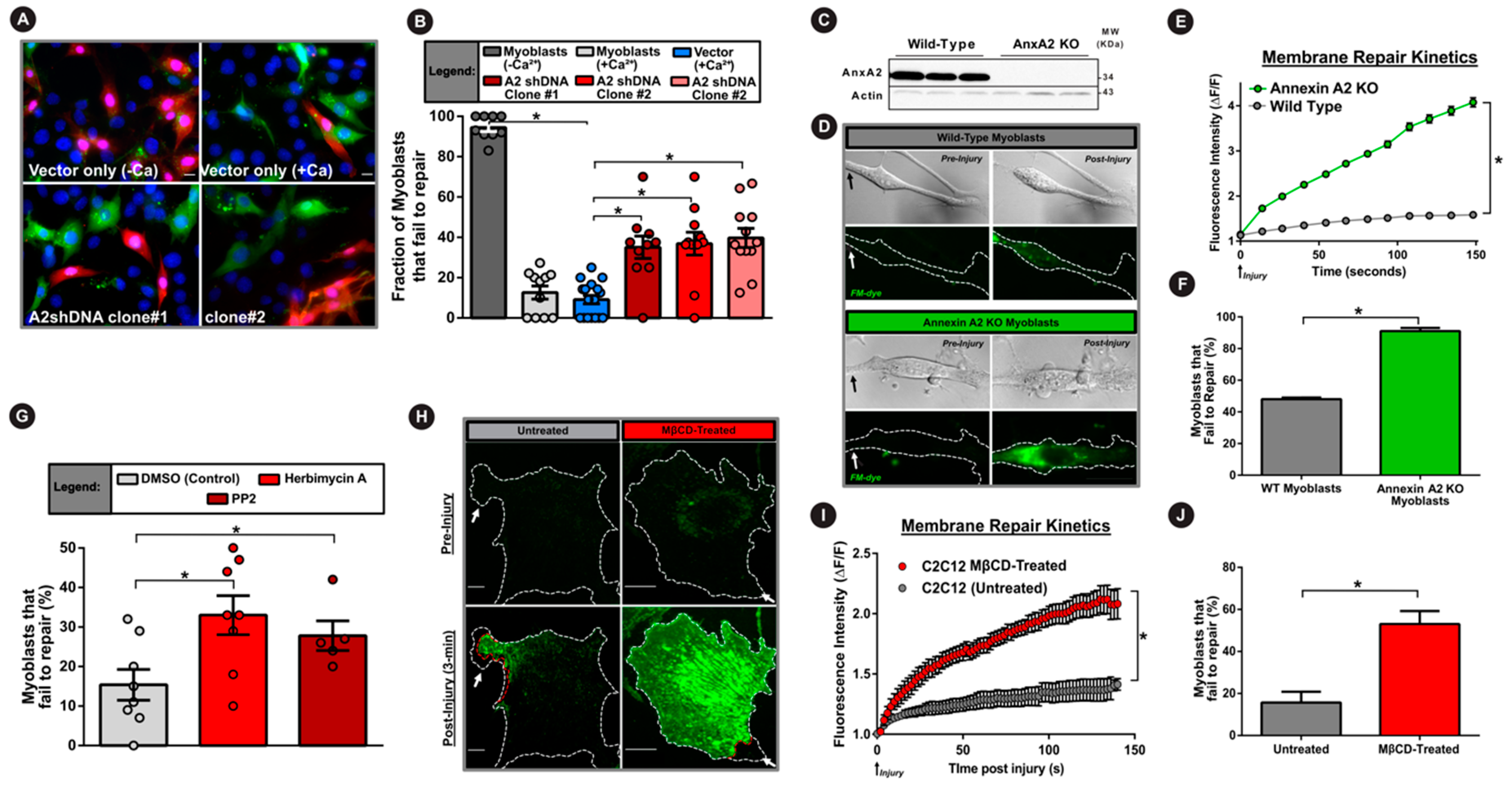
3.4. AnxA2 and Its Interaction with Dysferlin is Required for Plasma Membrane Repair
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McNeil, P.L.; Khakee, R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am. J. Pathol. 1992, 140, 1097–1109. [Google Scholar]
- Demonbreun, A.R.; McNally, E.M. Plasma Membrane Repair in Health and Disease. Curr. Top. Membr. 2016, 77, 67–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashir, R.; Britton, S.; Strachan, T.; Keers, S.; Vafiadaki, E.; Lako, M.; Richard, I.; Marchand, S.; Bourg, N.; Argov, Z.; et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat. Genet. 1998, 20, 37–42. [Google Scholar] [CrossRef]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.-C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef]
- Ho, M.; Post, C.M.; Donahue, L.R.; Lidov, H.G.; Bronson, R.T.; Goolsby, H.; Watkins, S.C.; Cox, G.A.; Brown, R.H. Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum. Mol. Genet. 2004, 13, 1999–2010. [Google Scholar] [CrossRef] [Green Version]
- Lek, A.; Evesson, F.J.; Sutton, R.B.; North, K.N.; Cooper, S.T. Ferlins: Regulators of Vesicle Fusion for Auditory Neurotransmission, Receptor Trafficking and Membrane Repair. Traffic 2011, 13, 185–194. [Google Scholar] [CrossRef]
- Südhof, T.C. A molecular machine for neurotransmitter release: Synaptotagmin and beyond. Nat. Med. 2013, 19, 1227–1231. [Google Scholar] [CrossRef]
- Bansal, D.; Campbell, K.P. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Boil. 2004, 14, 206–213. [Google Scholar] [CrossRef]
- McNeil, P.L.; Kirchhausen, T. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Boil. 2005, 6, 499–505. [Google Scholar] [CrossRef]
- Middel, V.; Zhou, L.; Takamiya, M.; Beil, T.; Shahid, M.; Roostalu, U.; Grabher, C.; Rastegar, S.; Reischl, M.; Nienhaus, G.U.; et al. Dysferlin-mediated phosphatidylserine sorting engages macrophages in sarcolemma repair. Nat. Commun. 2016, 7, 12875. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Defour, A.; Van Der Meulen, J.H.; Bhat, R.; Bigot, A.; Bashir, R.; Nagaraju, K.; Jaiswal, J.K. Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion. Cell Death Dis. 2014, 5, e1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDade, J.R.; Archambeau, A.; Michele, D.E. Rapid actin-cytoskeleton–dependent recruitment of plasma membrane–derived dysferlin at wounds is critical for muscle membrane repair. FASEB J. 2014, 28, 3660–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Weisleder, N.; Ko, J.-K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane Repair Defects in Muscular Dystrophy Are Linked to Altered Interaction between MG53, Caveolin-3, and Dysferlin. J. Boil. Chem. 2009, 284, 15894–15902. [Google Scholar] [CrossRef] [Green Version]
- Roostalu, U.; Strahle, U. In Vivo Imaging of Molecular Interactions at Damaged Sarcolemma. Dev. Cell 2012, 22, 515–529. [Google Scholar] [CrossRef] [Green Version]
- McDade, J.R.; Michele, D.E. Membrane damage-induced vesicle-vesicle fusion of dysferlin-containing vesicles in muscle cells requires microtubules and kinesin. Hum. Mol. Genet. 2013, 23, 1677–1686. [Google Scholar] [CrossRef] [Green Version]
- Scheffer, L.L.; Sreetama, S.C.; Sharma, N.; Medikayala, S.; Brown, K.J.; Defour, A.; Jaiswal, J.K. Mechanism of Ca²⁺-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun. 2014, 5, 5646. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Medikayala, S.; Defour, A.; Rayavarapu, S.; Brown, K.J.; Hathout, Y.; Jaiswal, J.K. Use of Quantitative Membrane Proteomics Identifies a Novel Role of Mitochondria in Healing Injured Muscles*. J. Boil. Chem. 2012, 287, 30455–30467. [Google Scholar] [CrossRef] [Green Version]
- Swaggart, K.A.; Demonbreun, A.R.; Vo, A.H.; Swanson, K.E.; Kim, E.Y.; Fahrenbach, J.P.; Holley-Cuthrell, J.; Eskin, A.; Chen, Z.; Squire, K.; et al. Annexin A6 modifies muscular dystrophy by mediating sarcolemmal repair. Proc. Natl. Acad. Sci. USA 2014, 111, 6004–6009. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.-K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 Nucleates Assembly Of Cell Membrane Repair Machinery. Biophys. J. 2009, 96, 361a. [Google Scholar] [CrossRef] [Green Version]
- Sønder, S.L.; Boye, T.L.; Tölle, R.; Dengjel, J.; Maeda, K.; Jäättelä, M.; Simonsen, A.C.; Jaiswal, J.K.; Nylandsted, J. Annexin A7 is required for ESCRT III-mediated plasma membrane repair. Sci. Rep. 2019, 9, 6726. [Google Scholar] [CrossRef] [PubMed]
- Boye, T.L.; Maeda, K.; Pezeshkian, W.; Sønder, S.L.; Haeger, S.C.; Gerke, V.; Simonsen, A.C.; Nylandsted, J. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 2017, 8, 1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defour, A.; Medikayala, S.; Van Der Meulen, J.H.; Hogarth, M.; Holdreith, N.; Malatras, A.; Duddy, W.J.; Boehler, J.; Nagaraju, K.; Jaiswal, J.K. Annexin A2 links poor myofiber repair with inflammation and adipogenic replacement of the injured muscle. Hum. Mol. Genet. 2017, 26, 1979–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, J.K.; Nylandsted, J. S100 and annexin proteins identify cell membrane damage as the Achilles heel of metastatic cancer cells. Cell Cycle 2015, 14, 502–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, J.K.; Lauritzen, S.P.; Scheffer, L.; Sakaguchi, M.; Bunkenborg, J.; Simon, S.M.; Kallunki, T.; Jäättelä, M.; Nylandsted, J. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouter, A.; Gounou, C.; Bérat, R.; Tan, S.; Gallois, B.; Granier, T.; D’Estaintot, B.L.; Pöschl, E.; Brachvogel, B.; Brisson, A. Annexin-A5 assembled into two-dimensional arrays promotes cell membrane repair. Nat. Commun. 2011, 2, 270. [Google Scholar] [CrossRef] [Green Version]
- Draeger, A.; Monastyrskaya, K.; Babiychuk, E.B. Plasma membrane repair and cellular damage control: The annexin survival kit. Biochem. Pharmacol. 2011, 81, 703–712. [Google Scholar] [CrossRef]
- Gerke, V.; Creutz, C.E.; E Moss, S. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Boil. 2005, 6, 449–461. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Blebbing confers resistance against cell lysis. Cell Death Differ. 2010, 18, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Potez, S.; Luginbühl, M.; Monastyrskaya, K.; Hostettler, A.; Draeger, A.; Babiychuk, E.B. Tailored Protection against Plasmalemmal Injury by Annexins with Different Ca2+ Sensitivities. J. Boil. Chem. 2011, 286, 17982–17991. [Google Scholar] [CrossRef] [Green Version]
- Monastyrskaya, K.; Babiychuk, E.B.; Hostettler, A.; Wood, P.; Grewal, T.; Draeger, A. Plasma Membrane-associated Annexin A6 Reduces Ca2+Entry by Stabilizing the Cortical Actin Cytoskeleton. J. Boil. Chem. 2009, 284, 17227–17242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, S.R.; E Moss, S. Annexins in the secretory pathway. Cell. Mol. Life Sci. 1997, 53, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Leikina, E.; Defour, A.; Melikov, K.; Van Der Meulen, J.H.; Nagaraju, K.; Bhuvanendran, S.; Gebert, C.; Pfeifer, K.; Chernomordik, L.V.; Jaiswal, J.K. Annexin A1 Deficiency does not Affect Myofiber Repair but Delays Regeneration of Injured Muscles. Sci. Rep. 2015, 5, 18246. [Google Scholar] [CrossRef] [PubMed]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H. Dysferlin Interacts with Annexins A1 and A2 and Mediates Sarcolemmal Wound-healing. J. Boil. Chem. 2003, 278, 50466–50473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.; Utokaparch, S.; Sharma, A.; Yu, C.; Abraham, T.; Borchers, C.; Bernatchez, P. Proteomic identification of dysferlin-interacting protein complexes in human vascular endothelium. Biochem. Biophys. Res. Commun. 2011, 415, 263–269. [Google Scholar] [CrossRef]
- De Morrée, A.; Hensbergen, P.J.; Van Haagen, H.H.H.B.M.; Dragan, I.; Deelder, A.M.; AC’t Hoen, P.; Frants, R.R.; Van Der Maarel, S.M. Proteomic Analysis of the Dysferlin Protein Complex Unveils Its Importance for Sarcolemmal Maintenance and Integrity. PLoS ONE 2010, 5, e13854. [Google Scholar] [CrossRef] [Green Version]
- Morgan, J.; Beauchamp, J.; Pagel, C.; Peckham, M.; Ataliotis, P.; Jat, P.; Noble, M.; Farmer, K.; Partridge, T. Myogenic Cell Lines Derived from Transgenic Mice Carrying a Thermolabile T Antigen: A Model System for the Derivation of Tissue-Specific and Mutation-Specific Cell Lines. Dev. Boil. 1994, 162, 486–498. [Google Scholar] [CrossRef]
- Defour, A.; Sreetama, S.C.; Jaiswal, J.K. Imaging cell membrane injury and subcellular processes involved in repair. J. Vis. Exp. 2014, 2014, e51106. [Google Scholar] [CrossRef]
- Jaiswal, J.K.; Fix, M.; Takano, T.; Nedergaard, M.; Simon, S.M. Resolving vesicle fusion from lysis to monitor calcium-triggered lysosomal exocytosis in astrocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 14151–14156. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, J.K.; Simon, S.M. Imaging single events at the cell membrane. Nat. Methods 2007, 3, 92–98. [Google Scholar] [CrossRef]
- Deora, A.B.; Kreitzer, G.; Jacovina, A.T.; Hajjar, K.A. An Annexin 2 Phosphorylation Switch Mediates p11-dependent Translocation of Annexin 2 to the Cell Surface. J. Boil. Chem. 2004, 279, 43411–43418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Foley, K.; Huang, L.; Leubner, A.; Mo, G.; Olino, K.; Edil, B.H.; Mizuma, M.; Sharma, R.; Le, D.T.; et al. Tyrosine 23 Phosphorylation-Dependent Cell-Surface Localization of Annexin A2 Is Required for Invasion and Metastases of Pancreatic Cancer. PLoS ONE 2011, 6, e19390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babiychuk, E.B.; Draeger, A. Annexins in cell membrane dynamics. Ca(2+)-regulated association of lipid microdomains. J. Cell Biol. 2000, 150, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Chintagari, N.R.; Jin, N.; Wang, P.; Narasaraju, T.A.; Chen, J.; Liu, L. Effect of Cholesterol Depletion on Exocytosis of Alveolar Type II Cells. Am. J. Respir. Cell Mol. Boil. 2006, 34, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Chasserot-Golaz, S.; Vitale, N.; Umbrecht-Jenck, E.; Knight, D.; Gerke, V.; Bader, M.-F. Annexin 2 Promotes the Formation of Lipid Microdomains Required for Calcium-regulated Exocytosis of Dense-Core Vesicles. Mol. Boil. Cell 2005, 16, 1108–1119. [Google Scholar] [CrossRef] [Green Version]
- Han, W.-Q.; Xia, M.; Xu, M.; Boini, K.M.; Ritter, J.K.; Li, N.-J.; Li, P.-L. Lysosome fusion to the cell membrane is mediated by the dysferlin C2A domain in coronary arterial endothelial cells. J. Cell Sci. 2012, 125, 1225–1234. [Google Scholar] [CrossRef] [Green Version]
- Feuk-Lagerstedt, E.; Movitz, C.; Pellmé, S.; Dahlgren, C.; Karlsson, A. Lipid raft proteome of the human neutrophil azurophil granule. Proteomics 2007, 7, 194–205. [Google Scholar] [CrossRef]
- Sreetama, S.C.; Chandra, G.; Van Der Meulen, J.H.; Ahmad, M.M.; Suzuki, P.; Bhuvanendran, S.; Nagaraju, K.; Hoffman, E.P.; Jaiswal, J.K. Membrane Stabilization by Modified Steroid Offers a Potential Therapy for Muscular Dystrophy Due to Dysferlin Deficit. Mol. Ther. 2018, 26, 2231–2242. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Deviez, D.J.; Howes, M.T.; Laval, S.H.; Bushby, K.; Hancock, J.F.; Parton, R.G. Caveolin Regulates Endocytosis of the Muscle Repair Protein, Dysferlin. J. Boil. Chem. 2007, 283, 6476–6488. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, C.; Hayashi, Y.K.; Ogawa, M.; Aoki, M.; Murayama, K.; Nishino, I.; Nonaka, I.; Arahata, K.; Brown, R.H. The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum. Mol. Genet. 2001, 10, 1761–1766. [Google Scholar] [CrossRef]
- Jaiswal, J.K. Calcium — how and why? J. Biosci. 2001, 26, 357–363. [Google Scholar] [CrossRef] [PubMed]
- McNeil, P.L.; Steinhardt, R.A. Plasma Membrane Disruption: Repair, Prevention, Adaptation. Annu. Rev. Cell Dev. Boil. 2003, 19, 697–731. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeil, A.K.; Rescher, U.; Gerke, V.; McNeil, P.L. Requirement for Annexin A1 in Plasma Membrane Repair. J. Boil. Chem. 2006, 281, 35202–35207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emans, N.; Gorvel, J.P.; Walter, C.; Gerke, V.; Kellner, R.; Griffiths, G.; Gruenberg, J. Annexin II is a major component of fusogenic endosomal vesicles. J. Cell Boil. 1993, 120, 1357–1369. [Google Scholar] [CrossRef]
- Gabel, M.; Chasserot-Golaz, S. Annexin A2, an essential partner of the exocytotic process in chromaffin cells. J. Neurochem. 2016, 137, 890–896. [Google Scholar] [CrossRef] [Green Version]
- Morel, E.; Gruenberg, J. Annexin A2 Binding to Endosomes and Functions in Endosomal Transport Are Regulated by Tyrosine 23 Phosphorylation. J. Boil. Chem. 2008, 284, 1604–1611. [Google Scholar] [CrossRef] [Green Version]
- Bjorge, J.D.; Jakymiw, A.; Fujita, D.J. Selected glimpses into the activation and function of Src kinase. Oncogene 2000, 19, 5620–5635. [Google Scholar] [CrossRef] [Green Version]
- Ayala-Sanmartin, J. Cholesterol Enhances Phospholipid Binding and Aggregation of Annexins by Their Core Domain. Biochem. Biophys. Res. Commun. 2001, 283, 72–79. [Google Scholar] [CrossRef]
- Codding, S.J.; Marty, N.; Abdullah, N.; Johnson, C.P. Dysferlin Binds SNAREs (Soluble N-Ethylmaleimide-sensitive Factor (NSF) Attachment Protein Receptors) and Stimulates Membrane Fusion in a Calcium-sensitive Manner*. J. Boil. Chem. 2016, 291, 14575–14584. [Google Scholar] [CrossRef] [Green Version]
- Cenacchi, G.; Fanin, M.; De Giorgi, L.B.; Angelini, C. Ultrastructural changes in dysferlinopathy support defective membrane repair mechanism. J. Clin. Pathol. 2005, 58, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagliani, R.; Magri, F.; Toscano, A.; Merlini, L.; Fortunato, F.; Lamperti, C.; Rodolico, C.; Prelle, A.; Sironi, M.; Aguennouz, M.; et al. Mutation finding in patients with dysferlin deficiency and role of the dysferlin interacting proteins annexin A1 and A2 in muscular dystrophies. Hum. Mutat. 2005, 26, 283. [Google Scholar] [CrossRef] [PubMed]
- Kesari, A.; Fukuda, M.; Knoblach, S.; Bashir, R.; Nader, G.A.; Rao, D.A.; Nagaraju, K.; Hoffman, E.P. Dysferlin Deficiency Shows Compensatory Induction of Rab27A/Slp2a That May Contribute to Inflammatory Onset. Am. J. Pathol. 2008, 173, 1476–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogarth, M.W.; Defour, A.; Lazarski, C.; Gallardo, E.; Manera, J.D.; Partridge, T.A.; Nagaraju, K.; Jaiswal, J.K. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat. Commun. 2019, 10, 2430. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bittel, D.C.; Chandra, G.; Tirunagri, L.M.S.; Deora, A.B.; Medikayala, S.; Scheffer, L.; Defour, A.; Jaiswal, J.K. Annexin A2 Mediates Dysferlin Accumulation and Muscle Cell Membrane Repair. Cells 2020, 9, 1919. https://doi.org/10.3390/cells9091919
Bittel DC, Chandra G, Tirunagri LMS, Deora AB, Medikayala S, Scheffer L, Defour A, Jaiswal JK. Annexin A2 Mediates Dysferlin Accumulation and Muscle Cell Membrane Repair. Cells. 2020; 9(9):1919. https://doi.org/10.3390/cells9091919
Chicago/Turabian StyleBittel, Daniel C., Goutam Chandra, Laxmi M. S. Tirunagri, Arun B. Deora, Sushma Medikayala, Luana Scheffer, Aurelia Defour, and Jyoti K. Jaiswal. 2020. "Annexin A2 Mediates Dysferlin Accumulation and Muscle Cell Membrane Repair" Cells 9, no. 9: 1919. https://doi.org/10.3390/cells9091919