Involvement of JNK1 in Neuronal Polarization During Brain Development

 , , , ,
, , , ,  and
and 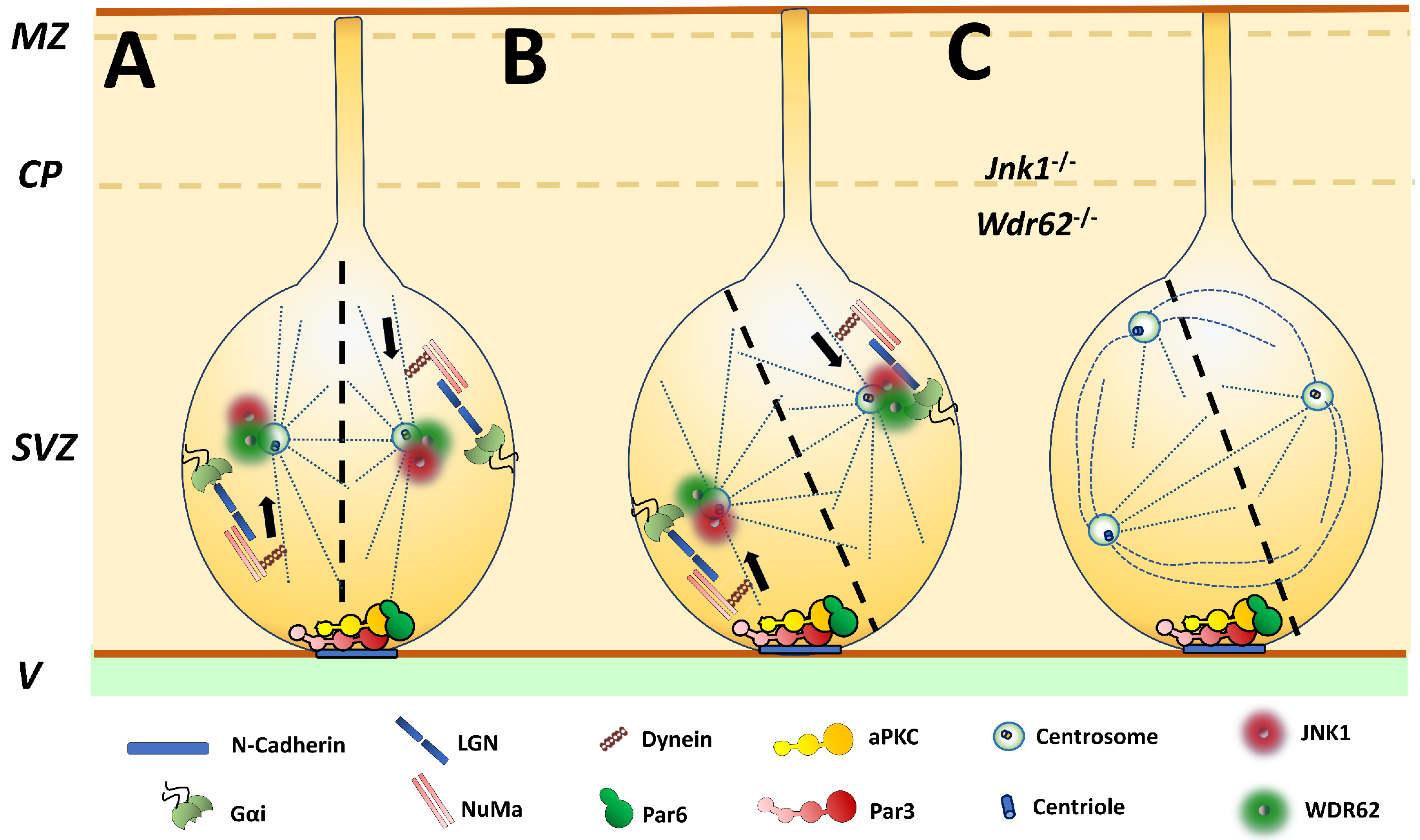{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. The JNKs Signaling Cascade
1.2. Cytosolic Substrates of JNK in Developing Neurons
1.3. JNKs Have Specific Functions. JNK1 is Essential for Proper CNS Development
1.4. JNKs Regulate Programmed Cell Death During Brain Development
2. Polarization of Cell Division: JNK1 and Asymmetric Division of Neural Stem Cells
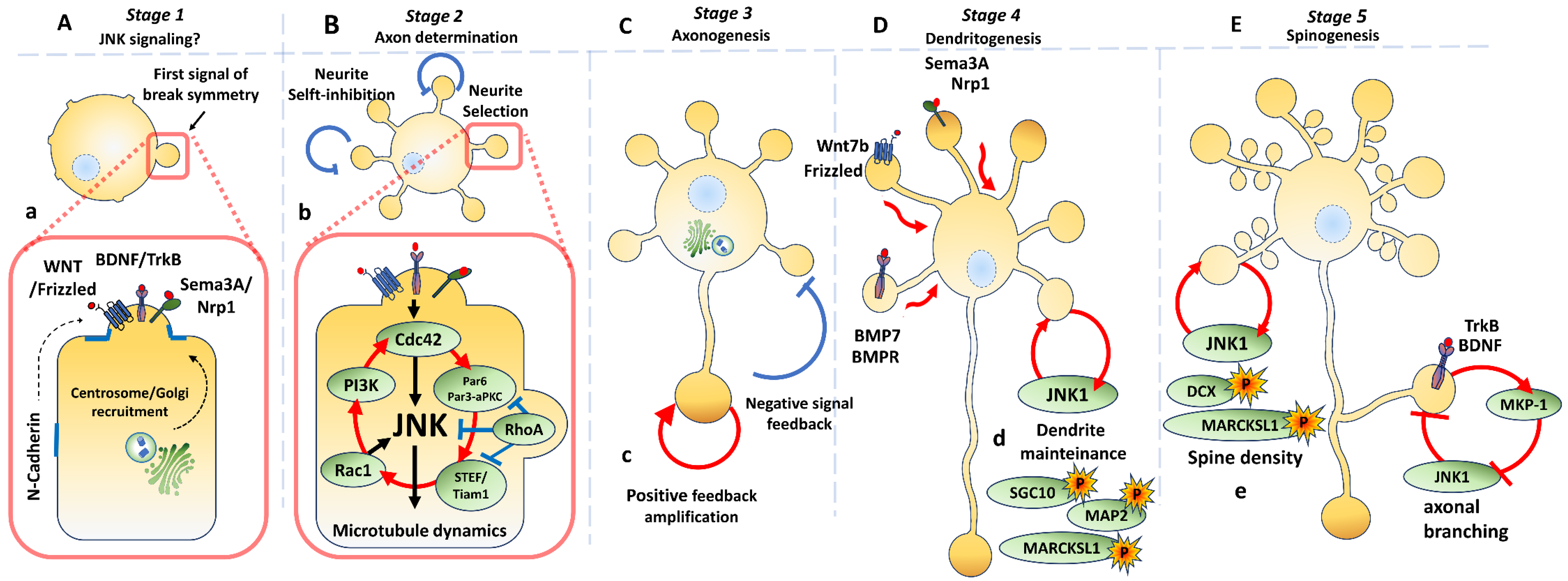
3. Neuronal Polarization
3.1. Molecular Signalling Along Different Stages
3.2. Axon Determination
3.2.1. Ras Homologous (Rho) GTPase Family
3.2.2. WNT/Frizzled/Disheveled Pathway
3.3. Axonogenesis
3.4. Dendritic Organization
3.4.1. Dendritogenesis
3.4.2. Dendrite Maintenance
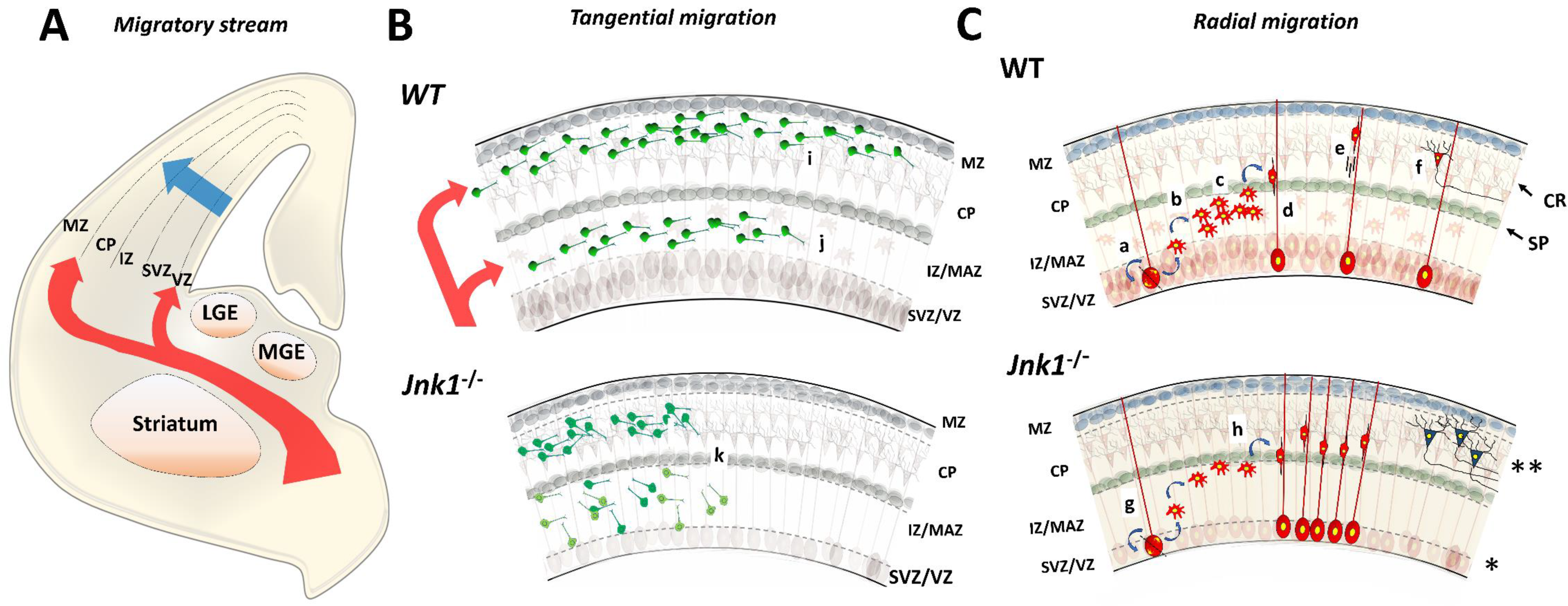
4. Neuronal Migration
4.1. Tangential Migration
4.2. Radial Migration
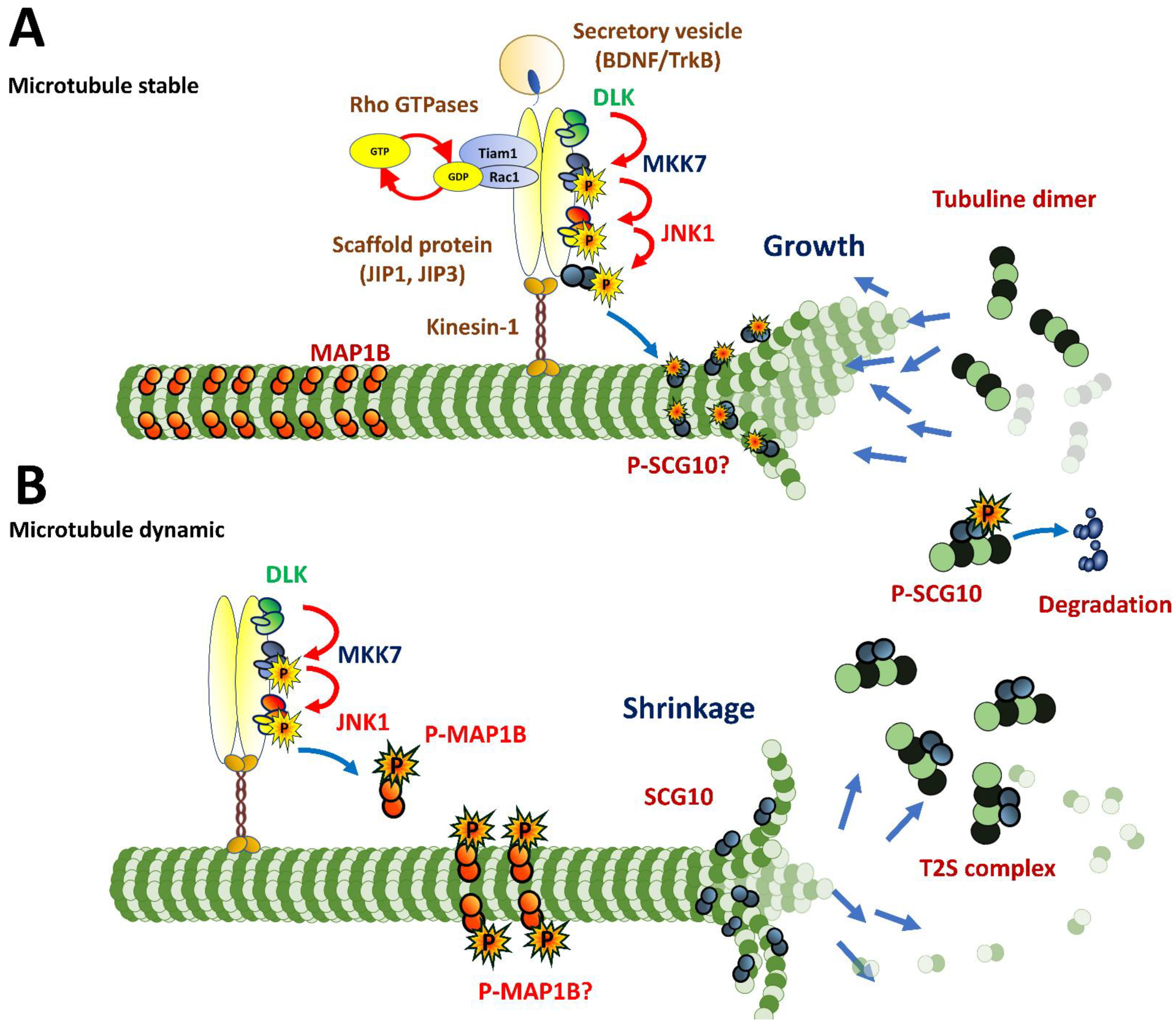
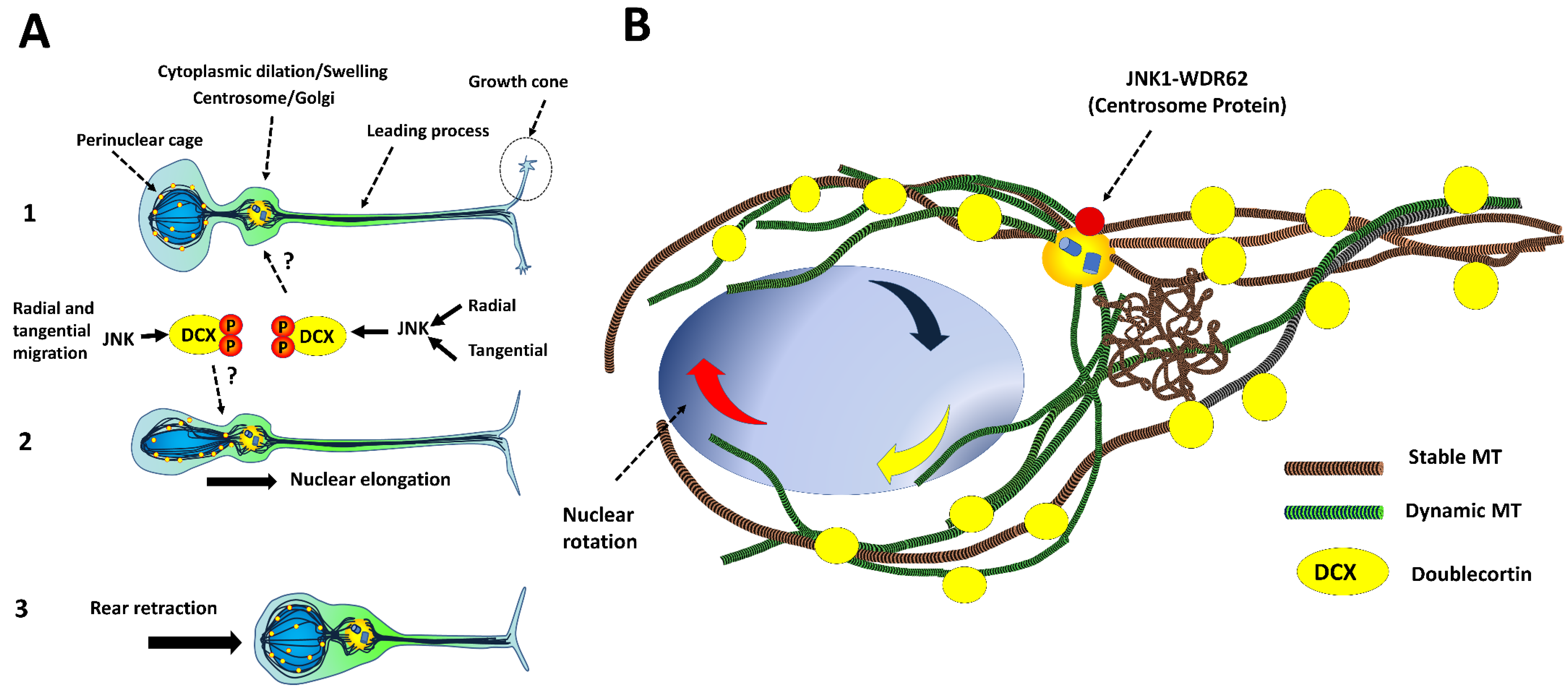
4.3. Intracellular Dynamics of Microtubules in Migratory Neurons
- (a)
- DCX decorates microtubules in the perinuclear cage and the growth cone of the leading process [36].
- (b)
- (c)
- Phospho-DCX, phosphor-SGC10 and phospho-JNK overlap in the intermediate zone in developing cortices. Jnk1−/− exhibits reductions in phosphorylation rates at Ser73 of SGC10 in the cortical layers [115].
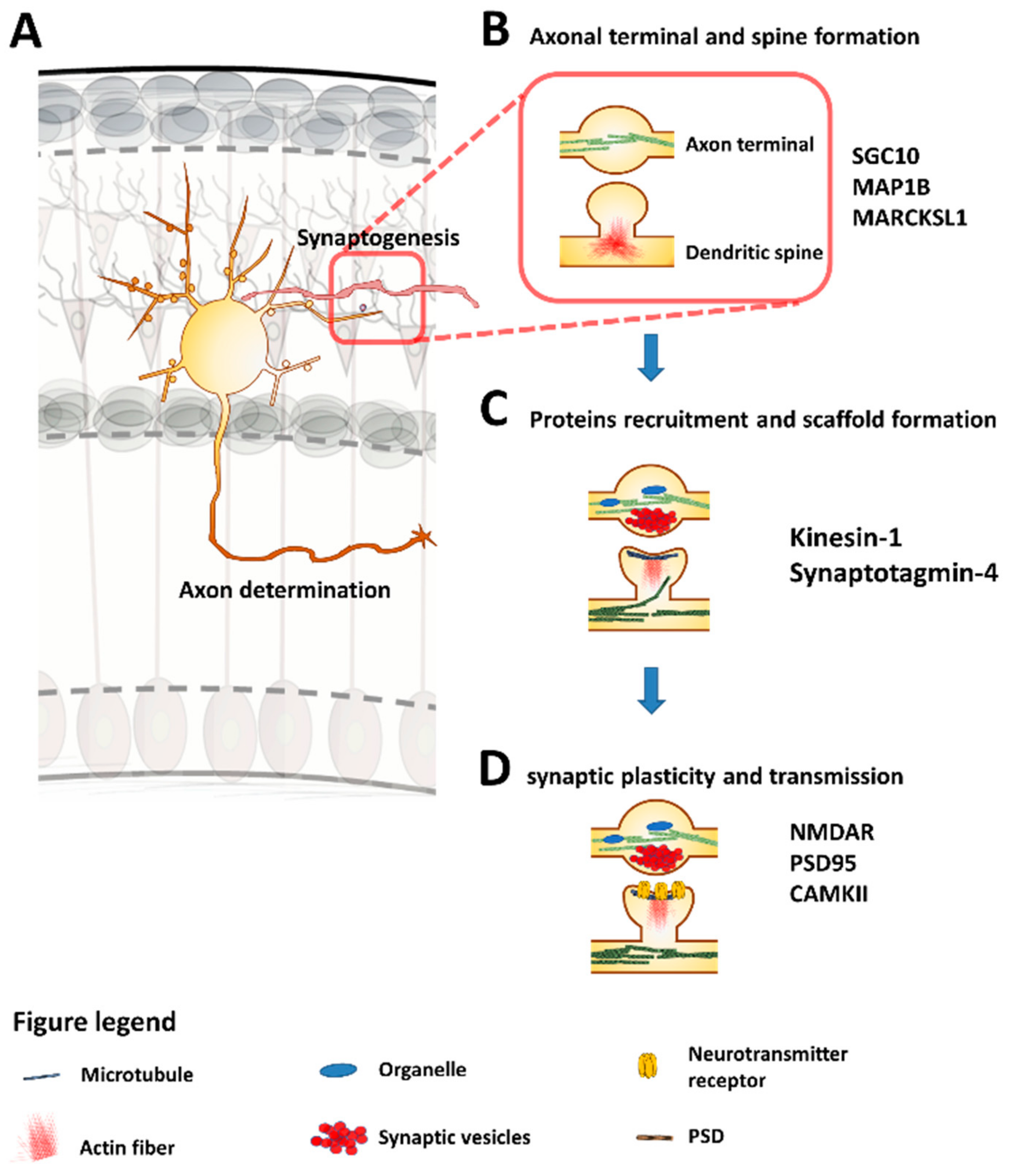
5. Synaptogenesis
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| aPKC | atypical Protein Kinase C |
| BDNF | Brain-Derived Neurotrophic Factor |
| BMP7 | Bone Morphogenic Protein 7 |
| BMPR | Bone Morphogenic Protein Receptors |
| Cdc42 | Cell Division Cycle 42 |
| CP | cortical plate |
| CR | Cajal-Retzius cell |
| DCX | Doublecortin |
| DLK | Dual Leucine Zipper Kinase 1 |
| Frizzled | Wnt receptor |
| GE | Ganglionic eminence |
| Gαi | subunit of heterotrimeric G protein |
| IZ | Intermedial zone |
| JIP | JNK (c-Jun N-terminal kinase)-interacting proteins |
| JNK1 | c-Jun N-terminal kinase 1 |
| LGE | lateral ganglionic eminence |
| LGN | Leu-Gly-Asn Partner of Inscuteable (Pins) |
| MAP1B | Microtubule-associated Protein 1B |
| MAP2 | Microtubule-associated Protein 2 |
| MAZ | multipolar accumulation zone |
| MGE | medial ganglionic eminence |
| MKK7 | Mitogen-Activated Protein Kinase Kinase 7. |
| MZ | Marginal zone |
| NRP1 | Neuropilin-1 |
| NSC | Neural Stem Cells |
| Numa | Nuclear Mitotic Apparatus Protein |
| Par3 | Partitioning Defective 3n(C.Elegans) Homolog |
| Par6 | Partitioning Defective 6 (C.Elegans) Homolog |
| PI3K | Phosphatidylinositol-4: 5-Bisphosphate 3-Kinase |
| Rac1 | Rac Family Small GTPase 1 |
| RacGEFs | Guanine Nucleotide Exchange Factors |
| RG | Radial Glial cells |
| RhoA | Ras Homolog Family Member A |
| SCG10 | Superior Cervical Ganglia Neural Specific-10 |
| Sema 3A | Semaphorina3A |
| SP | subplate |
| STEF | Sif- And Tiam1-Like Exchange Factor |
| SVZ | subventricular zone |
| Tiam1 | Lymphoma Invasion-and Metastasis-Inducing Protein 1 |
| TrkB | Tirosine Kinase Receptor B |
| V | ventricle |
| VZ | ventricular zone |
| WDR62 | WD40-(tryptophan[W]-aspartic[D] dipeptide 40)-Repeated Domain 62 protein. |
| Wnt | Wingless Proteins family member. |
References
- Sabapathy, K. Role of the JNK Pathway in Human Diseases; Elsevier: Amsterdam, The Netherlands, 2012; pp. 145–169. [Google Scholar]
- Sun, Y.; Liu, W.-Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Coffey, E.T. Nuclear and cytosolic JNK signalling in neurons. Nat. Rev. Neurosci. 2014, 15, 285–299. [Google Scholar] [CrossRef]
- Cui, J.; Zhang, M.; Zhang, Y.-Q.; Xu, Z. JNK pathway: diseases and therapeutic potential. Acta Pharmacol. Sin. 2007, 28, 601–608. [Google Scholar] [CrossRef]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Boil. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Boil. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Nikoloudaki, G.; Brooks, S.; Peidl, A.P.; Tinney, D.; Hamilton, D.W. JNK Signaling as a Key Modulator of Soft Connective Tissue Physiology, Pathology, and Healing. Int. J. Mol. Sci. 2020, 21, 1015. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, F.; Wang, Y.; Sun, Y.; Xu, Z. Microcephaly-Associated Protein WDR62 Regulates Neurogenesis through JNK1 in the Developing Neocortex. Cell Rep. 2014, 6, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Walters, G.B.; Gustafsson, O.; Sveinbjornsson, G.; Eiriksdottir, V.K.; Agustsdottir, A.B.; Jonsdottir, G.A.; Steinberg, S.; Gunnarsson, A.F.; Magnusson, M.I.; Unnsteinsdottir, U.; et al. MAP1B mutations cause intellectual disability and extensive white matter deficit. Nat. Commun. 2018, 9, 3456. [Google Scholar] [CrossRef]
- Reiner, O. LIS1 and DCX: Implications for Brain Development and Human Disease in Relation to Microtubules. Science 2013, 2013, 1–17. [Google Scholar] [CrossRef]
- Auladell, C.; De Lemos, L.; Verdaguer, E.; Ettcheto, M.; Busquets, O.; Lazarowski, A.; Beas-Zarate, C.; Olloquequi, J.; Folch, J.; Camins, A. Role of JNK isoforms in the kainic acid experimental model of epilepsy and neurodegeneration. Front. Biosci. 2017, 22, 795–814. [Google Scholar] [CrossRef]
- Mukaetova-Ladinska, E.B.; Arnold, H.; Jaros, E.; Perry, R.; Perry, E. Depletion of MAP2 expression and laminar cytoarchitectonic changes in dorsolateral prefrontal cortex in adult autistic individuals. Neuropathol. Appl. Neurobiol. 2004, 30, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Busquets, O.; Ettcheto, M.; Verdaguer, E.; Castro-Torres, R.D.; Auladell, C.; Beas-Zárate, C.; Folch, J.; Camins, A.; Dario, R. JNK1 inhibition by Licochalcone A leads to neuronal protection against excitotoxic insults derived of kainic acid. Neuropharmacology 2018, 131, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Castro-Torres, R.D.; Landa, J.; Rabaza, M.; Busquets, O.; Olloquequi, J.; Ettcheto, M.; Beas-Zarate, C.; Folch, J.; Camins, A.; Auladell, C.; et al. JNK Isoforms Are Involved in the Control of Adult Hippocampal Neurogenesis in Mice, Both in Physiological Conditions and in an Experimental Model of Temporal Lobe Epilepsy. Mol. Neurobiol. 2019, 56, 5856–5865. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.T.; Hongisto, V.; Dickens, M.; Davis, R.J.; Courtney, M.J. Dual Roles for c-Jun N-Terminal Kinase in Developmental and Stress Responses in Cerebellar Granule Neurons. J. Neurosci. 2000, 20, 7602–7613. [Google Scholar] [CrossRef] [PubMed]
- De Lemos, L.; Junyent, F.; Camins, A.; Castro-Torres, R.D.; Folch, J.; Olloquequi, J.; Beas-Zarate, C.; Verdaguer, E.; Auladell, C.; Auladell, C. Neuroprotective Effects of the Absence of JNK1 or JNK3 Isoforms on Kainic Acid-Induced Temporal Lobe Epilepsy-Like Symptoms. Mol. Neurobiol. 2017, 55, 4437–4452. [Google Scholar] [CrossRef]
- Nadal-Ribelles, M.; Solé, C.; Martínez-Cebrián, G.; Posas, F.; De Nadal, E. Shaping the Transcriptional Landscape through MAPK Signaling. In Gene Expression and Control; IntechOpen: London, UK, 2019. [Google Scholar]
- Tredici, G.; Miloso, M.; Scuteri, A.; Foudah, D. MAPKs as mediators of cell fate determination: an approach to neurodegenerative diseases. Curr. Med. Chem. 2008, 15, 538–548. [Google Scholar] [CrossRef]
- Morrison, D.K.; Davis, R.J. Regulation of MAP Kinase Signaling Modules by Scaffold Proteins in Mammals. Annu. Rev. Cell Dev. Boil. 2003, 19, 91–118. [Google Scholar] [CrossRef]
- Treiber, D.K.; Shah, N.P. Ins and outs of kinase DFG motifs. Chem. Boil. 2013, 20, 745–746. [Google Scholar] [CrossRef]
- Mishra, P.; Günther, S. New insights into the structural dynamics of the kinase JNK3. Sci. Rep. 2018, 8, 9435. [Google Scholar] [CrossRef]
- Tönges, L.; Planchamp, V.; Koch, J.C.; Herdegen, T.; Bähr, M.; Lingor, P. JNK Isoforms Differentially Regulate Neurite Growth and Regeneration in Dopaminergic Neurons In Vitro. J. Mol. Neurosci. 2011, 45, 284–293. [Google Scholar] [CrossRef]
- Mehan, S.; Meena, H.; Sharma, D.; Sankhla, R. JNK: A Stress-Activated Protein Kinase Therapeutic Strategies and Involvement in Alzheimer’s and Various Neurodegenerative Abnormalities. J. Mol. Neurosci. 2010, 43, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.M.; Moser, R.; Régulier, E.; Breuillaud, L.; Dixon, M.; Beesen, A.A.; Elliston, L.; Santos, M.D.F.S.; Kim, J.; Jones, L.; et al. MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington’s disease via additive effects of JNK and p38 inhibition. J. Neurosci. 2013, 33, 2313–2325. [Google Scholar] [CrossRef] [PubMed]
- Perrin, V.; Dufour, N.; Raoul, C.; Hassig, R.; Brouillet, E.; Aebischer, P.; Luthi-Carter, R.; Déglon, N. Implication of the JNK pathway in a rat model of Huntington’s disease. Exp. Neurol. 2009, 215, 191–200. [Google Scholar] [CrossRef]
- Morfini, G.; Pigino, G.; Szebenyi, G.; You, Y.; Pollema, S.; Brady, S.T. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat. Neurosci. 2006, 9, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Pirvola, U.; Xing-Qun, L.; Virkkala, J.; Saarma, M.; Murakata, C.; Camoratto, A.M.; Walton, K.M.; Ylikoski, J. Rescue of Hearing, Auditory Hair Cells, and Neurons by CEP-1347/KT7515, an Inhibitor of c-Jun N-Terminal Kinase Activation. J. Neurosci. 2000, 20, 43–50. [Google Scholar] [CrossRef]
- Qu, C.; Li, W.; Shao, Q.; Dwyer, T.; Huang, H.; Yang, T.; Liu, G. c-Jun N-terminal Kinase 1 (JNK1) Is Required for Coordination of Netrin Signaling in Axon Guidance*. J. Boil. Chem. 2012, 288, 1883–1895. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, S.A.; Duprez, L.; Hagens, O.; Waetzig, V.; Menzel, C.; Herdegen, T.; Schweiger, S.; Dan, B.; Vamos, E.; Ropers, H.-H.; et al. Truncation of the CNS-expressed JNK3 in a patient with a severe developmental epileptic encephalopathy. Qual. Life Res. 2005, 118, 559–567. [Google Scholar] [CrossRef]
- Kunde, S.-A.; Rademacher, N.; Tzschach, A.; Wiedersberg, E.; Ullmann, R.; Kalscheuer, V.M.; Shoichet, S.A. Characterisation of de novo MAPK10/JNK3 truncation mutations associated with cognitive disorders in two unrelated patients. Qual. Life Res. 2013, 132, 461–471. [Google Scholar] [CrossRef]
- Winchester, C.L.; Ohzeki, H.; Vouyiouklis, D.A.; Thompson, R.; Penninger, J.M.; Yamagami, K.; Norrie, J.D.; Hunter, R.; Pratt, J.A.; Morris, B.J. Converging evidence that sequence variations in the novel candidate gene MAP2K7 (MKK7) are functionally associated with schizophrenia. Hum. Mol. Genet. 2012, 21, 4910–4921. [Google Scholar] [CrossRef]
- McCarthy, S.E.; Wellcome Trust Case Control Consortium; Makarov, V.; Kirov, G.; Addington, A.M.; McClellan, J.; Yoon, S.; Perkins, D.O.; Dickel, D.E.; Kusenda, M.; et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 2009, 41, 1223–1227. [Google Scholar] [CrossRef]
- Mohammad, H.; Marchisella, F.; Ortega-Martinez, S.; Hollos, P.; Eerola, K.; Komulainen, E.; Kulesskaya, N.; Freemantle, E.; Fagerholm, V.; Savontous, E.; et al. JNK1 controls adult hippocampal neurogenesis and imposes cell-autonomous control of anxiety behaviour from the neurogenic niche. Mol. Psychiatry 2016, 23, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Busquets, O.; Ettcheto, M.; Eritja, À.; Espinosa-Jiménez, T.; Verdaguer, E.; Olloquequi, J.; Beas-Zarate, C.; Castro-Torres, R.D.; Casadesús, G.; Auladell, C.; et al. c-Jun N-terminal Kinase 1 ablation protects against metabolic-induced hippocampal cognitive impairments. J. Mol. Med. 2019, 97, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.H.; Utton, M.A.; Gibb, G.M.; Yates, A.; Anderton, B.H. Stress-Activated Protein Kinase/c-Jun N-Terminal Kinase Phosphorylates ? Protein. J. Neurochem. 2002, 68, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Gdalyahu, A.; Ghosh, I.; Levy, T.; Sapir, T.; Sapoznik, S.; Fishler, Y.; Azoulai, D.; Reiner, O. DCX, a new mediator of the JNK pathway. EMBO J. 2004, 23, 823–832. [Google Scholar] [CrossRef]
- Neidhart, S.; Antonsson, B.; Gillieron, C.; Vilbois, F.; Grenningloh, G.; Arkinstall, S. c-Jun N-terminal kinase-3 (JNK3)/stress-activated protein kinase-β (SAPKβ) binds and phosphorylates the neuronal microtubule regulator SCG10. FEBS Lett. 2001, 508, 259–264. [Google Scholar] [CrossRef]
- Kawasaki, A.; Okada, M.; Tamada, A.; Okuda, S.; Nozumi, M.; Ito, Y.; Kobayashi, D.; Yamasaki, T.; Yokoyama, R.; Shibata, T.; et al. Growth Cone Phosphoproteomics Reveals that GAP-43 Phosphorylated by JNK Is a Marker of Axon Growth and Regeneration. iScience 2018, 4, 190–203. [Google Scholar] [CrossRef]
- Feltrin, D.; Fusco, L.; Witte, H.; Moretti, F.; Martin, K.; Letzelter, M.; Fluri, E.; Scheiffele, P.; Pertz, O. Growth Cone MKK7 mRNA Targeting Regulates MAP1b-Dependent Microtubule Bundling to Control Neurite Elongation. PLoS Boil. 2012, 10, e1001439. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Hochedlinger, K.; Nam, S.Y.; Bauer, A.; Karin, M.; Wagner, E.F. Distinct Roles for JNK1 and JNK2 in Regulating JNK Activity and c-Jun-Dependent Cell Proliferation. Mol. Cell 2004, 15, 713–725. [Google Scholar] [CrossRef]
- Chang, L.; Jones, Y.; Ellisman, M.H.; Goldstein, L.S.; Karin, M. JNK1 Is Required for Maintenance of Neuronal Microtubules and Controls Phosphorylation of Microtubule-Associated Proteins. Dev. Cell 2003, 4, 521–533. [Google Scholar] [CrossRef]
- Sabapathy, K.; Jochum, W.; Hochedlinger, K.; Chang, L.; Karin, M.; Wagner, E.F. Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech. Dev. 1999, 89, 115–124. [Google Scholar] [CrossRef]
- Kuan, C.-Y.; Yang, D.D.; Roy, D.R.; Davis, R.J.; Rakic, P.; Flavell, R.A. The Jnk1 and Jnk2 Protein Kinases Are Required for Regional Specific Apoptosis during Early Brain Development. Neuron 1999, 22, 667–676. [Google Scholar] [CrossRef]
- Weston, C.R.; Wong, A.; Hall, J.P.; Goad, M.E.; Flavell, R.A.; Davis, R.J. JNK initiates a cytokine cascade that causes Pax2 expression and closure of the optic fissure. Genes Dev. 2003, 17, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.D.; Kuan, C.-Y.; Whitmarsh, A.J.; Rinócn, M.; Zheng, T.S.; Davis, R.J.; Rakic, P.; Flavell, R.A.; Rincón, M. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997, 389, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Kuan, C.-Y.; Whitmarsh, A.J.; Yang, D.D.; Liao, G.; Schloemer, A.J.; Dong, C.; Bao, J.; Banasiak, K.J.; Haddad, G.G.; Flavell, R.A.; et al. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 15184–15189. [Google Scholar] [CrossRef]
- Mondal, T.; Bag, I.; Sncvl, P.; Garikapati, K.R.; Bhadra, U.; Bhadra, M.P. Two way controls of apoptotic regulators consign DmArgonaute-1 a better clasp on it. PLoS ONE 2018, 13, e0190548. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, C.; Wu, C.; Yang, Y.; Li, W.; Xue, L. The canonical Wg signaling modulates Bsk-mediated cell death in Drosophila. Cell Death Dis. 2015, 6, e1713. [Google Scholar] [CrossRef]
- Götz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Boil. 2005, 6, 777–788. [Google Scholar] [CrossRef]
- Culurgioni, S.; Mapelli, M. Going vertical: functional role and working principles of the protein Inscuteable in asymmetric cell divisions. Cell. Mol. Life Sci. 2013, 70, 4039–4046. [Google Scholar] [CrossRef]
- Mapelli, M.; Gonzalez, C. On the inscrutable role of Inscuteable: structural basis and functional implications for the competitive binding of NuMA and Inscuteable to LGN. Open Boil. 2012, 2, 120102. [Google Scholar] [CrossRef][Green Version]
- MacCorkle-Chosnek, R.A.; VanHooser, A.; Goodrich, D.E.; Brinkley, B.; Tan, T.-H. Cell Cycle Regulation of c-Jun N-Terminal Kinase Activity at the Centrosomes. Biochem. Biophys. Res. Commun. 2001, 289, 173–180. [Google Scholar] [CrossRef]
- Konno, D.; Shioi, G.; Shitamukai, A.; Mori, A.; Kiyonari, H.; Miyata, T.; Matsuzaki, F. Neuroepithelial progenitors undergo LGN-dependent planar divisions to maintain self-renewability during mammalian neurogenesis. Nature 2007, 10, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, S.-L.; Yang, M.; Herrlinger, S.; Shao, Q.; Collar, J.L.; Fierro, E.; Shi, Y.; Liu, A.; Lu, H.; et al. Modeling microcephaly with cerebral organoids reveals a WDR62–CEP170–KIF2A pathway promoting cilium disassembly in neural progenitors. Nat. Commun. 2019, 10, 2612. [Google Scholar] [CrossRef] [PubMed]
- Shohayeb, B.; Lim, N.R.; Ho, U.; Xu, Z.; Dottori, M.; Quinn, L.; Ng, D.C.H. The Role of WD40-Repeat Protein 62 (MCPH2) in Brain Growth: Diverse Molecular and Cellular Mechanisms Required for Cortical Development. Mol. Neurobiol. 2017, 55, 5409–5424. [Google Scholar] [CrossRef] [PubMed]
- Westerlund, N.; Zdrojewska, J.; Padzik, A.; Komulainen, E.; Björkblom, B.; Rannikko, E.; Tararuk, T.; García-Frigola, C.; Sandholm, J.; Nguyen, L.; et al. Phosphorylation of SCG10/stathmin-2 determines multipolar stage exit and neuronal migration rate. Nat. Neurosci. 2011, 14, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Lim, N.R.; Yeap, Y.Y.C.; Zhao, T.T.; Yip, Y.Y.; Wong, S.C.; Xu, D.; Ang, C.-S.; Williamson, N.A.; Xu, Z.; Bogoyevitch, M.A.; et al. Opposing roles for JNK and Aurora A in regulating the association of WDR62 with spindle microtubules. J. Cell Sci. 2014, 128, 527–540. [Google Scholar] [CrossRef]
- Conde, C.; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef]
- Schelski, M.; Bradke, F. Neuronal polarization: From spatiotemporal signaling to cytoskeletal dynamics. Mol. Cell. Neurosci. 2017, 84, 11–28. [Google Scholar] [CrossRef]
- Yogev, S.; Shen, K. Establishing Neuronal Polarity with Environmental and Intrinsic Mechanisms. Neuron 2017, 96, 638–650. [Google Scholar] [CrossRef]
- Dotti, C.; Sullivan, C.; Banker, G. The establishment of polarity by hippocampal neurons in culture. J. Neurosci. 1988, 8, 1454–1468. [Google Scholar] [CrossRef]
- Da Silva, J.S.; Dotti, C.G. Breaking the neuronal sphere: regulation of the actin cytoskeleton in neuritogenesis. Nat. Rev. Neurosci. 2002, 3, 694–704. [Google Scholar] [CrossRef]
- Poulain, F.E.; Sobel, A. The microtubule network and neuronal morphogenesis: Dynamic and coordinated orchestration through multiple players. Mol. Cell. Neurosci. 2010, 43, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Toriyama, M.; Sakumura, Y. Systems biology of symmetry breaking during neuronal polarity formation. Dev. Neurobiol. 2011, 71, 584–593. [Google Scholar] [CrossRef] [PubMed]
- De Anda, F.C.; Pollarolo, G.; Da Silva, J.S.; Camoletto, P.G.; Feiguin, F.; Dotti, C.G. Centrosome localization determines neuronal polarity. Nature 2005, 436, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Linstedt, A. Golgi Positioning. Cold Spring Harb. Perspect. Boil. 2011, 3, a005322. [Google Scholar] [CrossRef]
- Gärtner, A.; Fornasiero1, E.F.; Munck, S.; Vennekens, K.; Seuntjens, E.; Huttner, W.B.; Valtorta, F.; Dotti, C.G. N-cadherin specifies first asymmetry in developing neurons. EMBO J. 2012, 31, 1893–1903. [Google Scholar] [CrossRef]
- Levkovitz, Y.; Baraban, J.M. A Dominant Negative Egr Inhibitor Blocks Nerve Growth Factor-Induced Neurite Outgrowth by Suppressing c-Jun Activation: Role of an Egr/c-Jun Complex. J. Neurosci. 2002, 22, 3845–3854. [Google Scholar] [CrossRef]
- York, R.D.; Yao, H.; Dillon, T.; Ellig, C.L.; Eckert, S.P.; McCleskey, E.W.; Stork, P.A. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 1998, 392, 622–626. [Google Scholar] [CrossRef]
- Yu, Y.-M.; Han, P.-L.; Lee, J.-K. JNK pathway is required for retinoic acid-induced neurite outgrowth of human neuroblastoma, SH-SY5Y. NeuroReport 2003, 14, 941–945. [Google Scholar] [CrossRef]
- Bodmer, D.; Gloddek, B.; Ryan, A.F.; Huverstuhl, J.; Brors, D. Inhibition of the c-Jun N-Terminal Kinase Signaling Pathway Influences Neurite Outgrowth of Spiral Ganglion Neurons In Vitro. Laryngoscope 2002, 112, 2057–2061. [Google Scholar] [CrossRef]
- Eom, D.-S.; Choi, W.-S.; Ji, S.; Cho, J.W.; Oh, Y.J. Activation of c-Jun N-terminal kinase is required for neurite outgrowth of dopaminergic neuronal cells. NeuroReport 2005, 16, 823–828. [Google Scholar] [CrossRef]
- Takano, T.; Funahashi, Y.; Kaibuchi, K. Neuronal Polarity: Positive and Negative Feedback Signals. Front. Cell Dev. Boil. 2019, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Hapak, S.M.; Rothlin, C.V.; Ghosh, S. PAR3–PAR6–atypical PKC polarity complex proteins in neuronal polarization. Cell. Mol. Life Sci. 2018, 75, 2735–2761. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.; Macara, I.G. The PAR Proteins: Fundamental Players in Animal Cell Polarization. Dev. Cell 2007, 13, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.H.; Duellberg, C.; Mieck, C.; Loose, M.; Hippenmeyer, S. Cell Polarity in Cerebral Cortex Development—Cellular Architecture Shaped by Biochemical Networks. Front. Cell. Neurosci. 2017, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C. Cytoskeletal Symmetry Breaking and Chirality: From Reconstituted Systems to Animal Development. Symmetry 2015, 7, 2062–2107. [Google Scholar] [CrossRef]
- Matsuo, N.; Hoshino, M.; Yoshizawa, M.; Nabeshima, Y.-I. Characterization of STEF, a Guanine Nucleotide Exchange Factor for Rac1, Required for Neurite Growth. J. Boil. Chem. 2001, 277, 2860–2868. [Google Scholar] [CrossRef]
- Coso, O.A.; Chiariello, M.; Yu, J.-C.; Teramoto, H.; Crespo, P.; Xu, N.; Miki, T.; Gutkind, J.S. The small GTP-binding proteins Rac1 and Cdc42regulate the activity of the JNK/SAPK signaling pathway. Cell 1995, 81, 1137–1146. [Google Scholar] [CrossRef]
- Ciani, L.; Salinas, P.C. c-Jun N-terminal kinase (JNK) cooperates with Gsk3β to regulate Dishevelled-mediated microtubule stability. BMC Cell Boil. 2007, 8, 27. [Google Scholar] [CrossRef]
- Slater, P.G.; Ramírez, V.T.; Gonzalez-Billault, C.; Varela-Nallar, L.; Inestrosa, N.C. Frizzled-5 Receptor Is Involved in Neuronal Polarity and Morphogenesis of Hippocampal Neurons. PLoS ONE 2013, 8, e78892. [Google Scholar] [CrossRef]
- Oliva, A.A.; Atkins, C.M.; Copenagle, L.; Banker, G.A. Activated c-Jun N-Terminal Kinase Is Required for Axon Formation. J. Neurosci. 2006, 26, 9462–9470. [Google Scholar] [CrossRef]
- Meur, A.M.-L. JNK gives axons a second chance. J. Neurosci. 2006, 26, 12104–12105. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, C.; Schnapp, B.; Banker, G.A. A Change in the Selective Translocation of the Kinesin-1 Motor Domain Marks the Initial Specification of the Axon. Neuron 2006, 49, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Demura, T.; Morita, M.; Banker, G.A.; Tanii, T.; Nakamura, S. Differential neurite outgrowth is required for axon specification by cultured hippocampal neurons. J. Neurochem. 2012, 123, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wen, Q.; Zhang, X.-J.; Kan, Q.-C. Specific effects of c-Jun NH2-terminal kinase-interacting protein 1 in neuronal axons. Neural Regen. Res. 2016, 11, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Padzik, A.; Deshpande, P.; Hollos, P.; Franker, M.; Rannikko, E.; Cai, D.; Prus, P.; Mågård, M.; Westerlund, N.; Verhey, K.J.; et al. KIF5C S176 Phosphorylation Regulates Microtubule Binding and Transport Efficiency in Mammalian Neurons. Front. Cell. Neurosci. 2016, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Verhey, K.J.; Meyer, D.; Deehan, R.; Blenis, J.; Schnapp, B.J.; Rapoport, T.A.; Margolis, B. Cargo of Kinesin Identified as Jip Scaffolding Proteins and Associated Signaling Molecules. J. Cell Boil. 2001, 152, 959–970. [Google Scholar] [CrossRef]
- Marinissen, M.J.; Gutkind, J.S. Scaffold proteins dictate Rho GTPase-signaling specificity. Trends Biochem. Sci. 2005, 30, 423–426. [Google Scholar] [CrossRef]
- Bailador, F.D.; Jones, E.V.; Whitmarsh, A.J. The JIP1 Scaffold Protein Regulates Axonal Development in Cortical Neurons. Curr. Boil. 2008, 18, 221–226. [Google Scholar] [CrossRef]
- Hirai, S.-I.; Banba, Y.; Satake, T.; Ohno, S. Axon Formation in Neocortical Neurons Depends on Stage-Specific Regulation of Microtubule Stability by the Dual Leucine Zipper Kinase–c-Jun N-Terminal Kinase Pathway. J. Neurosci. 2011, 31, 6468–6480. [Google Scholar] [CrossRef]
- Sun, T.; Yu, N.; Zhai, L.-K.; Li, N.; Zhang, C.; Zhou, L.; Huang, Z.; Jiang, X.; Shen, Y.; Chen, Z.-Y. c-Jun NH2-terminal Kinase (JNK)-interacting Protein-3 (JIP3) Regulates Neuronal Axon Elongation in a Kinesin- and JNK-dependent Manner*. J. Boil. Chem. 2013, 288, 14531–14543. [Google Scholar] [CrossRef]
- Barnat, M.; Enslen, H.; Propst, F.; Davis, R.J.; Soares, S.; Nothias, F. Distinct Roles of c-Jun N-Terminal Kinase Isoforms in Neurite Initiation and Elongation during Axonal Regeneration. J. Neurosci. 2010, 30, 7804–7816. [Google Scholar] [CrossRef]
- Hirai, S.-I.; Cui, D.F.; Miyata, T.; Ogawa, M.; Kiyonari, H.; Suda, Y.; Aizawa, S.; Banba, Y.; Ohno, S. The c-Jun N-Terminal Kinase Activator Dual Leucine Zipper Kinase Regulates Axon Growth and Neuronal Migration in the Developing Cerebral Cortex. J. Neurosci. 2006, 26, 11992–12002. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.; Kawasaki, H.; Arakawa, S.; Shimizu, K.; Shimizu, S.; Reiner, O.; Okano, H.; Nishina, S.; Azuma, N.; Penninger, J.M.; et al. Stress-Activated Protein Kinase MKK7 Regulates Axon Elongation in the Developing Cerebral Cortex. J. Neurosci. 2011, 31, 16872–16883. [Google Scholar] [CrossRef] [PubMed]
- Nouar, R.; Breuzard, G.; Bastonero, S.; Gorokhova, S.; Barbier, P.; Devred, F.; Kovacic, H.; Peyrot, V. Direct evidence for the interaction of stathmin along the length and the plus end of microtubules in cells. FASEB J. 2016, 30, 3202–3215. [Google Scholar] [CrossRef]
- De Anda, F.C.; Rosário, A.L.; Durak, O.; Tran, T.; Gräff, J.; Meletis, K.; Rei, D.; Soda, T.; Madabhushi, R.; Ginty, D.D.; et al. Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat. Neurosci. 2012, 15, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Podkowa, M.; Zhao, X.; Chow, C.-W.; Coffey, E.T.; Davis, R.J.; Attisano, L. Microtubule Stabilization by Bone Morphogenetic Protein Receptor-Mediated Scaffolding of c-Jun N-Terminal Kinase Promotes Dendrite Formation. Mol. Cell. Boil. 2010, 30, 2241–2250. [Google Scholar] [CrossRef]
- Ferrari, M.E.; Bernis, M.E.; McLeod, F.; Podpolny, M.; Coullery, R.P.; Casadei, I.M.; Salinas, P.C.; Rosso, S.B. Wnt7b signalling through Frizzled-7 receptor promotes dendrite development by coactivating CaMKII and JNK. J. Cell Sci. 2018, 131, jcs216101. [Google Scholar] [CrossRef]
- Waetzig, V.; Zhao, Y.; Herdegen, T. The bright side of JNKs—Multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog. Neurobiol. 2006, 80, 84–97. [Google Scholar] [CrossRef]
- Komulainen, E.; Zdrojewska, J.; Freemantle, E.; Mohammad, H.; Kulesskaya, N.; Deshpande, P.; Marchisella, F.; Mysore, R.; Hollos, P.; Michelsen, K.A.; et al. JNK1 controls dendritic field size in L2/3 and L5 of the motor cortex, constrains soma size, and influences fine motor coordination. Front. Cell. Neurosci. 2014, 8, 8. [Google Scholar] [CrossRef]
- Björkblom, B.; Östman, N.; Hongisto, V.; Komarovski, V.; Filén, J.-J.; Nyman, T.A.; Kallunki, T.; Courtney, M.J.; Coffey, E.T. Constitutively Active Cytoplasmic c-Jun N-Terminal Kinase 1 Is a Dominant Regulator of Dendritic Architecture: Role of Microtubule-Associated Protein 2 as an Effector. J. Neurosci. 2005, 25, 6350–6361. [Google Scholar] [CrossRef]
- Jeanneteau, F.; Deinhardt, K.; Miyoshi, G.; Bennett, A.M.; Chao, M.V. The MAP kinase phosphatase MKP-1 regulates BDNF-induced axon branching. Nat. Neurosci. 2010, 13, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.S. Molecular Genetics of Neuronal Migration Disorders. Curr. Neurol. Neurosci. Rep. 2011, 11, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.V.; Nabeshima, Y.-I.; Kawauchi, T. Morphological and Molecular Basis of Cytoplasmic Dilation and Swelling in Cortical Migrating Neurons. Brain Sci. 2017, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Marín, O.; Valiente, M.; Ge, X.; Tsai, L.-H. Guiding Neuronal Cell Migrations. Cold Spring Harb. Perspect. Boil. 2010, 2, a001834. [Google Scholar] [CrossRef]
- Hatanaka, Y.; Zhu, Y.; Torigoe, M.; Kita, Y.; Murakami, F. From migration to settlement: the pathways, migration modes and dynamics of neurons in the developing brain. Proc. Jpn. Acad. Ser. B 2016, 92, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.K.; Cunningham, J.G.; Smith, S.E.; Snow, J.P.; Smoot, C.A.; Tucker, E.S. JNK signaling is required for proper tangential migration and laminar allocation of cortical interneurons. Development 2020, 147, dev180646. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.K.; Meechan, D.W.; Adney, D.R.; Tucker, E.S. Cortical Interneurons Require Jnk1 to Enter and Navigate the Developing Cerebral Cortex. J. Neurosci. 2014, 34, 7787–7801. [Google Scholar] [CrossRef] [PubMed]
- Bellion, A.; Baudoin, J.-P.; Alvarez, C.; Bornens, M.; Métin, C. Nucleokinesis in Tangentially Migrating Neurons Comprises Two Alternating Phases: Forward Migration of the Golgi/Centrosome Associated with Centrosome Splitting and Myosin Contraction at the Rear. J. Neurosci. 2005, 25, 5691–5699. [Google Scholar] [CrossRef]
- Higginbotham, H.R.; Gleeson, J.G. The centrosome in neuronal development. Trends Neurosci. 2007, 30, 276–283. [Google Scholar] [CrossRef]
- Ohtaka-Maruyama, C.; Okado, H. Molecular Pathways Underlying Projection Neuron Production and Migration during Cerebral Cortical Development. Front. Mol. Neurosci. 2015, 9, 447. [Google Scholar] [CrossRef]
- Mizutani, K.-I. Physiological significance of multipolar cells generated from neural stem cells and progenitors for the establishment of neocortical cytoarchitecture. Genes Cells 2017, 23, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Suzuki, H.; Mikoshiba, K.; Ohshima, T.; Hirai, S.-I. JNK phosphorylates Ser332 of doublecortin and regulates its function in neurite extension and neuronal migration. Dev. Neurobiol. 2010, 70, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Tararuk, T.; Östman, N.; Li, W.; Bjoörkblom, B.; Padzik, A.; Zdrojewska, J.; Hongisto, V.; Herdegen, T.; Konopka, W.; Courtney, M.J.; et al. JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J. Cell Boil. 2006, 173, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Coker, N.K.; Tucker, E.S. JNK signaling controls branching, nucleokinesis, and positioning of centrosomes and primary cilia in migrating cortical interneurons. bioRxiv 2020. [Google Scholar] [CrossRef]
- Petzoldt, A.G.; Sigrist, S.J. Synaptogenesis. Curr. Boil. 2014, 24, R1076–R1080. [Google Scholar] [CrossRef]
- Yoshihara, Y.; De Roo, M.; Muller, D. Dendritic spine formation and stabilization. Curr. Opin. Neurobiol. 2009, 19, 146–153. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Moser, H.W. Dendritic anomalies in disorders associated with mental retardation. Cereb. Cortex 2000, 10, 981–991. [Google Scholar] [CrossRef]
- Hotulainen, P.; Hoogenraad, C.C. Actin in dendritic spines: connecting dynamics to function. J. Cell Boil. 2010, 189, 619–629. [Google Scholar] [CrossRef]
- Biggi, S.; Buccarello, L.; Sclip, A.; Lippiello, P.; Tonna, N.; Rumio, C.; Di Marino, D.; Miniaci, M.C.; Borsello, T. Evidence of Presynaptic Localization and Function of the c-Jun N-Terminal Kinase. Neural Plast. 2017, 2017, 1–14. [Google Scholar] [CrossRef]
- Komulainen, E.; Varidaki, A.; Kulesskaya, N.; Mohammad, H.; Sourander, C.; Rauvala, H.; Coffey, E.T. Impact of JNK and Its Substrates on Dendritic Spine Morphology. Cells 2020, 9, 440. [Google Scholar] [CrossRef]
- Bodaleo, F.J.; Montenegro-Venegas, C.; Henríquez, D.R.; Court, F.A.; Gonzalez-Billault, C. Microtubule-associated protein 1B (MAP1B)-deficient neurons show structural presynaptic deficiencies in vitro and altered presynaptic physiology. Sci. Rep. 2016, 6, 30069. [Google Scholar] [CrossRef] [PubMed]
- Grenningloh, G.; Soehrman, S.; Bondallaz, P.; Ruchti, E.; Cadas, H. Role of the microtubule destabilizing proteins SCG10 and stathmin in neuronal growth. J. Neurobiol. 2003, 58, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Okada, M.; Honda, A.; Ito, Y.; Tamada, A.; Endo, N.; Igarashi, M. Phosphorylation sites of microtubule-associated protein 1B (MAP 1B) are involved in axon growth and regeneration. Mol. Brain 2019, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Björkblom, B.; Padzik, A.; Mohammad, H.; Westerlund, N.; Komulainen, E.; Hollos, P.; Parviainen, L.; Papageorgiou, T.; Iljin, K.; Kallioniemi, O.; et al. c-Jun N-Terminal Kinase Phosphorylation of MARCKSL1 Determines Actin Stability and Migration in Neurons and in Cancer Cells. Mol. Cell. Boil. 2012, 32, 3513–3526. [Google Scholar] [CrossRef] [PubMed]
- El Amri, M.; Fitzgerald, U.; Schlosser, G. MARCKS and MARCKS-like proteins in development and regeneration. J. Biomed. Sci. 2018, 25, 43. [Google Scholar] [CrossRef] [PubMed]
- Tortosa, E.; Montenegro-Venegas, C.; Benoist, M.; Härtel, S.; González-Billault, C.; Esteban, J.A.; Avila, J. Microtubule-associated Protein 1B (MAP1B) Is Required for Dendritic Spine Development and Synaptic Maturation. J. Boil. Chem. 2011, 286, 40638–40648. [Google Scholar] [CrossRef]
- Bharat, V.; Siebrecht, M.; Burk, K.; Ahmed, S.; Reissner, C.; Kohansal-Nodehi, M.; Steubler, V.; Zweckstetter, M.; Ting, J.T.; Dean, C. Capture of Dense Core Vesicles at Synapses by JNK-Dependent Phosphorylation of Synaptotagmin-4. Cell Rep. 2017, 21, 2118–2133. [Google Scholar] [CrossRef]
- Corlew, R.J.; Brasier, D.J.; Feldman, D.E.; Philpot, B.D. Presynaptic NMDA receptors: newly appreciated roles in cortical synaptic function and plasticity. Neuroscience 2008, 14, 609–625. [Google Scholar] [CrossRef]
- Deng, M.; Chen, S.-R.; Chen, H.; Luo, Y.; Dong, Y.; Pan, H.-L. Mitogen-activated protein kinase signaling mediates opioid-induced presynaptic NMDA receptor activation and analgesic tolerance. J. Neurochem. 2018, 148, 275–290. [Google Scholar] [CrossRef]
- Jordan, B.A.; Fernholz, B.D.; Boussac, M.; Xu, C.; Grigorean, G.; Ziff, E.B.; Neubert, T.A. Identification and Verification of Novel Rodent Postsynaptic Density Proteins. Mol. Cell. Proteom. 2004, 3, 857–871. [Google Scholar] [CrossRef]
- Reiner, O.; Coquelle, F.M.; Peter, B.; Levy, T.; Kaplan, A.; Sapir, T.; Orr, I.; Barkai, N.; Eichele, G.; Bergman, S. The evolving doublecortin (DCX) superfamily. BMC Genom. 2006, 7, 188. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.; Kashiwagi, Y.; Kuriu, T.; Iwasaki, H.; Tanaka, T.; Koizumi, H.; Gleeson, J.G.; Okabe, S. Doublecortin-like kinase enhances dendritic remodelling and negatively regulates synapse maturation. Nat. Commun. 2013, 4, 1440. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Futai, K.; Jo, J.; Hayashi, Y.; Cho, K.; Sheng, M. Synaptic Accumulation of PSD-95 and Synaptic Function Regulated by Phosphorylation of Serine-295 of PSD-95. Neuron 2007, 56, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.M.; Lin, D.-T.; Nuriya, M.; Huganir, R.L. Rapid and bi-directional regulation of AMPA receptor phosphorylation and trafficking by JNK. EMBO J. 2008, 27, 361–372. [Google Scholar] [CrossRef]
- Sung, Y.-J.; Wu, F.; Schacher, S.; Ambron, R.T. Synaptogenesis Regulates Axotomy-Induced Activation of c-Jun–Activator Protein-1 Transcription. J. Neurosci. 2006, 26, 6439–6449. [Google Scholar] [CrossRef]
- Morel, C.; Sherrin, T.; Kennedy, N.J.; Forest, K.H.; Barutcu, S.A.; Robles, M.; Carpenter-Hyland, E.; Alfulaij, N.; Standen, C.L.; Nichols, R.A.; et al. JIP1-Mediated JNK Activation Negatively Regulates Synaptic Plasticity and Spatial Memory. J. Neurosci. 2018, 38, 3708–3728. [Google Scholar] [CrossRef]
- Sherrin, T.; Blank, T.; Todorovic, C. c-Jun N-terminal kinases in memory and synaptic plasticity. Rev. Neurosci. 2011, 22, 403–410. [Google Scholar] [CrossRef]
- Hollos, P.; John, J.M.; Lehtonen, J.V.; Coffey, E.T. Optogenetic Control of Spine-Head JNK Reveals a Role in Dendritic Spine Regression. eneuro 2020, 7. [Google Scholar] [CrossRef]
- Kutlu, M.G.; Gould, T.J. Nicotinic modulation of hippocampal cell signaling and associated effects on learning and memory. Physiol. Behav. 2016, 155, 162–171. [Google Scholar] [CrossRef]
- Leach, P.T.; Kenney, J.W.; Gould, T.J. Stronger learning recruits additional cell-signaling cascades: c-Jun-N-terminal kinase 1 (JNK1) is necessary for expression of stronger contextual fear conditioning. Neurobiol. Learn. Mem. 2014, 118, 162–166. [Google Scholar] [CrossRef][Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro-Torres, R.D.; Busquets, O.; Parcerisas, A.; Verdaguer, E.; Olloquequi, J.; Ettcheto, M.; Beas-Zarate, C.; Folch, J.; Camins, A.; Auladell, C. Involvement of JNK1 in Neuronal Polarization During Brain Development. Cells 2020, 9, 1897. https://doi.org/10.3390/cells9081897
Castro-Torres RD, Busquets O, Parcerisas A, Verdaguer E, Olloquequi J, Ettcheto M, Beas-Zarate C, Folch J, Camins A, Auladell C. Involvement of JNK1 in Neuronal Polarization During Brain Development. Cells. 2020; 9(8):1897. https://doi.org/10.3390/cells9081897
Chicago/Turabian StyleCastro-Torres, Rubén Darío, Oriol Busquets, Antoni Parcerisas, Ester Verdaguer, Jordi Olloquequi, Miren Ettcheto, Carlos Beas-Zarate, Jaume Folch, Antoni Camins, and Carme Auladell. 2020. "Involvement of JNK1 in Neuronal Polarization During Brain Development" Cells 9, no. 8: 1897. https://doi.org/10.3390/cells9081897
APA StyleCastro-Torres, R. D., Busquets, O., Parcerisas, A., Verdaguer, E., Olloquequi, J., Ettcheto, M., Beas-Zarate, C., Folch, J., Camins, A., & Auladell, C. (2020). Involvement of JNK1 in Neuronal Polarization During Brain Development. Cells, 9(8), 1897. https://doi.org/10.3390/cells9081897