The Lipid Receptor G2A (GPR132) Mediates Macrophage Migration in Nerve Injury-Induced Neuropathic Pain

 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals
2.3. Spared Nerve Injury (SNI)
2.4. Behavioral Experiments
2.5. Tissue Isolation
2.6. qRT-PCR
2.7. Immunohistochemistry
2.8. ELISA and Multiplex Assays
2.9. Differentiation and Stimulation of Bone Marrow-Derived Macrophages
2.10. Proteome Anaylsis
2.10.1. Sample Preparation for LC-MS2
2.10.2. High-pH Reverse Phase Fractionation
2.10.3. LC-MS3
2.11. Western Blot
2.12. Fluorescence-Activated Cell Sorting (FACS) Analysis
2.13. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) for the Determination of Lipid Mediators
2.14. Data Analysis and Statistics
3. Results
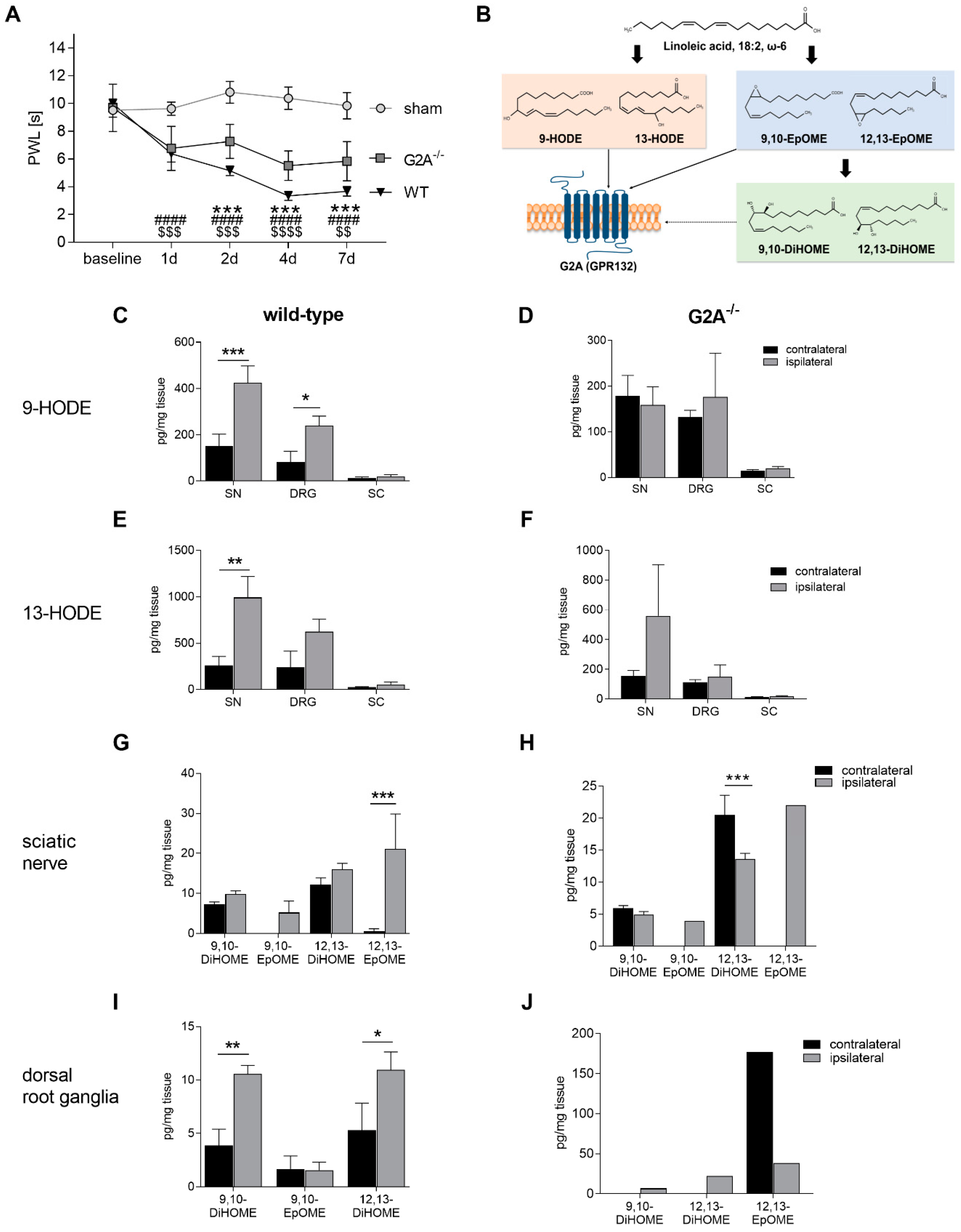
3.1. Loss of G2A Alleviates Mechanical Hypersensitivity and Alters Lipid Signaling
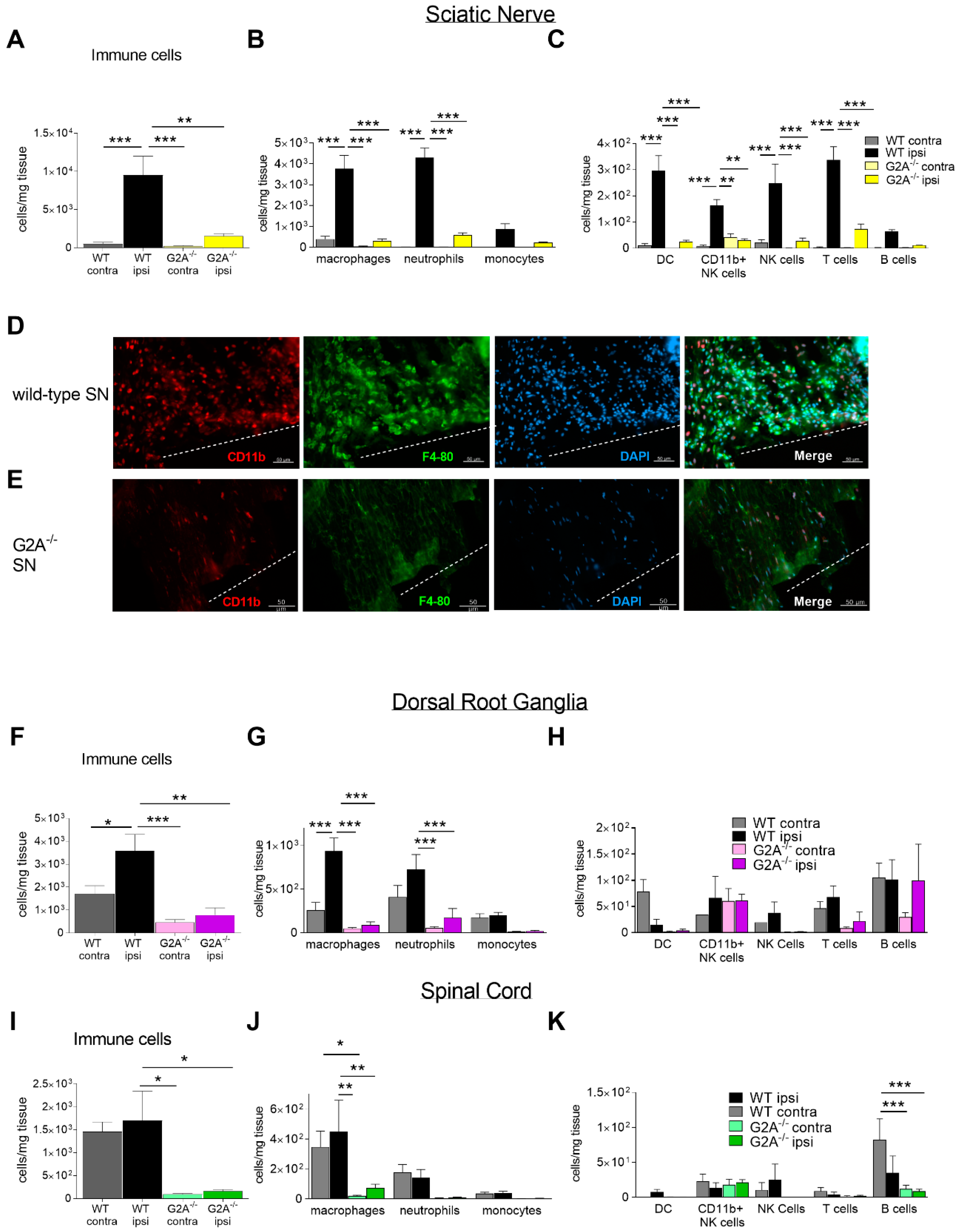
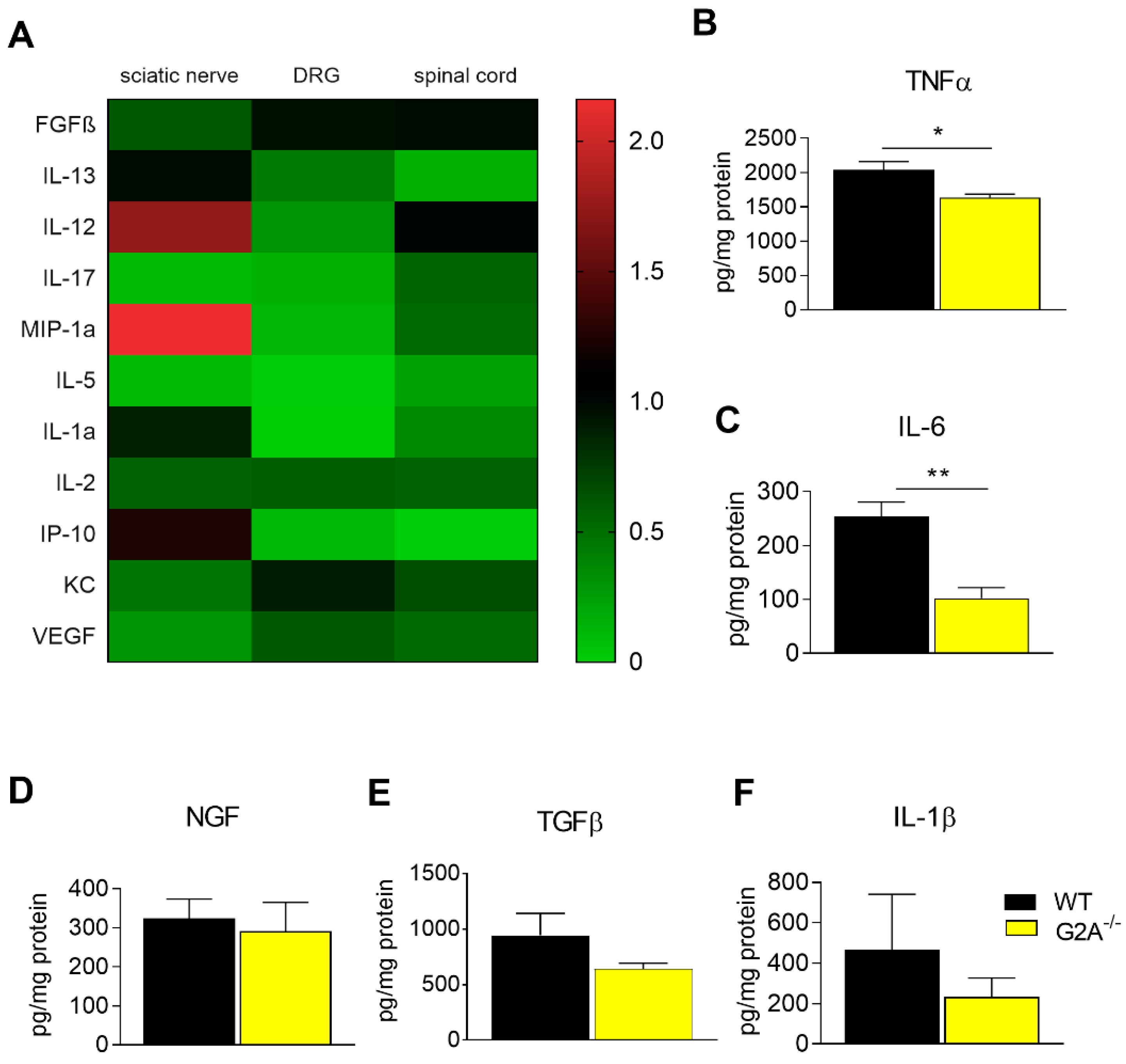
3.2. G2A-Dependent Alterations of Immune Cell Recruitment and Cytokine Synthesis in Nociceptive Tissue
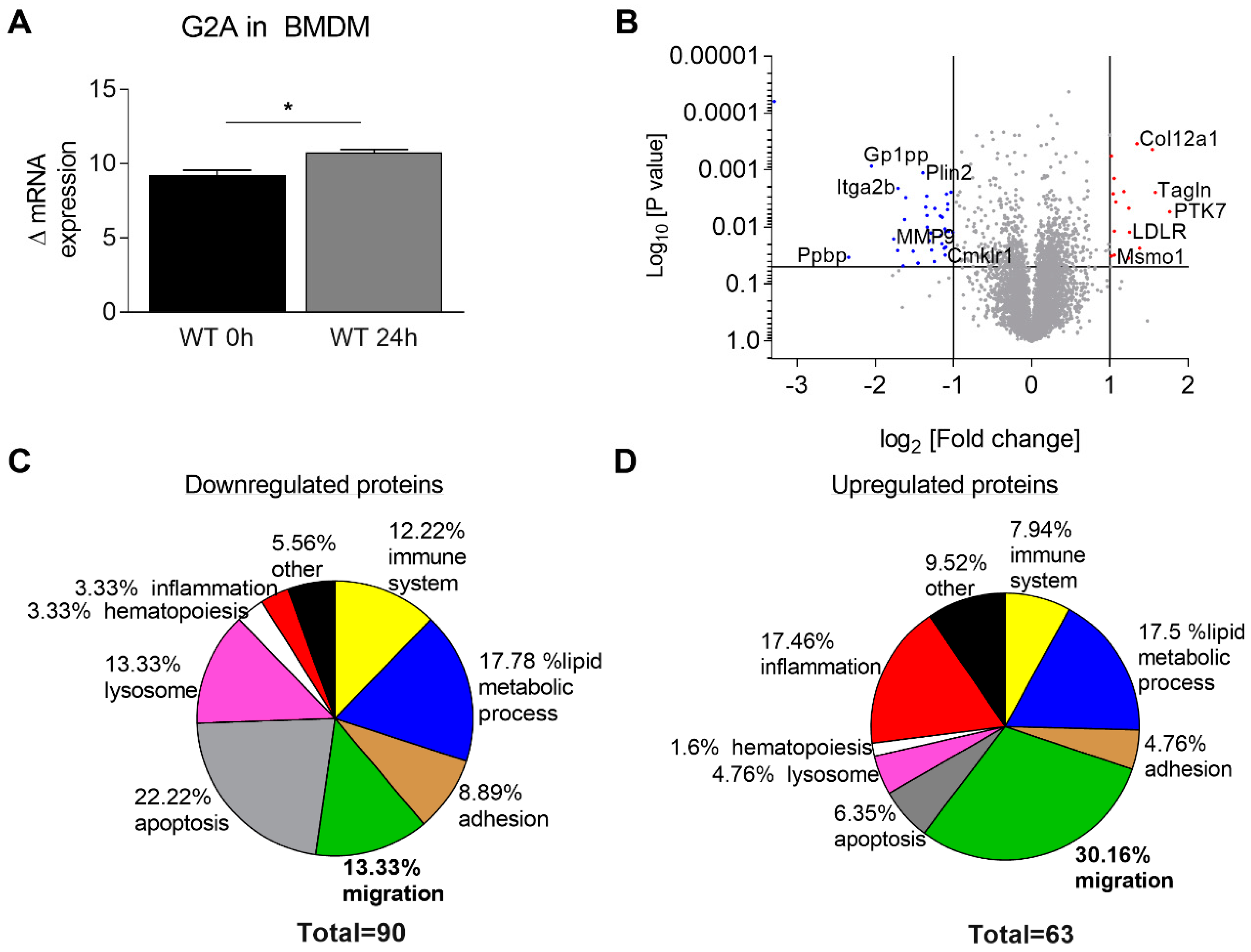
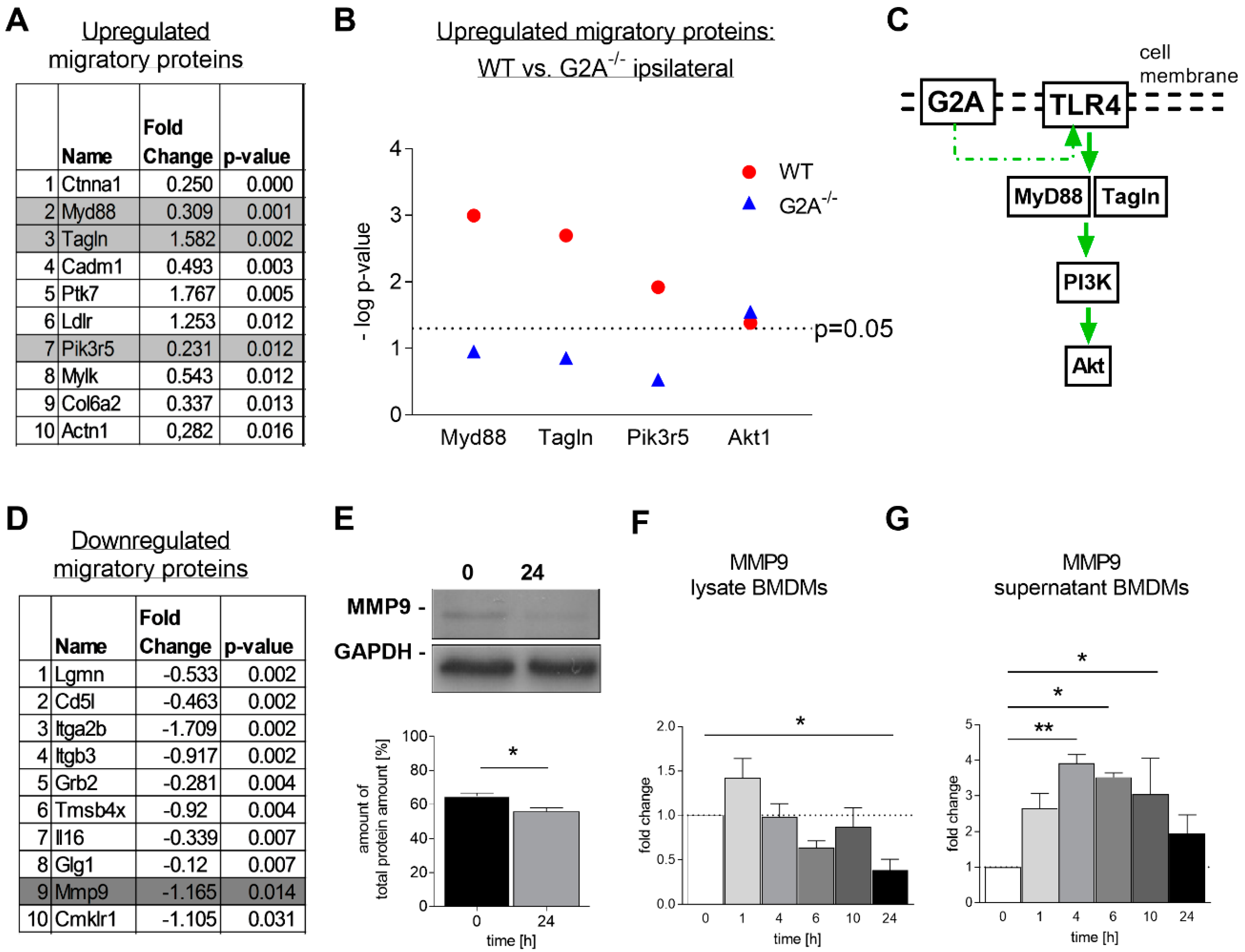
3.3. Migration of Macrophages is Impaired by Loss of G2A Receptor
3.4. Is Macrophage Migration Affected by MMP9 Regulation?
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Costigan, M.; Scholz, J.; Woolf, C.J. Neuropathic Pain. Annu. Rev. Neursci. 2009, 32, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Von Hehn, C.A.; Baron, R.; Woolf, C.J. Deconstructing the Neuropathic Pain Phenotype to Reveal Neural Mechanisms. Neuron 2012, 73, 638–652. [Google Scholar] [CrossRef]
- Liedgens, H.; Obradovic, M.; de Courcy, J.; Holbrook, T.; Jakubanis, R. A burden of illness study for neuropathic pain in Europe. Clinicoecon Outcomes Res. 2016, 8, 113–126. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, A.B.; Dworkin, R.H. Treatment of neuropathic pain. Am. J. Med. 2009, 122, S22–S32. [Google Scholar] [CrossRef] [PubMed]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef]
- Vallejo, R.; Tilley, D.M.; Vogel, L.; Benyamin, R. The role of glia and the immune system in the development and maintenance of neuropathic pain. Pain Pract. 2010, 10, 167–184. [Google Scholar] [CrossRef]
- White, F.A.; Feldman, P.; Miller, R.J. Chemokine signaling and the management of neuropathic pain. Mol. Interv. 2009, 9, 188–195. [Google Scholar] [CrossRef]
- Ji, R.R.; Nackley, A.; Huh, Y.; Terrando, N.; Maixner, W. Neuroinflammation and Central Sensitization in Chronic and Widespread Pain. Anesthesiology 2018. [Google Scholar] [CrossRef]
- Kiguchi, N.; Kobayashi, Y.; Kishioka, S. Chemokines and cytokines in neuroinflammation leading to neuropathic pain. Curr. Opin. Pharmacol. 2012, 12, 55–61. [Google Scholar] [CrossRef]
- Ji, R.R.; Kohno, T.; Moore, K.A.; Woolf, C.J. Central sensitization and LTP. Trends Neurosci. 2003, 26, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Julius, D.; Basbaum, A.I. Molecular mechanisms of nociception. Nature 2001, 413, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J.; Walters, E.T. Common patterns of plasticity contributing to nociceptive sensitization in mammals and Aplysia. Trends Neurosci. 1991, 14, 74–78. [Google Scholar] [CrossRef]
- Campbell, J.N.; Meyer, R.A. Mechanisms of neuropathic pain. Neuron 2006, 52, 77–92. [Google Scholar] [CrossRef]
- Osthues, T.; Sisignano, M. Oxidized Lipids in Persistent Pain States. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Hohmann, S.W.; Angioni, C.; Tunaru, S.; Lee, S.; Woolf, C.J.; Offermanns, S.; Geisslinger, G.; Scholich, K.; Sisignano, M. The G2A receptor (GPR132) contributes to oxaliplatin-induced mechanical pain hypersensitivity. Sci. Rep. 2017, 7, 446. [Google Scholar] [CrossRef]
- Patwardhan, A.M.; Scotland, P.E.; Akopian, A.N.; Hargreaves, K.M. Activation of TRPV1 in the spinal cord by oxidized linoleic acid metabolites contributes to inflammatory hyperalgesia. Proc. Natl. Acad. Sci. USA 2009, 106, 18820–18824. [Google Scholar] [CrossRef]
- Kwan, H.-Y.; Huang, Y.; Yao, X. TRP channels in endothelial function and dysfunction. Biochim. Biophys. 2007, 1772, 907–914. [Google Scholar] [CrossRef]
- Murakami, N.; Yokomizo, T.; Okuno, T.; Shimizu, T. G2A is a proton-sensing G-protein-coupled receptor antagonized by lysophosphatidylcholine. J. Biol. Chem. 2004, 279, 42484–42491. [Google Scholar] [CrossRef]
- Radu, C.G.; Yang, L.V.; Riedinger, M.; Au, M.; Witte, O.N. T cell chemotaxis to lysophosphatidylcholine through the G2A receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 245–250. [Google Scholar] [CrossRef]
- Foster, J.R.; Ueno, S.; Chen, M.X.; Harvey, J.; Dowell, S.J.; Irving, A.J.; Brown, A.J. N-Palmitoylglycine and other N-acylamides activate the lipid receptor G2A/GPR132. Pharmacol. Res. Perspect. 2019, 7, e00542. [Google Scholar] [CrossRef] [PubMed]
- Kern, K.; Schäfer, S.M.G.; Cohnen, J.; Pierre, S.; Osthues, T.; Tarighi, N.; Hohmann, S.; Ferreiros, N.; Brüne, B.; Weigert, A.; et al. The G2A Receptor Controls Polarization of Macrophage by Determining Their Localization Within the Inflamed Tissue. Front. Immunol. 2018, 9, 2261. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.V.; Radu, C.G.; Wang, L.; Riedinger, M.; Witte, O.N. Gi-independent macrophage chemotaxis to lysophosphatidylcholine via the immunoregulatory GPCR G2A. Blood 2005, 105, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Peter, C.; Waibel, M.; Radu, C.G.; Yang, L.V.; Witte, O.N.; Schulze-Osthoff, K.; Wesselborg, S.; Lauber, K. Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J. Biol. Chem. 2008, 283, 5296–5305. [Google Scholar] [CrossRef] [PubMed]
- Le, L.Q.; Kabarowski, J.H.; Weng, Z.; Satterthwaite, A.B.; Harvill, E.T.; Jensen, E.R.; Miller, J.F.; Witte, O.N. Mice lacking the orphan G protein-coupled receptor G2A develop a late-onset autoimmune syndrome. Immunity 2001, 14, 561–571. [Google Scholar] [CrossRef]
- Andrews, N.A.; Latremoliere, A.; Basbaum, A.I.; Mogil, J.S.; Porreca, F.; Rice, A.S.; Woolf, C.J.; Currie, G.L.; Dworkin, R.H.; Eisenach, J.C.; et al. Ensuring transparency and minimization of methodologic bias in preclinical pain research. Pain 2016, 157, 901–909. [Google Scholar] [CrossRef]
- Decosterd, I.; Woolf, C.J. Spared nerve injury. Pain 2000, 87, 149–158. [Google Scholar] [CrossRef]
- Sisignano, M.; Park, C.K.; Angioni, C.; Zhang, D.D.; von Hehn, C.; Cobos, E.J.; Ghasemlou, N.; Xu, Z.Z.; Kumaran, V.; Lu, R.; et al. 5,6-EET is released upon neuronal activity and induces mechanical pain hypersensitivity via TRPA1 on central afferent terminals. J. Neurosci. 2012, 32, 6364–6372. [Google Scholar] [CrossRef]
- Kallenborn-Gerhardt, W.; Hohmann, S.W.; Syhr, K.M.; Schroder, K.; Sisignano, M.; Weigert, A.; Lorenz, J.E.; Lu, R.; Brune, B.; Brandes, R.P.; et al. Nox2-dependent signaling between macrophages and sensory neurons contributes to neuropathic pain hypersensitivity. Pain 2014, 155, 2161–2170. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schmitz, K.; Wilken-Schmitz, A.; Vasic, V.; Brunkhorst, R.; Schmidt, M.; Tegeder, I. Progranulin deficiency confers resistance to autoimmune encephalomyelitis in mice. Cell Mol. Immunol. 2019. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Münch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Schuh, C.D.; Brenneis, C.; Zhang, D.D.; Angioni, C.; Schreiber, Y.; Ferreiros-Bouzas, N.; Pierre, S.; Henke, M.; Linke, B.; Nusing, R.; et al. Prostacyclin regulates spinal nociceptive processing through cyclic adenosine monophosphate-induced translocation of glutamate receptors. Anesthesiology 2014, 120, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Berkelman, T.; Yadav, G.; Hammond, M. A defined methodology for reliable quantification of Western blot data. Mol. Biotechnol. 2013, 55, 217–226. [Google Scholar] [CrossRef]
- Weigert, A.; Weichand, B.; Sekar, D.; Sha, W.; Hahn, C.; Mora, J.; Ley, S.; Essler, S.; Dehne, N.; Brüne, B. HIF-1α is a negative regulator of plasmacytoid DC development in vitro and in vivo. Blood 2012, 120, 3001–3006. [Google Scholar] [CrossRef] [PubMed]
- Sisignano, M.; Angioni, C.; Park, C.K.; Meyer Dos Santos, S.; Jordan, H.; Kuzikov, M.; Liu, D.; Zinn, S.; Hohman, S.W.; Schreiber, Y.; et al. Targeting CYP2J to reduce paclitaxel-induced peripheral neuropathic pain. Proc. Natl. Acad. Sci. USA 2016, 113, 12544–12549. [Google Scholar] [CrossRef]
- Justus, C.R.; Dong, L.; Yang, L.V. Acidic tumor microenvironment and pH-sensing G protein-coupled receptors. Front. Physiol. 2013, 4, 354. [Google Scholar] [CrossRef]
- Scholz, J.; Woolf, C.J. The neuropathic pain triad. Nat. Neurosci. 2007, 10, 1361–1368. [Google Scholar] [CrossRef]
- Costigan, M.; Moss, A.; Latremoliere, A.; Johnston, C.; Verma-Gandhu, M.; Herbert, T.A.; Barrett, L.; Brenner, G.J.; Vardeh, D.; Woolf, C.J.; et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J. Neurosci. 2009, 29, 14415–14422. [Google Scholar] [CrossRef] [PubMed]
- Sisignano, M.; Bennett, D.L.; Geisslinger, G.; Scholich, K. TRP-channels as key integrators of lipid pathways in nociceptive neurons. Prog. Lipid Res. 2014, 53, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Lahvic, J.L.; Ammerman, M.; Li, P.; Blair, M.C.; Stillman, E.R.; Fast, E.M.; Robertson, A.L.; Christodoulou, C.; Perlin, J.R.; Yang, S.; et al. Specific oxylipins enhance vertebrate hematopoiesis via the receptor GPR132. Proc. Natl. Acad. Sci. USA 2018, 115, 9252–9257. [Google Scholar] [CrossRef]
- Inceoglu, B.; Jinks, S.L.; Schmelzer, K.R.; Waite, T.; Kim, I.H.; Hammock, B.D. Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. 2006, 79, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, B.; Angioni, C.; Osthues, T.; Toewe, A.; Thomas, D.; Pierre, S.C.; Geisslinger, G.; Scholich, K.; Sisignano, M. The oxidized linoleic acid metabolite 12,13-DiHOME mediates thermal hyperalgesia during inflammatory pain. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2018, 1863, 669–678. [Google Scholar] [CrossRef]
- Vega-Avelaira, D.; Géranton, S.M.; Fitzgerald, M. Differential regulation of immune responses and macrophage/neuron interactions in the dorsal root ganglion in young and adult rats following nerve injury. Mol. Pain 2009, 5, 70. [Google Scholar] [CrossRef]
- Cobos, E.J.; Nickerson, C.A.; Gao, F.; Chandran, V.; Bravo-Caparrós, I.; González-Cano, R.; Riva, P.; Andrews, N.A.; Latremoliere, A.; Seehus, C.R.; et al. Mechanistic Differences in Neuropathic Pain Modalities Revealed by Correlating Behavior with Global Expression Profiling. Cell Rep. 2018, 22, 1301–1312. [Google Scholar] [CrossRef]
- Kwon, M.J.; Kim, J.; Shin, H.; Jeong, S.R.; Kang, Y.M.; Choi, J.Y.; Hwang, D.H.; Kim, B.G. Contribution of Macrophages to Enhanced Regenerative Capacity of Dorsal Root Ganglia Sensory Neurons by Conditioning Injury. J. Neurosci. 2013, 33, 15095–15108. [Google Scholar] [CrossRef]
- Lindborg, J.A.; Niemi, J.P.; Howarth, M.A.; Liu, K.W.; Moore, C.Z.; Mahajan, D.; Zigmond, R.E. Molecular and cellular identification of the immune response in peripheral ganglia following nerve injury. J. Neuroinflamm. 2018, 15, 1–17. [Google Scholar] [CrossRef]
- Dubový, P.; Klusáková, I.; Hradilová-Svíženská, I.; Joukal, M.; Boadas-Vaello, P. Activation of Astrocytes and Microglial Cells and CCL2/CCR2 Upregulation in the Dorsolateral and Ventrolateral Nuclei of Periaqueductal Gray and Rostral Ventromedial Medulla Following Different Types of Sciatic Nerve Injury. Front. Cell Neurosci. 2018, 12, 40. [Google Scholar] [CrossRef]
- Ji, R.R.; Xu, Z.Z.; Gao, Y.J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug. Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Hamill, K.J.; Hiroyasu, S.; Colburn, Z.T.; Ventrella, R.V.; Hopkinson, S.B.; Skalli, O.; Jones, J.C.R. Alpha actinin-1 regulates cell-matrix adhesion organization in keratinocytes: Consequences for skin cell motility. J. Investig. Dermatol. 2015, 135, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Hägerbrand, K.; Westlund, J.; Yrlid, U.; Agace, W.; Johansson-Lindbom, B. MyD88 Signaling Regulates Steady-State Migration of Intestinal CD103+ Dendritic Cells Independently of TNF-α and the Gut Microbiota. J. Immunol. 2015, 195, 2888–2899. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Vermeulen, S.J.; Nollet, F.; Teugels, E.; Krist’l, M.V.; Malfait, F.; Philippé, J.; Speleman, F.; Bracke, M.E.; van Roy, F.M.; Mareel, M.M. The αE-catenin gene (CTNNA1) acts as an invasion-suppressor gene in human colon cancer cells. Oncogene 1999, 18, 905–915. [Google Scholar] [CrossRef]
- Chauhan, S.K.; Lee, H.K.; Lee, H.S.; Park, E.Y.; Jeong, E.; Dana, R. PTK7+ Mononuclear Cells Express VEGFR2 and Contribute to Vascular Stabilization by Upregulating Angiopoietin-1. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1606–1615. [Google Scholar] [CrossRef]
- Lee, H.K.; Chauhan, S.K.; Kay, E.; Dana, R. Flt-1 regulates vascular endothelial cell migration via a protein tyrosine kinase-7-dependent pathway. Blood 2011, 117, 5762–5771. [Google Scholar] [CrossRef]
- Kleinsimon, S.; Longmuss, E.; Rolff, J.; Jäger, S.; Eggert, A.; Delebinski, C.; Seifert, G. GADD45A and CDKN1A are involved in apoptosis and cell cycle modulatory effects of viscumTT with further inactivation of the STAT3 pathway. Sci. Rep. 2018. [Google Scholar] [CrossRef]
- Chwieralski, C.E.; Welte, T.; Bühling, F. Cathepsin-regulated apoptosis. Apoptosis 2006, 11, 143–149. [Google Scholar] [CrossRef]
- Webb, A.H.; Gao, B.T.; Goldsmith, Z.K.; Irvine, A.S.; Saleh, N.; Lee, R.P.; Lendermon, J.B.; Bheemreddy, R.; Zhang, Q.; Brennan, R.C.; et al. Inhibition of MMP-2 and MMP-9 decreases cellular migration, and angiogenesis in in vitro models of retinoblastoma. BMC Cancer 2017, 17, 434. [Google Scholar] [CrossRef]
- Weng, Z.; Fluckiger, A.-C.; Nisitani, S.; Wahl, M.I.; Le, L.Q.; Hunter, C.A.; Fernal, A.A.; Le Beau, M.M.; Witte, O.N. A DNA damage and stress inducible G protein-coupled receptor blocks cells in G2/M. Proc. Natl. Acad. Sci. USA 1998, 95, 12334–12339. [Google Scholar] [CrossRef] [PubMed]
- Laedermann, C.J.; Pertin, M.; Suter, M.R.; Decosterd, I. Voltage-gated sodium channel expression in mouse DRG after SNI leads to re-evaluation of projections of injured fibers. Mol. Pain 2014, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Chamessian, A.; Zhang, Y.-Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Gattlen, C.; Clarke, C.B.; Piller, N.; Kirschmann, G.; Pertin, M.; Decosterd, I.; Gosselin, R.-D.; Suter, M.R. Spinal Cord T-Cell Infiltration in the Rat Spared Nerve Injury Model: A Time Course Study. Int. J. Mol. Sci. 2016, 17, 352. [Google Scholar] [CrossRef]
- Frasch, S.C.; McNamee, E.N.; Kominsky, D.; Jedlicka, P.; Jakubzick, C.; Zemski Berry, K.; Mack, M.; Furuta, G.T.; Lee, J.J.; Henson, P.M.; et al. G2A Signaling Dampens Colitic Inflammation via Production of IFN-gamma. J. Immunol. 2016, 197, 1425–1434. [Google Scholar] [CrossRef]
- Parks, B.W.; Black, L.L.; Zimmerman, K.A.; Metz, A.E.; Steele, C.; Murphy-Ullrich, J.E.; Kabarowski, J.H. CD36, but not G2A, modulates efferocytosis, inflammation, and fibrosis following bleomycin-induced lung injury. J. Lipid Res. 2013, 54, 1114–1123. [Google Scholar] [CrossRef]
- Ji, R.R.; Xu, Z.Z.; Wang, X.; Lo, E.H. Matrix metalloprotease regulation of neuropathic pain. Trends Pharmacol. Sci. 2009, 30, 336–340. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Wu, T.-T.; Chen, T.-L.; Chen, R.-M. Lipopolysaccharide triggers macrophage activation of inflammatory cytokine expression, chemotaxis, phagocytosis, and oxidative ability via a toll-like receptor 4-dependent pathway: Validated by RNA interference. Toxicol. Lett. 2009, 191, 195–202. [Google Scholar] [CrossRef]
- Kim, H.-R.; Lee, H.-S.; Lee, K.-S.; Jung, I.D.; Kwon, M.-S.; Kim, C.-H.; Kim, S.-M.; Yoon, M.-H.; Park, Y.-M.; Lee, S.-M.; et al. An Essential Role for TAGLN2 in Phagocytosis of Lipopolysaccharide-activated Macrophages. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Jackson, L.; Cady, C.T.; Cambier, J.C. TLR4-mediated signaling induces MMP9-dependent cleavage of B cell surface CD23. J. Immunol. 2009, 183, 2585–2592. [Google Scholar] [CrossRef]
- Üçeyler, N.; Rogausch, J.P.; Toyka, K.V.; Sommer, C. Differential expression of cytokines in painful and painless neuropathies. Neurology 2007, 69, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.L.; Lim, M.; Doshi, T.L. Targeting cytokines for treatment of neuropathic pain. Scand. J. Pain 2017, 17, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-Q.; Liu, Z.; Liu, Z.-H.; Chen, S.-P.; Li, M.; Shahveranov, A.; Ye, D.-W.; Tian, Y.-K. Interleukin-6: An emerging regulator of pathological pain. J. Neuroinflamm. 2016, 13, 141. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Gereau, R.W. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J. Neurosci. 2006, 26, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Bolick, D.T.; Skaflen, M.D.; Johnson, L.E.; Kwon, S.-C.; Howatt, D.; Daugherty, A.; Ravichandran, K.S.; Hedrick, C.C. G2A Deficiency in Mice Promotes Macrophage Activation and Atherosclerosis. Circ. Res. 2008, 104, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Obinata, H.; Ogawa, A.; Kishi, M.; Tatei, K.; Ishikawa, O.; Izumi, T. G2A plays proinflammatory roles in human keratinocytes under oxidative stress as a receptor for 9-hydroxyoctadecadienoic acid. J. Investig. Dermatol. 2008, 128, 1123–1133. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osthues, T.; Zimmer, B.; Rimola, V.; Klann, K.; Schilling, K.; Mathoor, P.; Angioni, C.; Weigert, A.; Geisslinger, G.; Münch, C.; et al. The Lipid Receptor G2A (GPR132) Mediates Macrophage Migration in Nerve Injury-Induced Neuropathic Pain. Cells 2020, 9, 1740. https://doi.org/10.3390/cells9071740
Osthues T, Zimmer B, Rimola V, Klann K, Schilling K, Mathoor P, Angioni C, Weigert A, Geisslinger G, Münch C, et al. The Lipid Receptor G2A (GPR132) Mediates Macrophage Migration in Nerve Injury-Induced Neuropathic Pain. Cells. 2020; 9(7):1740. https://doi.org/10.3390/cells9071740
Chicago/Turabian StyleOsthues, Tabea, Béla Zimmer, Vittoria Rimola, Kevin Klann, Karin Schilling, Praveen Mathoor, Carlo Angioni, Andreas Weigert, Gerd Geisslinger, Christian Münch, and et al. 2020. "The Lipid Receptor G2A (GPR132) Mediates Macrophage Migration in Nerve Injury-Induced Neuropathic Pain" Cells 9, no. 7: 1740. https://doi.org/10.3390/cells9071740
APA StyleOsthues, T., Zimmer, B., Rimola, V., Klann, K., Schilling, K., Mathoor, P., Angioni, C., Weigert, A., Geisslinger, G., Münch, C., Scholich, K., & Sisignano, M. (2020). The Lipid Receptor G2A (GPR132) Mediates Macrophage Migration in Nerve Injury-Induced Neuropathic Pain. Cells, 9(7), 1740. https://doi.org/10.3390/cells9071740