Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications


 ,
,  and
and 
Abstract
1. Introduction
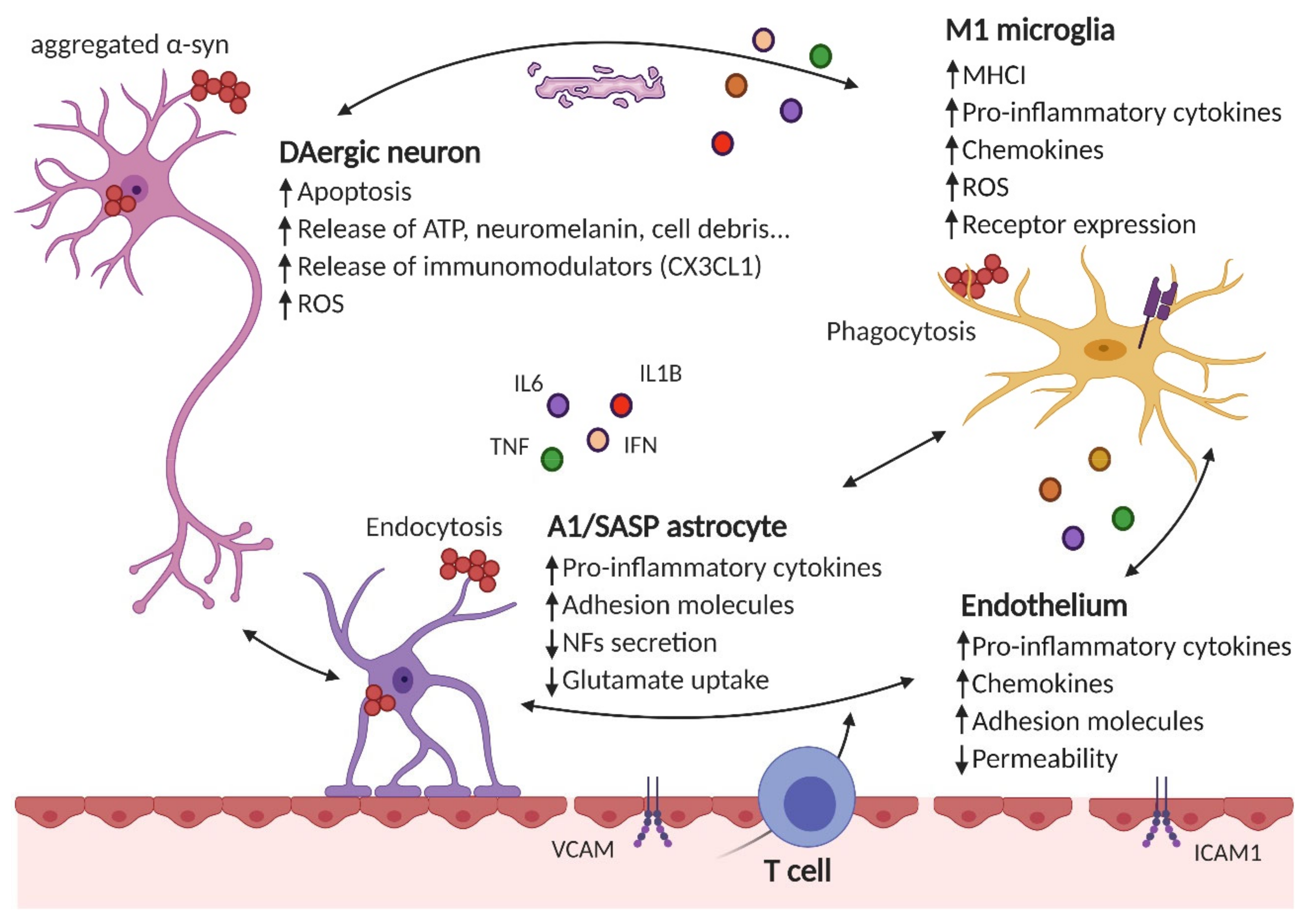
2. Role of Microglia
3. Role of Astrocytes
4. Endothelial Inflammation and Blood–Brain Barrier Permeability
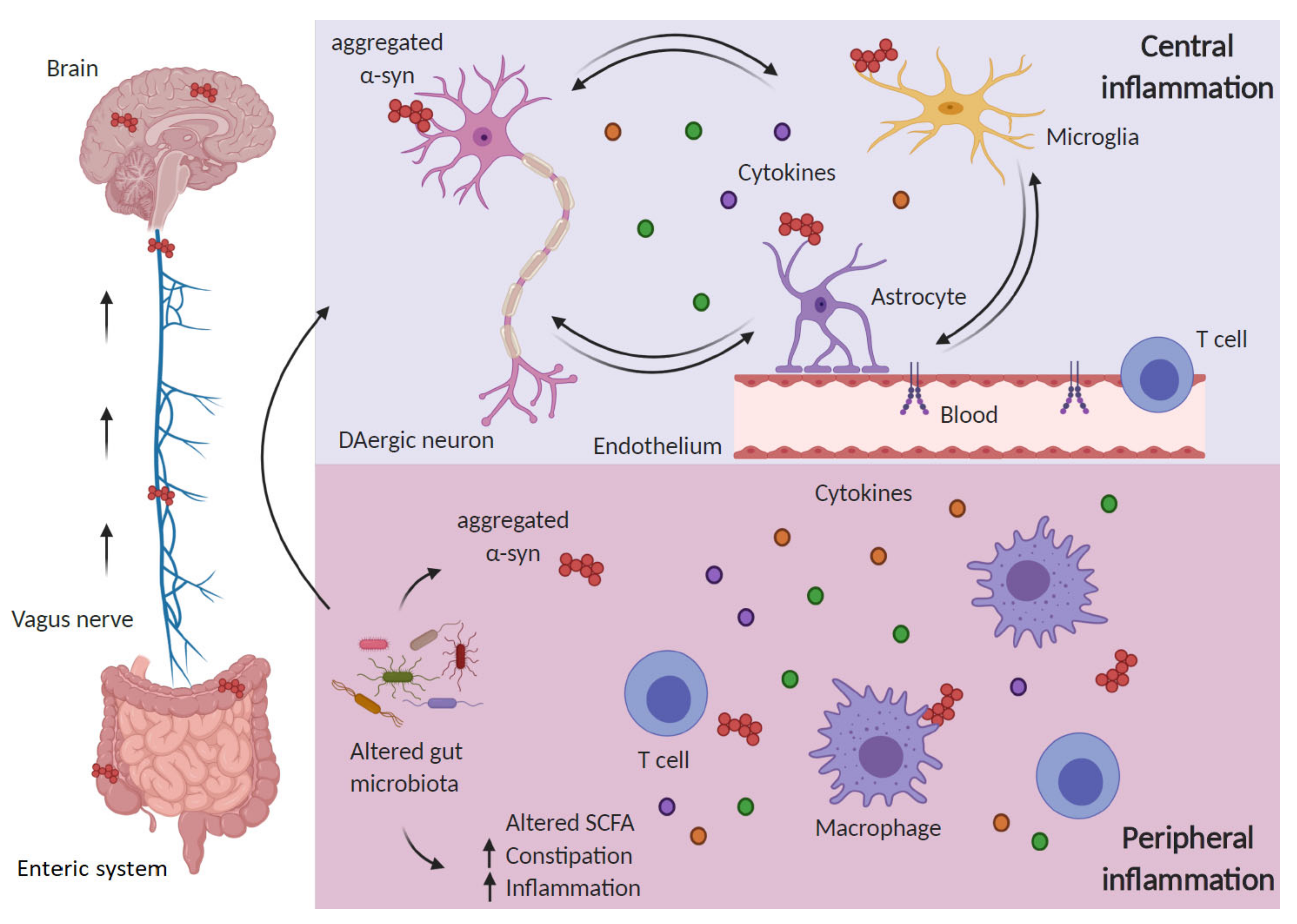
5. Role of Peripheral Inflammation: Novel Hypothesis for PD Origin and Spreading
6. Genomics of PD-Related Inflammation
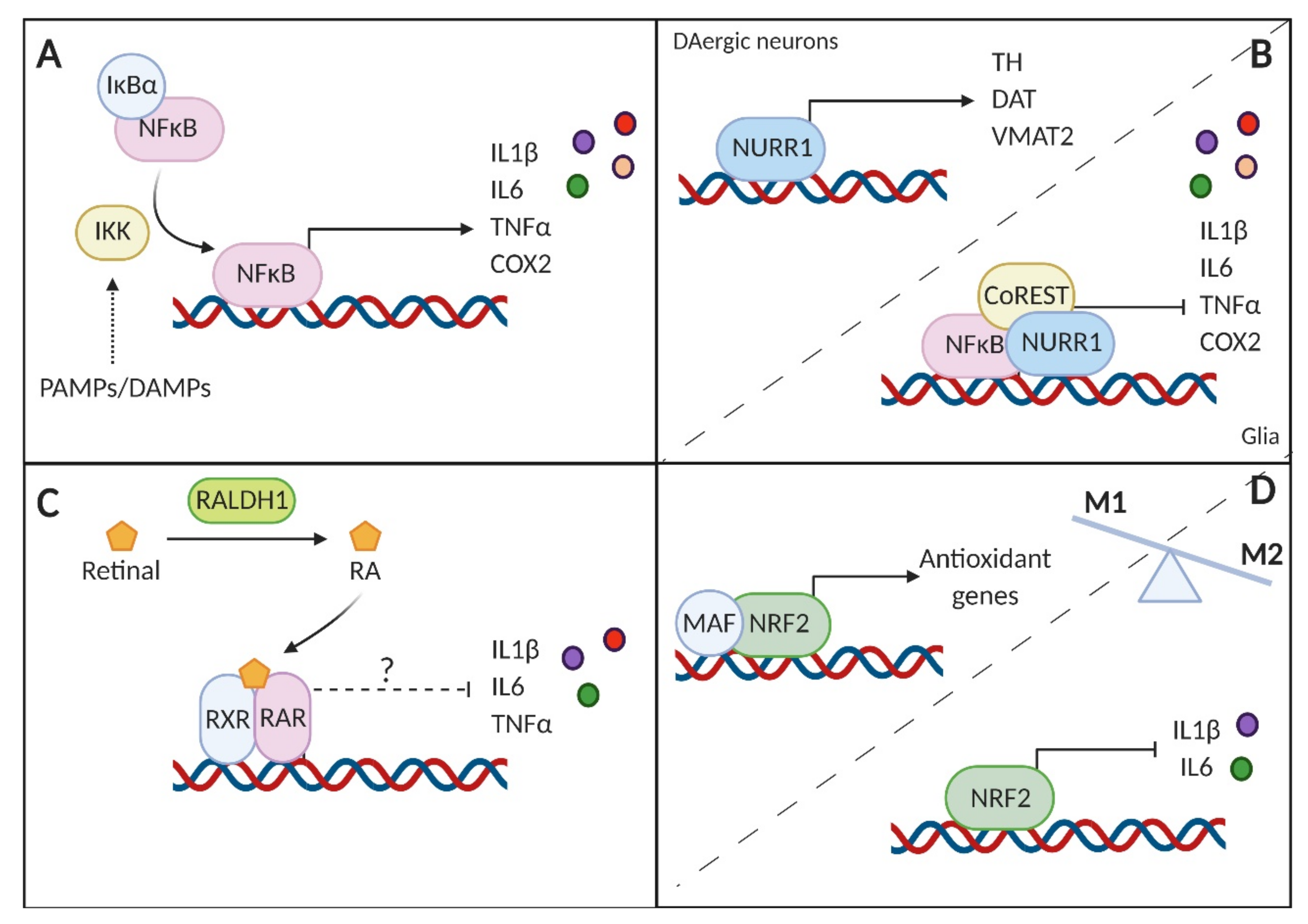
7. Transcriptional Control of Inflammation in PD
8. The Immunomodulatory Role of Dopamine
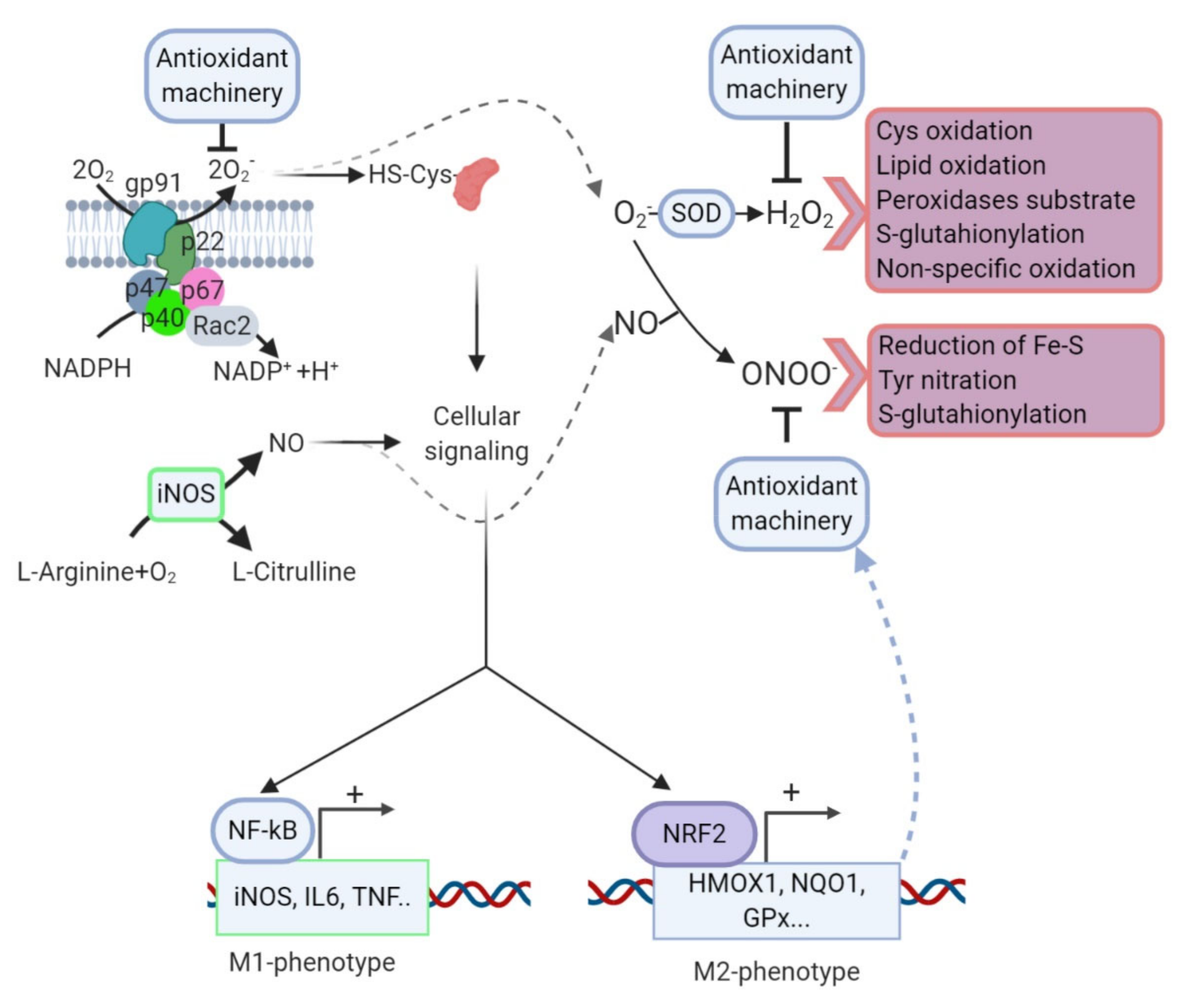
9. Oxidative Stress and Neuroinflammation: A Vicious Circle
10. Therapeutic Strategies Targeting Neuroinflammation in PD
11. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α-Syn | α-synuclein |
| 6-OHDA | 6-hydroxydopamine |
| BBB | blood–brain barrier |
| CNS | central nervous system |
| DAMPs | Damage-associated molecular patterns |
| EBV | Ebola virus |
| GSH | reduced glutathione |
| HSV-1 | herpes simplex virus |
| LPS | lipopolysaccharide |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridinium |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| NLRs | nucleotide-binding oligomerization domain-like receptors |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| NURR1 | Nuclear receptor related 1 |
| PAMPs | Pathogen-associated molecular patterns |
| PD | Parkinson’s disease |
| RA | retinoic acid |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SN | substantia nigra |
| TLR | toll-like receptor. |
References
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 322338. [Google Scholar] [CrossRef] [PubMed]
- Wirdefeldt, K.; Adami, H.O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur. J. Epidemiol. 2011, 26 (Suppl. 1), S1–S58. [Google Scholar] [CrossRef] [PubMed]
- Lutters, B.; Foley, P.; Koehler, P.J. The centennial lesson of encephalitis lethargica. Neurology 2018, 90, 563–567. [Google Scholar] [CrossRef]
- Limphaibool, N.; Iwanowski, P.; Holstad, M.J.V.; Kobylarek, D.; Kozubski, W. Infectious Etiologies of Parkinsonism: Pathomechanisms and Clinical Implications. Front. Neurol. 2019, 10, 652. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef]
- Maries, E.; Dass, B.; Collier, T.J.; Kordower, J.H.; Steece-Collier, K. The role of alpha-synuclein in Parkinson’s disease: Insights from animal models. Nat. Rev. Neurosci. 2003, 4, 727–738. [Google Scholar] [CrossRef]
- Stolzenberg, E.; Berry, D.; Yang, D.E.; Lee, E.Y.; Kroemer, A.; Kaufman, S.; Wong, G.C.L.; Oppenheim, J.J.; Sen, S.; Fishbein, T.; et al. A Role for Neuronal Alpha-Synuclein in Gastrointestinal Immunity. J. Innate Immun. 2017, 9, 456–463. [Google Scholar] [CrossRef]
- Chen, Z.; Trapp, B.D. Microglia and neuroprotection. J. Neurochem. 2016, 136 (Suppl. 1), 10–17. [Google Scholar] [CrossRef]
- Del Rio-Hortega Bereciartu, J. Pio del Rio-Hortega: The Revolution of Glia. Anat. Rec. 2020, 303, 1232–1241. [Google Scholar] [CrossRef]
- Le, W.; Wu, J.; Tang, Y. Protective Microglia and Their Regulation in Parkinson’s Disease. Front. Mol. Neurosci. 2016, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Romero-Ramos, M. Immune system responses in Parkinson’s disease: Early and dynamic. Eur. J. Neurosci. 2019, 49, 364–383. [Google Scholar] [CrossRef]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Tang, Y. Editorial: Microglial Polarization in the Pathogenesis and Therapeutics of Neurodegenerative Diseases. Front. Aging Neurosci. 2018, 10, 154. [Google Scholar] [CrossRef] [PubMed]
- Dubbelaar, M.L.; Kracht, L.; Eggen, B.J.L.; Boddeke, E. The Kaleidoscope of Microglial Phenotypes. Front. Immunol. 2018, 9, 1753. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Eugenin-von Bernhardi, L.; Eugenin, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef]
- He, M.; Dong, H.; Huang, Y.; Lu, S.; Zhang, S.; Qian, Y.; Jin, W. Astrocyte-Derived CCL2 is Associated with M1 Activation and Recruitment of Cultured Microglial Cells. Cell Physiol. Biochem. 2016, 38, 859–870. [Google Scholar] [CrossRef]
- Fakhoury, M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr. Neuropharmacol. 2018, 16, 508–518. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef]
- Salvi, V.; Sozio, F.; Sozzani, S.; Del Prete, A. Role of Atypical Chemokine Receptors in Microglial Activation and Polarization. Front. Aging Neurosci. 2017, 9, 148. [Google Scholar] [CrossRef] [PubMed]
- Rico, D.; Vaquerizas, J.M.; Dopazo, H.; Bosca, L. Identification of conserved domains in the promoter regions of nitric oxide synthase 2: Implications for the species-specific transcription and evolutionary differences. BMC Genom. 2007, 8, 271. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Oyarzabal, E.A.; Hong, J.S. Critical role of the Mac1/NOX2 pathway in mediating reactive microgliosis-generated chronic neuroinflammation and progressive neurodegeneration. Curr. Opin. Pharmacol. 2016, 26, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Back, M.; Yurdagul, A., Jr.; Tabas, I.; Oorni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Changes in cytokines and neurotrophins in Parkinson’s disease. J. Neural. Transm. Suppl. 2000, 277–290. [Google Scholar] [CrossRef]
- Tang, Y.; Li, T.; Li, J.; Yang, J.; Liu, H.; Zhang, X.J.; Le, W. Jmjd3 is essential for the epigenetic modulation of microglia phenotypes in the immune pathogenesis of Parkinson’s disease. Cell Death Differ. 2014, 21, 369–380. [Google Scholar] [CrossRef]
- Long-Smith, C.M.; Collins, L.; Toulouse, A.; Sullivan, A.M.; Nolan, Y.M. Interleukin-1beta contributes to dopaminergic neuronal death induced by lipopolysaccharide-stimulated rat glia in vitro. J. Neuroimmunol. 2010, 226, 20–26. [Google Scholar] [CrossRef]
- Tsutsumi, R.; Hori, Y.; Seki, T.; Kurauchi, Y.; Sato, M.; Oshima, M.; Hisatsune, A.; Katsuki, H. Involvement of exosomes in dopaminergic neurodegeneration by microglial activation in midbrain slice cultures. Biochem. Biophys Res. Commun. 2019, 511, 427–433. [Google Scholar] [CrossRef]
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia affect alpha-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol. Neurodegener. 2019, 14, 34. [Google Scholar] [CrossRef]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef] [PubMed]
- Na, S.J.; DiLella, A.G.; Lis, E.V.; Jones, K.; Levine, D.M.; Stone, D.J.; Hess, J.F. Molecular profiling of a 6-hydroxydopamine model of Parkinson’s disease. Neurochem. Res. 2010, 35, 761–772. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Glial reactions in Parkinson’s disease. Mov. Disord. 2008, 23, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Tofaris, G.K.; Garcia Reitbock, P.; Humby, T.; Lambourne, S.L.; O’Connell, M.; Ghetti, B.; Gossage, H.; Emson, P.C.; Wilkinson, L.S.; Goedert, M.; et al. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): Implications for Lewy body disorders. J. Neurosci. 2006, 26, 3942–3950. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef]
- Watson, M.B.; Richter, F.; Lee, S.K.; Gabby, L.; Wu, J.; Masliah, E.; Effros, R.B.; Chesselet, M.F. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp. Neurol. 2012, 237, 318–334. [Google Scholar] [CrossRef]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rabano, A.; Kirik, D.; Cuadrado, A. alpha-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef]
- Tapias, V.; Hu, X.; Luk, K.C.; Sanders, L.H.; Lee, V.M.; Greenamyre, J.T. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell. Mol. Life Sci. 2017, 74, 2851–2874. [Google Scholar] [CrossRef]
- Harms, A.S.; Delic, V.; Thome, A.D.; Bryant, N.; Liu, Z.; Chandra, S.; Jurkuvenaite, A.; West, A.B. alpha-Synuclein fibrils recruit peripheral immune cells in the rat brain prior to neurodegeneration. Acta Neuropathol. Commun. 2017, 5, 85. [Google Scholar] [CrossRef]
- Sznejder-Pacholek, A.; Joniec-Maciejak, I.; Wawer, A.; Ciesielska, A.; Mirowska-Guzel, D. The effect of alpha-synuclein on gliosis and IL-1alpha, TNFalpha, IFNgamma, TGFbeta expression in murine brain. Pharmacol. Rep. 2017, 69, 242–251. [Google Scholar] [CrossRef]
- Earls, R.H.; Menees, K.B.; Chung, J.; Barber, J.; Gutekunst, C.A.; Hazim, M.G.; Lee, J.K. Intrastriatal injection of preformed alpha-synuclein fibrils alters central and peripheral immune cell profiles in non-transgenic mice. J. Neuroinflamm. 2019, 16, 250. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, K.S.; Lee, S.B.; Ryu, J.S.; Chung, K.C.; Choo, Y.K.; Jou, I.; Kim, J.; Park, S.M. On the mechanism of internalization of alpha-synuclein into microglia: Roles of ganglioside GM1 and lipid raft. J. Neurochem. 2009, 110, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J. Neural Transm. 2000, 107, 335–341. [Google Scholar] [CrossRef]
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Parrella, E.; Bellucci, A.; Porrini, V.; Benarese, M.; Lanzillotta, A.; Faustini, G.; Longhena, F.; Abate, G.; Uberti, D.; Pizzi, M. NF-kappaB/c-Rel deficiency causes Parkinson’s disease-like prodromal symptoms and progressive pathology in mice. Transl. Neurodegener. 2019, 8, 16. [Google Scholar] [CrossRef]
- Shimoji, M.; Pagan, F.; Healton, E.B.; Mocchetti, I. CXCR4 and CXCL12 expression is increased in the nigro-striatal system of Parkinson’s disease. Neurotox. Res. 2009, 16, 318–328. [Google Scholar] [CrossRef]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Korzenik, J.R.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J. Immunol. 2010, 185, 5577–5585. [Google Scholar] [CrossRef]
- Lopez de Maturana, R.; Lang, V.; Zubiarrain, A.; Sousa, A.; Vazquez, N.; Gorostidi, A.; Aguila, J.; Lopez de Munain, A.; Rodriguez, M.; Sanchez-Pernaute, R. Mutations in LRRK2 impair NF-kappaB pathway in iPSC-derived neurons. J. Neuroinflamm. 2016, 13, 295. [Google Scholar] [CrossRef] [PubMed]
- Moussaud, S.; Jones, D.R.; Moussaud-Lamodiere, E.L.; Delenclos, M.; Ross, O.A.; McLean, P.J. Alpha-synuclein and tau: Teammates in neurodegeneration? Mol. Neurodegener. 2014, 9, 43. [Google Scholar] [CrossRef]
- Russo, I.; Bubacco, L.; Greggio, E. LRRK2 and neuroinflammation: Partners in crime in Parkinson’s disease? J. Neuroinflamm. 2014, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Perier, C.; Bove, J.; Vila, M. Mitochondria and programmed cell death in Parkinson’s disease: Apoptosis and beyond. Antioxid. Redox Signal. 2012, 16, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Suzumura, A. Neuron-microglia interaction in neuroinflammation. Curr. Protein Pept. Sci. 2013, 14, 16–20. [Google Scholar] [CrossRef]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Castro-Sanchez, S.; Garcia-Yague, A.J.; Lopez-Royo, T.; Casarejos, M.; Lanciego, J.L.; Lastres-Becker, I. Cx3cr1-deficiency exacerbates alpha-synuclein-A53T induced neuroinflammation and neurodegeneration in a mouse model of Parkinson’s disease. Glia 2018, 66, 1752–1762. [Google Scholar] [CrossRef]
- Nash, K.R.; Moran, P.; Finneran, D.J.; Hudson, C.; Robinson, J.; Morgan, D.; Bickford, P.C. Fractalkine over expression suppresses alpha-synuclein-mediated neurodegeneration. Mol. Ther. 2015, 23, 17–23. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Innamorato, N.G.; Jaworski, T.; Rabano, A.; Kugler, S.; Van Leuven, F.; Cuadrado, A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2014, 137, 78–91. [Google Scholar] [CrossRef]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef]
- Bolanos, J.P. Bioenergetics and redox adaptations of astrocytes to neuronal activity. J. Neurochem. 2016, 139 (Suppl. 2), 115–125. [Google Scholar] [CrossRef]
- Miklossy, J.; Doudet, D.D.; Schwab, C.; Yu, S.; McGeer, E.G.; McGeer, P.L. Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp. Neurol. 2006, 197, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Henning, J.; Strauss, U.; Wree, A.; Gimsa, J.; Rolfs, A.; Benecke, R.; Gimsa, U. Differential astroglial activation in 6-hydroxydopamine models of Parkinson’s disease. Neurosci. Res. 2008, 62, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef]
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W.; et al. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 2004, 127, 420–430. [Google Scholar] [CrossRef]
- Kim, K.S.; Kim, J.S.; Park, J.Y.; Suh, Y.H.; Jou, I.; Joe, E.H.; Park, S.M. DJ-1 associates with lipid rafts by palmitoylation and regulates lipid rafts-dependent endocytosis in astrocytes. Hum. Mol. Genet. 2013, 22, 4805–4817. [Google Scholar] [CrossRef]
- Waak, J.; Weber, S.S.; Waldenmaier, A.; Gorner, K.; Alunni-Fabbroni, M.; Schell, H.; Vogt-Weisenhorn, D.; Pham, T.T.; Reumers, V.; Baekelandt, V.; et al. Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J. 2009, 23, 2478–2489. [Google Scholar] [CrossRef]
- Ashley, A.K.; Hinds, A.I.; Hanneman, W.H.; Tjalkens, R.B.; Legare, M.E. DJ-1 mutation decreases astroglial release of inflammatory mediators. Neurotoxicology 2016, 52, 198–203. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Giasson, B.I.; Zhang, H.; Maguire, J.; Pelech, S.; McGeer, P.L. Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. FASEB J. 2006, 20, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous alpha-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017, 134, 789–808. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 2011, 34, 3–11. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Breidert, T.; Rousselet, E.; Hunot, S.; Hartmann, A.; Michel, P.P. The role of glial reaction and inflammation in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 214–228. [Google Scholar] [CrossRef]
- Wu, F.; Liu, L.; Zhou, H. Endothelial cell activation in central nervous system inflammation. J. Leukoc. Biol. 2017, 101, 1119–1132. [Google Scholar] [CrossRef]
- Nagyoszi, P.; Wilhelm, I.; Farkas, A.E.; Fazakas, C.; Dung, N.T.; Hasko, J.; Krizbai, I.A. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem. Int. 2010, 57, 556–564. [Google Scholar] [CrossRef]
- Perner, C.; Perner, F.; Gaur, N.; Zimmermann, S.; Witte, O.W.; Heidel, F.H.; Grosskreutz, J.; Prell, T. Plasma VCAM1 levels correlate with disease severity in Parkinson’s disease. J. Neuroinflamm. 2019, 16, 94. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.; Vaalburg, W.; Bart, J.; Willemsen, A.T.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef]
- Brochard, V.; Combadiere, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Pavlovic, D.; Dalkie, N.; Waldvogel, H.J.; O’Carroll, S.J.; Green, C.R.; Nicholson, L.F. Vascular degeneration in Parkinson’s disease. Brain Pathol. 2013, 23, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Carvey, P.M.; Zhao, C.H.; Hendey, B.; Lum, H.; Trachtenberg, J.; Desai, B.S.; Snyder, J.; Zhu, Y.G.; Ling, Z.D. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur. J. Neurosci. 2005, 22, 1158–1168. [Google Scholar] [CrossRef]
- Raasch, M.; Rennert, K.; Jahn, T.; Gartner, C.; Schonfelder, G.; Huber, O.; Seiler, A.E.; Mosig, A.S. An integrative microfluidically supported in vitro model of an endothelial barrier combined with cortical spheroids simulates effects of neuroinflammation in neocortex development. Biomicrofluidics 2016, 10, 044102. [Google Scholar] [CrossRef]
- Zhao, C.; Ling, Z.; Newman, M.B.; Bhatia, A.; Carvey, P.M. TNF-alpha knockout and minocycline treatment attenuates blood-brain barrier leakage in MPTP-treated mice. Neurobiol. Dis. 2007, 26, 36–46. [Google Scholar] [CrossRef]
- Santiago, J.A.; Bottero, V.; Potashkin, J.A. Biological and Clinical Implications of Comorbidities in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 394. [Google Scholar] [CrossRef]
- Bartels, A.L.; Willemsen, A.T.; Kortekaas, R.; de Jong, B.M.; de Vries, R.; de Klerk, O.; van Oostrom, J.C.; Portman, A.; Leenders, K.L. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J. Neural Transm. 2008, 115, 1001–1009. [Google Scholar] [CrossRef]
- Benner, E.J.; Banerjee, R.; Reynolds, A.D.; Sherman, S.; Pisarev, V.M.; Tsiperson, V.; Nemachek, C.; Ciborowski, P.; Przedborski, S.; Mosley, R.L.; et al. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE 2008, 3, e1376. [Google Scholar] [CrossRef] [PubMed]
- Cebrian, C.; Zucca, F.A.; Mauri, P.; Steinbeck, J.A.; Studer, L.; Scherzer, C.R.; Kanter, E.; Budhu, S.; Mandelbaum, J.; Vonsattel, J.P.; et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat. Commun. 2014, 5, 3633. [Google Scholar] [CrossRef] [PubMed]
- Pott Godoy, M.C.; Tarelli, R.; Ferrari, C.C.; Sarchi, M.I.; Pitossi, F.J. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain 2008, 131, 1880–1894. [Google Scholar] [CrossRef]
- Ferrari, C.C.; Pott Godoy, M.C.; Tarelli, R.; Chertoff, M.; Depino, A.M.; Pitossi, F.J. Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1beta in the substantia nigra. Neurobiol. Dis. 2006, 24, 183–193. [Google Scholar] [CrossRef]
- Brodacki, B.; Staszewski, J.; Toczylowska, B.; Kozlowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFalpha, and INFgamma concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef]
- Baba, Y.; Kuroiwa, A.; Uitti, R.J.; Wszolek, Z.K.; Yamada, T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Relat. Disord. 2005, 11, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, C.C.; Tarelli, R. Parkinson’s disease and systemic inflammation. Parkinsons Dis. 2011, 2011, 436813. [Google Scholar] [CrossRef]
- Filpa, V.; Moro, E.; Protasoni, M.; Crema, F.; Frigo, G.; Giaroni, C. Role of glutamatergic neurotransmission in the enteric nervous system and brain-gut axis in health and disease. Neuropharmacology 2016, 111, 14–33. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.L.; Bernstein, C.N. Brain-gut interactions in inflammatory bowel disease. Gastroenterology 2013, 144, 36–49. [Google Scholar] [CrossRef]
- Garrido-Gil, P.; Rodriguez-Perez, A.I.; Dominguez-Meijide, A.; Guerra, M.J.; Labandeira-Garcia, J.L. Bidirectional Neural Interaction Between Central Dopaminergic and Gut Lesions in Parkinson’s Disease Models. Mol. Neurobiol. 2018, 55, 7297–7316. [Google Scholar] [CrossRef]
- Selkrig, J.; Wong, P.; Zhang, X.; Pettersson, S. Metabolic tinkering by the gut microbiome: Implications for brain development and function. Gut Microbes 2014, 5, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, L.; Paudel, S.; Jin, L.; Jeyaseelan, S. The NLRP6 inflammasome in health and disease. Mucosal. Immunol. 2020, 13, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Yu, R.; Zhang, L.P.; Wen, S.Y.; Wang, S.J.; Zhang, X.Y.; Xu, Q.; Kong, L.D. Dietary fructose-induced gut dysbiosis promotes mouse hippocampal neuroinflammation: A benefit of short-chain fatty acids. Microbiome 2019, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Goto, S.; Tsuji, H.; Okuno, T.; Asahara, T.; Nomoto, K.; Shibata, A.; Fujisawa, Y.; Minato, T.; Okamoto, A.; et al. Intestinal Dysbiosis and Lowered Serum Lipopolysaccharide-Binding Protein in Parkinson’s Disease. PLoS ONE 2015, 10, e0142164. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef]
- Unger, M.M.; Spiegel, J.; Dillmann, K.U.; Grundmann, D.; Philippeit, H.; Burmann, J.; Fassbender, K.; Schwiertz, A.; Schafer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef]
- Devos, D.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; Bruley des Varannes, S.; Naveilhan, P.; Nguyen, J.M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48. [Google Scholar] [CrossRef]
- Schwiertz, A.; Spiegel, J.; Dillmann, U.; Grundmann, D.; Burmann, J.; Fassbender, K.; Schafer, K.H.; Unger, M.M. Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Parkinsonism Relat. Disord. 2018, 50, 104–107. [Google Scholar] [CrossRef]
- Mulak, A.; Koszewicz, M.; Panek-Jeziorna, M.; Koziorowska-Gawron, E.; Budrewicz, S. Fecal Calprotectin as a Marker of the Gut Immune System Activation Is Elevated in Parkinson’s Disease. Front. Neurosci. 2019, 13, 992. [Google Scholar] [CrossRef]
- Camacho-Soto, A.; Gross, A.; Searles Nielsen, S.; Dey, N.; Racette, B.A. Inflammatory bowel disease and risk of Parkinson’s disease in Medicare beneficiaries. Parkinsonism Relat. Disord. 2018, 50, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Peter, I.; Dubinsky, M.; Bressman, S.; Park, A.; Lu, C.; Chen, N.; Wang, A. Anti-Tumor Necrosis Factor Therapy and Incidence of Parkinson Disease Among Patients with Inflammatory Bowel Disease. JAMA Neurol. 2018, 75, 939–946. [Google Scholar] [CrossRef]
- Shannon, K.M.; Keshavarzian, A.; Dodiya, H.B.; Jakate, S.; Kordower, J.H. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord. 2012, 27, 716–719. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Horai, R.; Zarate-Blades, C.R.; Dillenburg-Pilla, P.; Chen, J.; Kielczewski, J.L.; Silver, P.B.; Jittayasothorn, Y.; Chan, C.C.; Yamane, H.; Honda, K.; et al. Microbiota-Dependent Activation of an Autoreactive T Cell Receptor Provokes Autoimmunity in an Immunologically Privileged Site. Immunity 2015, 43, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e1412. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Toth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabe de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rub, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Bjorklund, T.; Wang, Z.Y.; Roybon, L.; Melki, R.; Li, J.Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic alpha-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e627. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Horvath-Puho, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sorensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Rey, N.L.; Wesson, D.W.; Brundin, P. The olfactory bulb as the entry site for prion-like propagation in neurodegenerative diseases. Neurobiol. Dis. 2018, 109, 226–248. [Google Scholar] [CrossRef]
- Doty, R.L. Olfactory dysfunction in Parkinson disease. Nat. Rev. Neurol. 2012, 8, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Rolheiser, T.M.; Fulton, H.G.; Good, K.P.; Fisk, J.D.; McKelvey, J.R.; Scherfler, C.; Khan, N.M.; Leslie, R.A.; Robertson, H.A. Diffusion tensor imaging and olfactory identification testing in early-stage Parkinson’s disease. J. Neurol. 2011, 258, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; White, C.L., 3rd; Hladik, C.L.; Sabbagh, M.N.; Connor, D.J.; Shill, H.A.; Sue, L.I.; Sasse, J.; Bachalakuri, J.; Henry-Watson, J.; et al. Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol. 2009, 117, 169–174. [Google Scholar] [CrossRef]
- Rey, N.L.; Steiner, J.A.; Maroof, N.; Luk, K.C.; Madaj, Z.; Trojanowski, J.Q.; Lee, V.M.; Brundin, P. Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J. Exp. Med. 2016, 213, 1759–1778. [Google Scholar] [CrossRef]
- Vroon, A.; Drukarch, B.; Bol, J.G.; Cras, P.; Breve, J.J.; Allan, S.M.; Relton, J.K.; Hoogland, P.V.; Van Dam, A.M. Neuroinflammation in Parkinson’s patients and MPTP-treated mice is not restricted to the nigrostriatal system: Microgliosis and differential expression of interleukin-1 receptors in the olfactory bulb. Exp. Gerontol. 2007, 42, 762–771. [Google Scholar] [CrossRef]
- Pereira, P.A.B.; Aho, V.T.E.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. Oral and nasal microbiota in Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 38, 61–67. [Google Scholar] [CrossRef]
- Kustrimovic, N.; Marino, F.; Cosentino, M. Peripheral Immunity, Immunoaging and Neuroinflammation in Parkinson’s Disease. Curr. Med. Chem. 2019, 26, 3719–3753. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Li, J.Q.; Tan, L.; Yu, J.T. The role of the LRRK2 gene in Parkinsonism. Mol. Neurodegener. 2014, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Wallings, R.L.; Herrick, M.K.; Tansey, M.G. LRRK2 at the Interface Between Peripheral and Central Immune Function in Parkinson’s. Front. Neurosci. 2020, 14, 443. [Google Scholar] [CrossRef] [PubMed]
- Witoelar, A.; Jansen, I.E.; Wang, Y.; Desikan, R.S.; Gibbs, J.R.; Blauwendraat, C.; Thompson, W.K.; Hernandez, D.G.; Djurovic, S.; Schork, A.J.; et al. Genome-wide Pleiotropy Between Parkinson Disease and Autoimmune Diseases. JAMA Neurol. 2017, 74, 780–792. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.A.; Kannarkat, G.T.; Cintron, A.F.; Butkovich, L.M.; Fraser, K.B.; Chang, J.; Grigoryan, N.; Factor, S.A.; West, A.B.; Boss, J.M.; et al. LRRK2 levels in immune cells are increased in Parkinson’s disease. NPJ Parkinsons Dis. 2017, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Dzamko, N.L. LRRK2 and the Immune System. Adv. Neurobiol. 2017, 14, 123–143. [Google Scholar] [CrossRef]
- Hongge, L.; Kexin, G.; Xiaojie, M.; Nian, X.; Jinsha, H. The role of LRRK2 in the regulation of monocyte adhesion to endothelial cells. J. Mol. Neurosci. 2015, 55, 233–239. [Google Scholar] [CrossRef]
- Moehle, M.S.; Webber, P.J.; Tse, T.; Sukar, N.; Standaert, D.G.; DeSilva, T.M.; Cowell, R.M.; West, A.B. LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 2012, 32, 1602–1611. [Google Scholar] [CrossRef]
- Daher, J.P.; Volpicelli-Daley, L.A.; Blackburn, J.P.; Moehle, M.S.; West, A.B. Abrogation of alpha-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc. Natl. Acad. Sci. USA 2014, 111, 9289–9294. [Google Scholar] [CrossRef]
- Holmans, P.; Moskvina, V.; Jones, L.; Sharma, M.; International Parkinson’s Disease Genomics, C.; Vedernikov, A.; Buchel, F.; Saad, M.; Bras, J.M.; Bettella, F.; et al. A pathway-based analysis provides additional support for an immune-related genetic susceptibility to Parkinson’s disease. Hum. Mol. Genet. 2013, 22, 1039–1049. [Google Scholar] [CrossRef]
- Hamza, T.H.; Zabetian, C.P.; Tenesa, A.; Laederach, A.; Montimurro, J.; Yearout, D.; Kay, D.M.; Doheny, K.F.; Paschall, J.; Pugh, E.; et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 2010, 42, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Kannarkat, G.T.; Cook, D.A.; Lee, J.K.; Chang, J.; Chung, J.; Sandy, E.; Paul, K.C.; Ritz, B.; Bronstein, J.; Factor, S.A.; et al. Common Genetic Variant Association with Altered HLA Expression, Synergy with Pyrethroid Exposure, and Risk for Parkinson’s Disease: An Observational and Case-Control Study. NPJ Parkinsons Dis. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef]
- Napetschnig, J.; Wu, H. Molecular basis of NF-kappaB signaling. Annu Rev. Biophys. 2013, 42, 443–468. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Roy, A.; Liu, X.; Kordower, J.H.; Mufson, E.J.; Hartley, D.M.; Ghosh, S.; Mosley, R.L.; Gendelman, H.E.; Pahan, K. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18754–18759. [Google Scholar] [CrossRef]
- Mondal, S.; Roy, A.; Jana, A.; Ghosh, S.; Kordower, J.H.; Pahan, K. Testing NF-kappaB-based therapy in hemiparkinsonian monkeys. J. Neuroimmune Pharmacol. 2012, 7, 544–556. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef]
- Safe, S.; Jin, U.H.; Morpurgo, B.; Abudayyeh, A.; Singh, M.; Tjalkens, R.B. Nuclear receptor 4A (NR4A) family—Orphans no more. J. Steroid Biochem. Mol. Biol. 2016, 157, 48–60. [Google Scholar] [CrossRef]
- Zetterstrom, R.H.; Williams, R.; Perlmann, T.; Olson, L. Cellular expression of the immediate early transcription factors Nurr1 and NGFI-B suggests a gene regulatory role in several brain regions including the nigrostriatal dopamine system. Brain Res. Mol. Brain Res. 1996, 41, 111–120. [Google Scholar] [CrossRef]
- Le, W.D.; Xu, P.; Jankovic, J.; Jiang, H.; Appel, S.H.; Smith, R.G.; Vassilatis, D.K. Mutations in NR4A2 associated with familial Parkinson disease. Nat. Genet. 2003, 33, 85–89. [Google Scholar] [CrossRef]
- Le, W.; Pan, T.; Huang, M.; Xu, P.; Xie, W.; Zhu, W.; Zhang, X.; Deng, H.; Jankovic, J. Decreased NURR1 gene expression in patients with Parkinson’s disease. J. Neurol. Sci. 2008, 273, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Winner, B.; Carson, C.T.; Collier, J.G.; Boyer, L.; Rosenfeld, M.G.; Gage, F.H.; Glass, C.K. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 2009, 137, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Landreth, G.E. PPARs in the brain. Biochim. Biophys. Acta 2007, 1771, 1031–1045. [Google Scholar] [CrossRef]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 2011, 1812, 1007–1022. [Google Scholar] [CrossRef]
- Lee, E.; Hwang, I.; Park, S.; Hong, S.; Hwang, B.; Cho, Y.; Son, J.; Yu, J.W. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019, 26, 213–228. [Google Scholar] [CrossRef]
- Chen, L.; Xue, L.; Zheng, J.; Tian, X.; Zhang, Y.; Tong, Q. PPARss/delta agonist alleviates NLRP3 inflammasome-mediated neuroinflammation in the MPTP mouse model of Parkinson’s disease. Behav. Brain Res. 2019, 356, 483–489. [Google Scholar] [CrossRef]
- Tong, Q.; Wu, L.; Gao, Q.; Ou, Z.; Zhu, D.; Zhang, Y. PPARbeta/delta Agonist Provides Neuroprotection by Suppression of IRE1alpha-Caspase-12-Mediated Endoplasmic Reticulum Stress Pathway in the Rotenone Rat Model of Parkinson’s Disease. Mol. Neurobiol. 2016, 53, 3822–3831. [Google Scholar] [CrossRef]
- Ji, J.; Xue, T.F.; Guo, X.D.; Yang, J.; Guo, R.B.; Wang, J.; Huang, J.Y.; Zhao, X.J.; Sun, X.L. Antagonizing peroxisome proliferator-activated receptor gamma facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1-AMPK signaling pathway. Aging Cell 2018, 17, e12774. [Google Scholar] [CrossRef]
- Fujioka, S.; Niu, J.; Schmidt, C.; Sclabas, G.M.; Peng, B.; Uwagawa, T.; Li, Z.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J. NF-kappaB and AP-1 connection: Mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol. Cell Biol. 2004, 24, 7806–7819. [Google Scholar] [CrossRef]
- Le Menn, G.; Neels, J.G. Regulation of Immune Cell Function by PPARs and the Connection with Metabolic and Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 1575. [Google Scholar] [CrossRef]
- Xing, B.; Liu, M.; Bing, G. Neuroprotection with pioglitazone against LPS insult on dopaminergic neurons may be associated with its inhibition of NF-kappaB and JNK activation and suppression of COX-2 activity. J. Neuroimmunol. 2007, 192, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Targett-Adams, P.; McElwee, M.J.; Ehrenborg, E.; Gustafsson, M.C.; Palmer, C.N.; McLauchlan, J. A PPAR response element regulates transcription of the gene for human adipose differentiation-related protein. Biochim. Biophys. Acta 2005, 1728, 95–104. [Google Scholar] [CrossRef]
- Maden, M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 2007, 8, 755–765. [Google Scholar] [CrossRef]
- Galter, D.; Buervenich, S.; Carmine, A.; Anvret, M.; Olson, L. ALDH1 mRNA: Presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol. Dis. 2003, 14, 637–647. [Google Scholar] [CrossRef] [PubMed]
- van Neerven, S.; Nemes, A.; Imholz, P.; Regen, T.; Denecke, B.; Johann, S.; Beyer, C.; Hanisch, U.K.; Mey, J. Inflammatory cytokine release of astrocytes in vitro is reduced by all-trans retinoic acid. J. Neuroimmunol. 2010, 229, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Innamorato, N.G.; Martin-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef]
- von Otter, M.; Landgren, S.; Nilsson, S.; Celojevic, D.; Bergstrom, P.; Hakansson, A.; Nissbrandt, H.; Drozdzik, M.; Bialecka, M.; Kurzawski, M.; et al. Association of Nrf2-encoding NFE2L2 haplotypes with Parkinson’s disease. BMC Med. Genet. 2010, 11, 36. [Google Scholar] [CrossRef]
- von Otter, M.; Bergstrom, P.; Quattrone, A.; De Marco, E.V.; Annesi, G.; Soderkvist, P.; Wettinger, S.B.; Drozdzik, M.; Bialecka, M.; Nissbrandt, H.; et al. Genetic associations of Nrf2-encoding NFE2L2 variants with Parkinson’s disease—A multicenter study. BMC Med. Genet. 2014, 15, 131. [Google Scholar] [CrossRef]
- Czaniecki, C.; Ryan, T.; Stykel, M.G.; Drolet, J.; Heide, J.; Hallam, R.; Wood, S.; Coackley, C.; Sherriff, K.; Bailey, C.D.C.; et al. Axonal pathology in hPSC-based models of Parkinson’s disease results from loss of Nrf2 transcriptional activity at the Map1b gene locus. Proc. Natl. Acad. Sci. USA 2019, 116, 14280–14289. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Pacheco, R.; Lluis, C.; Ahern, G.P.; O’Connell, P.J. The emergence of neurotransmitters as immune modulators. Trends Immunol. 2007, 28, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.; Basu, B.; Chakroborty, D.; Dasgupta, P.S.; Basu, S. The immunoregulatory role of dopamine: An update. Brain Behav. Immun. 2010, 24, 525–528. [Google Scholar] [CrossRef]
- Cosentino, M.; Kustrimovic, N.; Ferrari, M.; Rasini, E.; Marino, F. cAMP levels in lymphocytes and CD4(+) regulatory T-cell functions are affected by dopamine receptor gene polymorphisms. Immunology 2018, 153, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Engler, H.; Doenlen, R.; Riether, C.; Engler, A.; Niemi, M.B.; Besedovsky, H.O.; del Rey, A.; Pacheco-Lopez, G.; Feldon, J.; Schedlowski, M. Time-dependent alterations of peripheral immune parameters after nigrostriatal dopamine depletion in a rat model of Parkinson’s disease. Brain Behav. Immun. 2009, 23, 518–526. [Google Scholar] [CrossRef]
- Pacheco, R.; Contreras, F.; Zouali, M. The dopaminergic system in autoimmune diseases. Front. Immunol. 2014, 5, 117. [Google Scholar] [CrossRef]
- Cosentino, M.; Fietta, A.M.; Ferrari, M.; Rasini, E.; Bombelli, R.; Carcano, E.; Saporiti, F.; Meloni, F.; Marino, F.; Lecchini, S. Human CD4+CD25+ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine/paracrine inhibitory functional loop. Blood 2007, 109, 632–642. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef]
- Zhu, J.; Hu, Z.; Han, X.; Wang, D.; Jiang, Q.; Ding, J.; Xiao, M.; Wang, C.; Lu, M.; Hu, G. Dopamine D2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of beta-arrestin2 and NLRP3. Cell Death Differ. 2018, 25, 2037–2049. [Google Scholar] [CrossRef]
- Alarcon, B.; Mestre, D.; Martinez-Martin, N. The immunological synapse: A cause or consequence of T-cell receptor triggering? Immunology 2011, 133, 420–425. [Google Scholar] [CrossRef]
- Nakano, K.; Higashi, T.; Takagi, R.; Hashimoto, K.; Tanaka, Y.; Matsushita, S. Dopamine released by dendritic cells polarizes Th2 differentiation. Int. Immunol. 2009, 21, 645–654. [Google Scholar] [CrossRef]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J. Neuroinflamm. 2018, 15, 205. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Contreras, F.; Prado, C.; Elgueta, D.; Franz, D.; Bernales, S.; Pacheco, R. Dopamine receptor D3 expressed on CD4+ T cells favors neurodegeneration of dopaminergic neurons during Parkinson’s disease. J. Immunol. 2013, 190, 5048–5056. [Google Scholar] [CrossRef] [PubMed]
- Montoya, A.; Elgueta, D.; Campos, J.; Chovar, O.; Falcon, P.; Matus, S.; Alfaro, I.; Bono, M.R.; Pacheco, R. Dopamine receptor D3 signalling in astrocytes promotes neuroinflammation. J. Neuroinflamm. 2019, 16, 258. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hu, Y.; Wang, B.; Li, S.; Ma, C.; Liu, X.; Moynagh, P.N.; Zhou, J.; Yang, S. Dopamine Uses the DRD5-ARRB2-PP2A Signaling Axis to Block the TRAF6-Mediated NF-kappaB Pathway and Suppress Systemic Inflammation. Mol. Cell 2020, 78, 42–56.e46. [Google Scholar] [CrossRef]
- Han, X.; Ni, J.; Wu, Z.; Wu, J.; Li, B.; Ye, X.; Dai, J.; Chen, C.; Xue, J.; Wan, R.; et al. Myeloid-specific dopamine D2 receptor signalling controls inflammation in acute pancreatitis via inhibiting M1 macrophage. Br. J. Pharmacol. 2020, 177, 2991–3008. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.S.; Pham, T.D.; Tamir, H.; Chen, J.J.; Gershon, M.D. Enteric dopaminergic neurons: Definition, developmental lineage, and effects of extrinsic denervation. J. Neurosci. 2004, 24, 1330–1339. [Google Scholar] [CrossRef]
- Goldstein, D.S. Dysautonomia in Parkinson disease. Compr. Physiol. 2014, 4, 805–826. [Google Scholar] [CrossRef]
- Natale, G.; Ryskalin, L.; Busceti, C.L.; Biagioni, F.; Fornai, F. The nature of catecholamine-containing neurons in the enteric nervous system in relationship with organogenesis, normal human anatomy and neurodegeneration. Arch. Ital. Biol. 2017, 155, 118–130. [Google Scholar] [CrossRef]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53 (Suppl. 3), S26–S38. [Google Scholar] [CrossRef]
- Bosco, D.A.; Fowler, D.M.; Zhang, Q.; Nieva, J.; Powers, E.T.; Wentworth, P., Jr.; Lerner, R.A.; Kelly, J.W. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat. Chem. Biol. 2006, 2, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Nayernia, Z.; Jaquet, V.; Krause, K.H. New insights on NOX enzymes in the central nervous system. Antioxid. Redox Signal. 2014, 20, 2815–2837. [Google Scholar] [CrossRef]
- Salmeen, A.; Barford, D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid. Redox Signal. 2005, 7, 560–577. [Google Scholar] [CrossRef]
- Rodriguez-Pallares, J.; Parga, J.A.; Munoz, A.; Rey, P.; Guerra, M.J.; Labandeira-Garcia, J.L. Mechanism of 6-hydroxydopamine neurotoxicity: The role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J. Neurochem. 2007, 103, 145–156. [Google Scholar] [CrossRef]
- Schober, A. Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res. 2004, 318, 215–224. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Qin, L.; Gao, H.M.; Wilson, B.; Ali, S.F.; Zhang, W.; Hong, J.S.; Liu, B. Neuroprotective effect of dextromethorphan in the MPTP Parkinson’s disease model: Role of NADPH oxidase. FASEB J. 2004, 18, 589–591. [Google Scholar] [CrossRef]
- Wu, D.C.; Teismann, P.; Tieu, K.; Vila, M.; Jackson-Lewis, V.; Ischiropoulos, H.; Przedborski, S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 6145–6150. [Google Scholar] [CrossRef]
- Choi, D.H.; Cristovao, A.C.; Guhathakurta, S.; Lee, J.; Joh, T.H.; Beal, M.F.; Kim, Y.S. NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012, 135, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Re, D.B.; Nagai, M.; Ischiropoulos, H.; Przedborski, S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12132–12137. [Google Scholar] [CrossRef]
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates alpha-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018, 14, 600–608. [Google Scholar] [CrossRef]
- Choi, S.H.; Aid, S.; Kim, H.W.; Jackson, S.H.; Bosetti, F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J. Neurochem. 2012, 120, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Cristovao, A.C.; Guhathakurta, S.; Bok, E.; Je, G.; Yoo, S.D.; Choi, D.H.; Kim, Y.S. NADPH oxidase 1 mediates alpha-synucleinopathy in Parkinson’s disease. J. Neurosci. 2012, 32, 14465–14477. [Google Scholar] [CrossRef]
- Saha, R.N.; Pahan, K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid. Redox Signal. 2006, 8, 929–947. [Google Scholar] [CrossRef]
- Min, K.J.; Pyo, H.K.; Yang, M.S.; Ji, K.A.; Jou, I.; Joe, E.H. Gangliosides activate microglia via protein kinase C and NADPH oxidase. Glia 2004, 48, 197–206. [Google Scholar] [CrossRef]
- Mander, P.; Brown, G.C. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: A dual-key mechanism of inflammatory neurodegeneration. J. Neuroinflamm. 2005, 2, 20. [Google Scholar] [CrossRef]
- Flood, P.M.; Qian, L.; Peterson, L.J.; Zhang, F.; Shi, J.S.; Gao, H.M.; Hong, J.S. Transcriptional Factor NF-kappaB as a Target for Therapy in Parkinson’s Disease. Parkinsons Dis. 2011, 2011, 216298. [Google Scholar] [CrossRef] [PubMed]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernandez-Ruiz, J.; Cuadrado, A. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.T.; Yu, A.C. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J. Neurotrauma 2001, 18, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Leal, M.C.; Casabona, J.C.; Puntel, M.; Pitossi, F.J. Interleukin-1beta and tumor necrosis factor-alpha: Reliable targets for protective therapies in Parkinson’s Disease? Front. Cell Neurosci. 2013, 7, 53. [Google Scholar] [CrossRef]
- Angelova, P.R.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Abramov, A.Y.; Shchepinov, M.S. Lipid peroxidation is essential for alpha-synuclein-induced cell death. J. Neurochem. 2015, 133, 582–589. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Rizor, A.; Pajarillo, E.; Johnson, J.; Aschner, M.; Lee, E. Astrocytic Oxidative/Nitrosative Stress Contributes to Parkinson’s Disease Pathogenesis: The Dual Role of Reactive Astrocytes. Antioxidants 2019, 8, 265. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, S.; Almeida, A.; Bolanos, J.P. Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem. J. 2012, 443, 3–11. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Razmpour, R.; Bernard, L.P. Glutathione and Parkinson’s disease: Is this the elephant in the room? Biomed. Pharmacother. 2008, 62, 236–249. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Garcia-Yague, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kugler, S.; Rabano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed]
- van Muiswinkel, F.L.; de Vos, R.A.; Bol, J.G.; Andringa, G.; Jansen Steur, E.N.; Ross, D.; Siegel, D.; Drukarch, B. Expression of NAD(P)H:quinone oxidoreductase in the normal and Parkinsonian substantia nigra. Neurobiol. Aging 2004, 25, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: Expanding frontiers of engagement. J. Neurochem. 2009, 110, 469–485. [Google Scholar] [CrossRef]
- Samii, A.; Etminan, M.; Wiens, M.O.; Jafari, S. NSAID use and the risk of Parkinson’s disease: Systematic review and meta-analysis of observational studies. Drugs Aging 2009, 26, 769–779. [Google Scholar] [CrossRef]
- Castano, A.; Herrera, A.J.; Cano, J.; Machado, A. The degenerative effect of a single intranigral injection of LPS on the dopaminergic system is prevented by dexamethasone, and not mimicked by rh-TNF-alpha, IL-1beta and IFN-gamma. J. Neurochem. 2002, 81, 150–157. [Google Scholar] [CrossRef]
- Cankaya, S.; Cankaya, B.; Kilic, U.; Kilic, E.; Yulug, B. The therapeutic role of minocycline in Parkinson’s disease. Drugs Context 2019, 8, 212553. [Google Scholar] [CrossRef]
- Liu, B.; Du, L.; Hong, J.S. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J. Pharmacol. Exp. Ther. 2000, 293, 607–617. [Google Scholar]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Krashia, P.; Cordella, A.; Nobili, A.; La Barbera, L.; Federici, M.; Leuti, A.; Campanelli, F.; Natale, G.; Marino, G.; Calabrese, V.; et al. Blunting neuroinflammation with resolvin D1 prevents early pathology in a rat model of Parkinson’s disease. Nat. Commun. 2019, 10, 3945. [Google Scholar] [CrossRef] [PubMed]
- Benner, E.J.; Mosley, R.L.; Destache, C.J.; Lewis, T.B.; Jackson-Lewis, V.; Gorantla, S.; Nemachek, C.; Green, S.R.; Przedborski, S.; Gendelman, H.E. Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 9435–9440. [Google Scholar] [CrossRef] [PubMed]
- Teema, A.M.; Zaitone, S.A.; Moustafa, Y.M. Ibuprofen or piroxicam protects nigral neurons and delays the development of l-dopa induced dyskinesia in rats with experimental Parkinsonism: Influence on angiogenesis. Neuropharmacology 2016, 107, 432–450. [Google Scholar] [CrossRef]
- Mandler, M.; Valera, E.; Rockenstein, E.; Weninger, H.; Patrick, C.; Adame, A.; Santic, R.; Meindl, S.; Vigl, B.; Smrzka, O.; et al. Next-generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014, 127, 861–879. [Google Scholar] [CrossRef]
- Swanson, C.R.; Joers, V.; Bondarenko, V.; Brunner, K.; Simmons, H.A.; Ziegler, T.E.; Kemnitz, J.W.; Johnson, J.A.; Emborg, M.E. The PPAR-gamma agonist pioglitazone modulates inflammation and induces neuroprotection in parkinsonian monkeys. J. Neuroinflamm. 2011, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Schintu, N.; Frau, L.; Ibba, M.; Caboni, P.; Garau, A.; Carboni, E.; Carta, A.R. PPAR-gamma-mediated neuroprotection in a chronic mouse model of Parkinson’s disease. Eur. J. Neurosci. 2009, 29, 954–963. [Google Scholar] [CrossRef]
- Zella, S.M.A.; Metzdorf, J.; Ciftci, E.; Ostendorf, F.; Muhlack, S.; Gold, R.; Tonges, L. Emerging Immunotherapies for Parkinson Disease. Neurol. Ther. 2019, 8, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Mante, M.; Crews, L.; Spencer, B.; Adame, A.; Patrick, C.; Trejo, M.; Ubhi, K.; Rohn, T.T.; et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS ONE 2011, 6, e19338. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.J.; Lee, H.J.; Rockenstein, E.; Ho, D.H.; Park, E.B.; Yang, N.Y.; Desplats, P.; Masliah, E.; Lee, S.J. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J. Neurosci. 2012, 32, 13454–13469. [Google Scholar] [CrossRef] [PubMed]
- Barichella, M.; Pacchetti, C.; Bolliri, C.; Cassani, E.; Iorio, L.; Pusani, C.; Pinelli, G.; Privitera, G.; Cesari, I.; Faierman, S.A.; et al. Probiotics and prebiotic fiber for constipation associated with Parkinson disease: An RCT. Neurology 2016, 87, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Brakedal, B.; Flones, I.; Reiter, S.F.; Torkildsen, O.; Dolle, C.; Assmus, J.; Haugarvoll, K.; Tzoulis, C. Glitazone use associated with reduced risk of Parkinson’s disease. Mov. Disord. 2017, 32, 1594–1599. [Google Scholar] [CrossRef]
- Brauer, R.; Bhaskaran, K.; Chaturvedi, N.; Dexter, D.T.; Smeeth, L.; Douglas, I. Glitazone Treatment and Incidence of Parkinson’s Disease among People with Diabetes: A Retrospective Cohort Study. PLoS Med. 2015, 12, e1001854. [Google Scholar] [CrossRef]
- Martin, H. Role of PPAR-gamma in inflammation. Prospects for therapeutic intervention by food components. Mutat. Res. 2009, 669, 1–7. [Google Scholar] [CrossRef]
- Hu, G.; Jousilahti, P.; Bidel, S.; Antikainen, R.; Tuomilehto, J. Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 2007, 30, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Driver, J.A.; Smith, A.; Buring, J.E.; Gaziano, J.M.; Kurth, T.; Logroscino, G. Prospective cohort study of type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 2008, 31, 2003–2005. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Park, Y.; Huang, X.; Hollenbeck, A.; Blair, A.; Schatzkin, A.; Chen, H. Diabetes and risk of Parkinson’s disease. Diabetes Care 2011, 34, 910–915. [Google Scholar] [CrossRef]
- Schernhammer, E.; Hansen, J.; Rugbjerg, K.; Wermuth, L.; Ritz, B. Diabetes and the risk of developing Parkinson’s disease in Denmark. Diabetes Care 2011, 34, 1102–1108. [Google Scholar] [CrossRef]
- Chung, S.J.; Jeon, S.; Yoo, H.S.; Kim, G.; Oh, J.S.; Kim, J.S.; Evans, A.C.; Sohn, Y.H.; Lee, P.H. Detrimental effect of type 2 diabetes mellitus in a large case series of Parkinson’s disease. Parkinsonism Relat. Disord. 2019, 64, 54–59. [Google Scholar] [CrossRef]
- Dehmer, T.; Heneka, M.T.; Sastre, M.; Dichgans, J.; Schulz, J.B. Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J. Neurochem. 2004, 88, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Carta, A.R.; Frau, L.; Pisanu, A.; Wardas, J.; Spiga, S.; Carboni, E. Rosiglitazone decreases peroxisome proliferator receptor-gamma levels in microglia and inhibits TNF-alpha production: New evidences on neuroprotection in a progressive Parkinson’s disease model. Neuroscience 2011, 194, 250–261. [Google Scholar] [CrossRef] [PubMed]
- NINDS Exploratory Trials in Parkinson Disease. Pioglitazone in early Parkinson’s disease: A phase 2, multicentre, double-blind, randomised trial. Lancet Neurol. 2015, 14, 795–803. [Google Scholar] [CrossRef]
- Kim, R.; Kim, H.J.; Kim, A.; Jang, M.; Kim, A.; Kim, Y.; Yoo, D.; Im, J.H.; Choi, J.H.; Jeon, B. Peripheral blood inflammatory markers in early Parkinson’s disease. J. Clin. Neurosci. 2018, 58, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.; Janelidze, S.; Surova, Y.; Widner, H.; Zetterberg, H.; Hansson, O. Cerebrospinal fluid concentrations of inflammatory markers in Parkinson’s disease and atypical parkinsonian disorders. Sci. Rep. 2018, 8, 13276. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular or Cellular Target | Pharmacological Intervention | Results | Reference |
|---|---|---|---|
| Glucocorticoid receptor | Dexamethasone | Prevention of the degenerative effect of intranigral LPS injection | [228] |
| Several G protein-coupled receptors | Resolvin D1 | Protection against nigrostriatal overexpression of α-Syn | [233] |
| Immunomodulator | Minocycline | Antiapoptotic, anti-inflammatory, and antioxidant effects on several PD models | [229] |
| COX2 | Nonsteroidal anti-inflammatory drugs | Prevention of inflammation and dyskinesia in the rotenone rat model | [235] |
| NLRP3 | MCC950 | Prevention of inflammation and dopaminergic death in mice submitted to preformed fibrils of α-Syn | [232] |
| T lymphocytes | Glatiramer acetate | Immunization strategy for suppression of microglial activation in (MPTP)-intoxicated mice | [234] |
| B lymphocytes | Short fragments of α-Syn | Vaccination against c-terminal fragment of α-Syn in murine models of PD and Dementia with Lewy bodies | [236] |
| NRF2 | Dimethyl fumarate | Antioxidant and neuroprotective effects on the mouse model of nigrostriatal overexpression of α-Syn | [224] |
| PPARγ | Pioglitazone | Reduction of dopaminergic cell deadnadn microgliosis in the MPTP intoxicated Reshus monkeys | [237] |
| Rosiglitazone | Improvement of motor activity and reduced neurodegeneration and inflammation in a chronic MPTP mouse model | [238] |
| Molecular or Cellular Target | Pharmacological Intervention | Results | Reference |
|---|---|---|---|
| TNF | Anti-TNF antibodies Retrospective cohort study | A higher incidence of PD was observed among patients with inflammatory bowel disease | [112] |
| GLP1 | Semaglutide (Phase 2) Not yet recruiting | neuroprotective and anti-inflammatory in idiopathic PD | NCT03659682 |
| Gut microbiota | Rifaximin (Phase 2) Recruiting | Change of gut microbiota. Blood biomarkers of neuroinflammation and exosomal alpha-synuclein | NCT03958708 |
| Gut microbiota | Fecal transplantation. Recruiting | Change in clinical symptoms | NCT03808389 |
| PPARγ | Pioglitazone. Retrospective cohort study | lower incidence of PD among diabetic patients | [243,244] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pajares, M.; I. Rojo, A.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. https://doi.org/10.3390/cells9071687
Pajares M, I. Rojo A, Manda G, Boscá L, Cuadrado A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells. 2020; 9(7):1687. https://doi.org/10.3390/cells9071687
Chicago/Turabian StylePajares, Marta, Ana I. Rojo, Gina Manda, Lisardo Boscá, and Antonio Cuadrado. 2020. "Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications" Cells 9, no. 7: 1687. https://doi.org/10.3390/cells9071687
APA StylePajares, M., I. Rojo, A., Manda, G., Boscá, L., & Cuadrado, A. (2020). Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells, 9(7), 1687. https://doi.org/10.3390/cells9071687