Fine Tuning of Calcium Constitutive Entry by Optogenetically-Controlled Membrane Polarization: Impact on Cell Migration

 , , , ,
, , , ,
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Culture and Transfection
2.2. Gene Constructs
2.3. Immunoblotting
2.4. Immunofluorescence Staining
2.5. Reverse Transcription and Polymerase Chain Reaction (RT-PCR)
2.6. Electrophysiology
2.7. Calcium Measurements
2.8. Fura-2 Quenching Assay
2.9. In Vitro Migration Assay
2.10. Statistical Analysis
3. Results
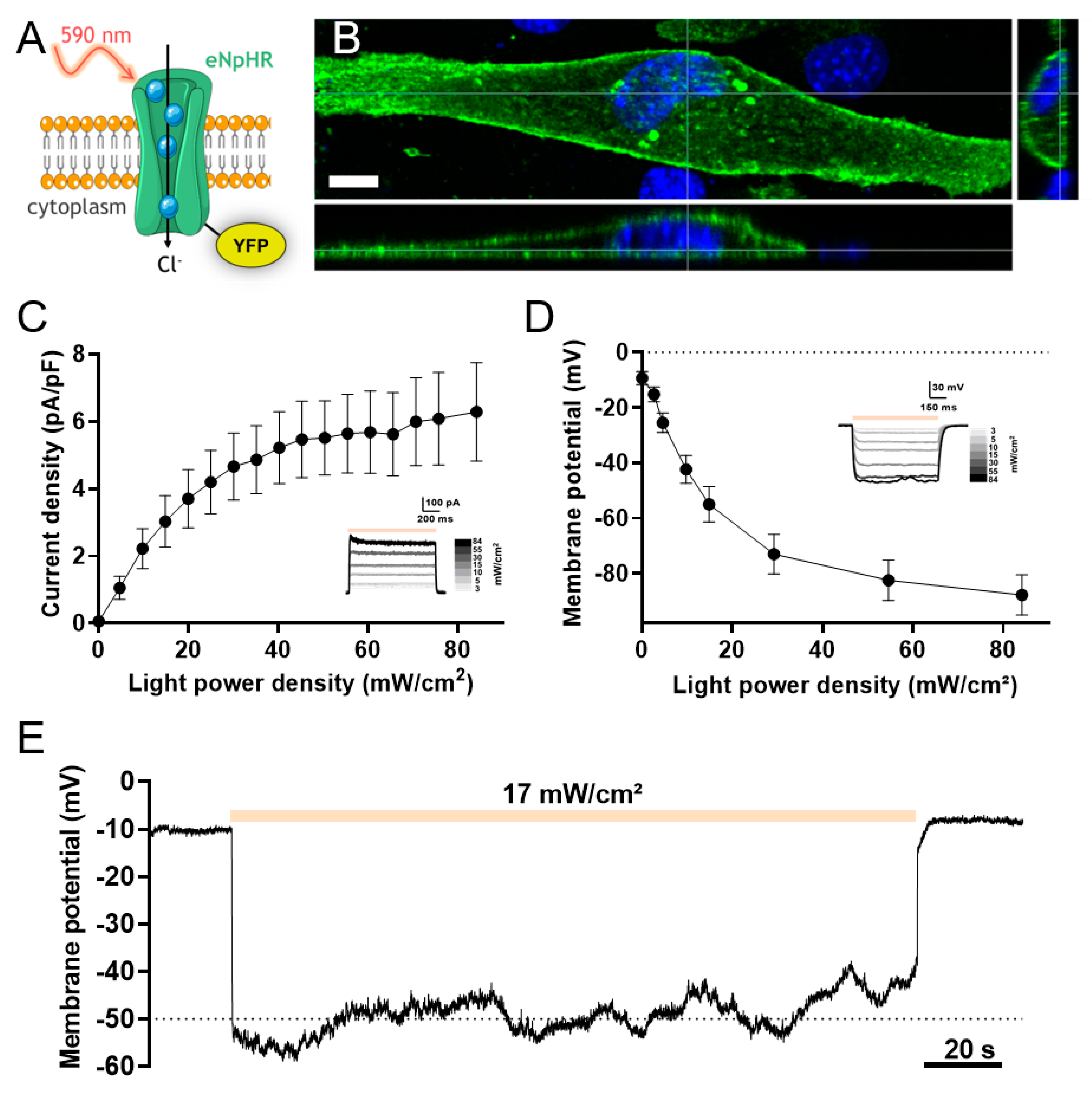
3.1. Light Activation of eNpHR Induces Membrane Polarization in C2C12 Myoblasts
3.2. Light-Activated Membrane Polarization Induces Calcium Elevation through Constitutive Ca2+ Entry
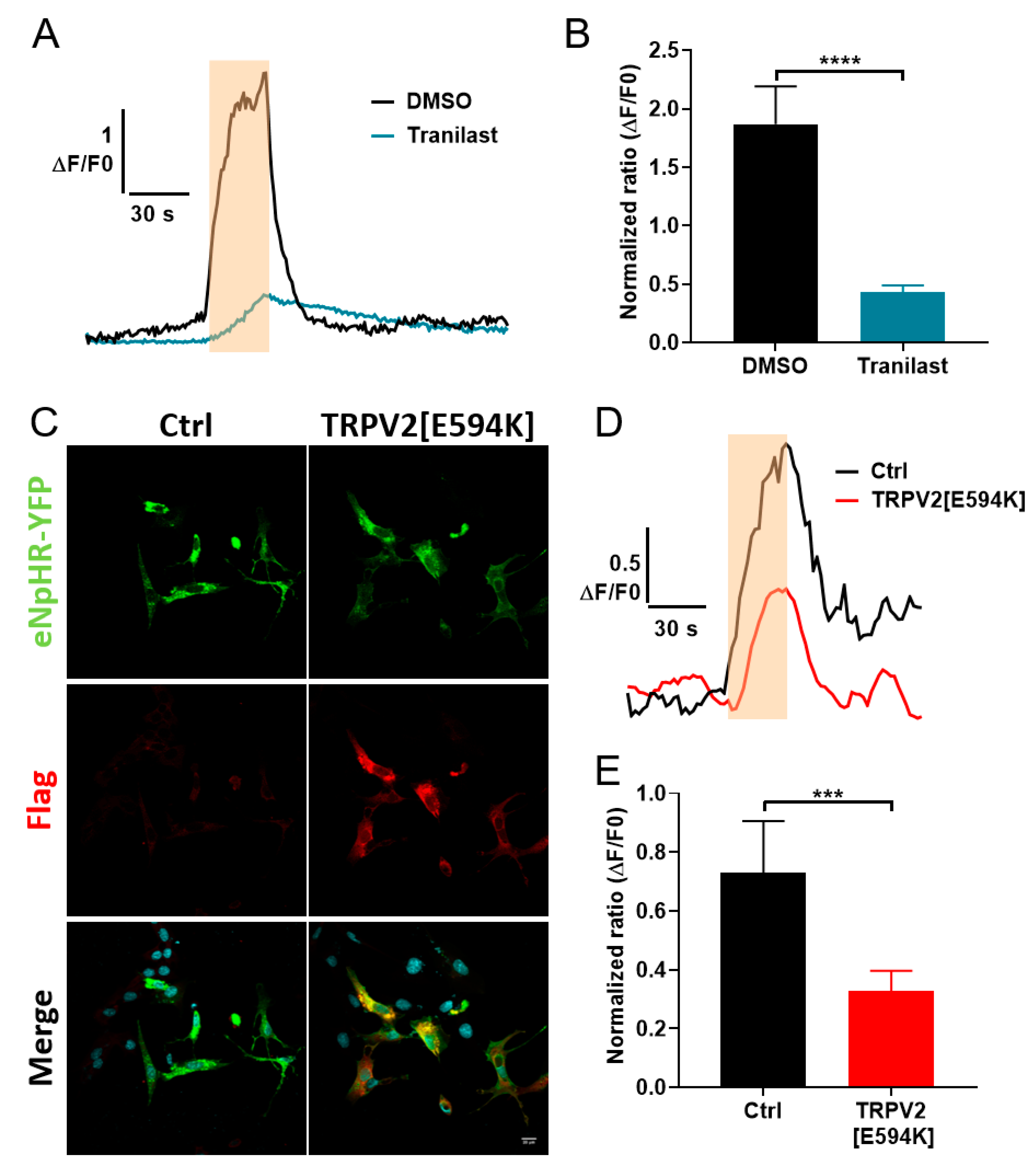
3.3. Expression of TRPV2 Channels in C2C12 Myoblasts
3.4. Involvement of TRPV2 in Calcium Response to Optical Stimulation
3.5. Optogenetic Control of TRPV2-Dependent C2C12 Myoblast Migration
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Tutdibi, O.; Brinkmeier, H.; Rüdel, R.; Föhr, K.J. Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J. Physiol. 1999, 515, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Sabourin, J.; Lamiche, C.; Vandebrouck, A.; Magaud, C.; Rivet, J.; Cognard, C.; Bourmeyster, N.; Constantin, B. Regulation of TRPC1 and TRPC4 cation channels requires an alpha1-syntrophin-dependent complex in skeletal mouse myotubes. J. Biol. Chem. 2009, 284, 36248–36261. [Google Scholar] [CrossRef] [PubMed]
- Aguettaz, E.; Lopez, J.J.; Krzesiak, A.; Lipskaia, L.; Adnot, S.; Hajjar, R.J.; Cognard, C.; Constantin, B.; Sebille, S. Axial stretch-dependent cation entry in dystrophic cardiomyopathy: Involvement of several TRPs channels. Cell Calcium 2016, 59, 145–155. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mignen, O.; Constantin, B.; Potier-Cartereau, M.; Penna, A.; Gautier, M.; Guéguinou, M.; Renaudineau, Y.; Shoji, K.; Félix, R.; Bayet, E.; et al. Constitutive calcium entry and cancer: Updated views and insights. Eur. Biophys. J. 2017, 46, 395–413. [Google Scholar] [CrossRef]
- Yang, M.; Brackenbury, W.J. Membrane potential and cancer progression. Front. Physiol 2013, 4, 185. [Google Scholar] [CrossRef]
- Bernheim, L.; Bader, C.R. Human myoblast differentiation: Ca(2+) channels are activated by K(+) channels. News Physiol. Sci. 2002, 17, 22–26. [Google Scholar] [CrossRef]
- Blackiston, D.J.; McLaughlin, K.A.; Levin, M. Bioelectric controls of cell proliferation. Cell Cycle 2009, 8, 3519–3528. [Google Scholar] [CrossRef]
- Sundelacruz, S.; Levin, M.; Kaplan, D.L. Role of membrane potential in the regulation of cell proliferation and differentiation. Stem Cell Rev. Rep. 2009, 5, 231–246. [Google Scholar] [CrossRef]
- Kim, E.Y.; Alvarez-Baron, C.P.; Dryer, S.E. Canonical transient receptor potential channel (TRPC)3 and TRPC6 associate with large-conductance Ca2+-activated K+ (BKCa) channels: Role in BKCa trafficking to the surface of cultured podocytes. Mol. Pharmacol. 2009, 75, 466–477. [Google Scholar] [CrossRef]
- Lallet-Daher, H.; Roudbaraki, M.; Bavencoffe, A.; Mariot, P.; Gackière, F.; Bidaux, G.; Urbain, R.; Gosset, P.; Delcourt, P.; Fleurisse, L.; et al. Intermediate-conductance Ca2+-activated K+ channels (IK Ca1) regulate human prostate cancer cell proliferation through a close control of calcium entry. Oncogene 2009, 28, 1792–1806. [Google Scholar] [CrossRef] [PubMed]
- Chantôme, A.; Potier-Cartereau, M.; Clarysse, L.; Fromont, G.; Marionneau-Lambot, S.; Guéguinou, M.; Pagès, J.-C.; Collin, C.; Oullier, T.; Girault, A.; et al. Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res. 2013, 73, 4852–4861. [Google Scholar] [CrossRef] [PubMed]
- Guéguinou, M.; Chantôme, A.; Fromont, G.; Bougnoux, P.; Vandier, C.; Potier-Cartereau, M. KCa and Ca(2+) channels: The complex thought. Biochim. Biophys. Acta 2014, 1843, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Gambade, A.; Zreika, S.; Guéguinou, M.; Chourpa, I.; Fromont, G.; Bouchet, A.M.; Burlaud-Gaillard, J.; Potier-Cartereau, M.; Roger, S.; Aucagne, V.; et al. Activation of TRPV2 and BKCa channels by the LL-37 enantiomers stimulates calcium entry and migration of cancer cells. Oncotarget 2016, 7, 23785–23800. [Google Scholar] [CrossRef]
- Payne, S.L.; Levin, M.; Oudin, M.J. Bioelectric control of metastasis in solid tumors. Bioelectricity 2019, 1, 114–130. [Google Scholar] [CrossRef]
- Chen, L.; Becker, T.M.; Koch, U.; Stauber, T. The LRRC8/VRAC anion channel facilitates myogenic differentiation of murine myoblasts by promoting membrane hyperpolarization. J. Biol. Chem. 2019, 294, 14279–14288. [Google Scholar] [CrossRef]
- Hu, H.-Z.; Gu, Q.; Wang, C.; Colton, C.K.; Tang, J.; Kinoshita-Kawada, M.; Lee, L.-Y.; Wood, J.D.; Zhu, M.X. 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J. Biol. Chem. 2004, 279, 35741–35748. [Google Scholar] [CrossRef]
- Blair, N.T.; Carvacho, I.; Chaudhuri, D.; Clapham, D.E.; DeCaen, P.; Delling, M.; Doerner, J.F.; Fan, L.; Ha, K.; Jordt, S.E.; et al. Transient receptor potential channels (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guid. Pharmacol. CITE 2019, 2019. [Google Scholar] [CrossRef]
- Boyden, E.S.; Zhang, F.; Bamberg, E.; Nagel, G.; Deisseroth, K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 2005, 8, 1263–1268. [Google Scholar] [CrossRef]
- Jia, Z.; Valiunas, V.; Lu, Z.; Bien, H.; Liu, H.; Wang, H.-Z.; Rosati, B.; Brink, P.R.; Cohen, I.S.; Entcheva, E. Stimulating cardiac muscle by light: Cardiac optogenetics by cell delivery. Circ. Arrhythmia Electrophysiol. 2011, 4, 753–760. [Google Scholar] [CrossRef]
- Bruegmann, T.; Boyle, P.M.; Vogt, C.C.; Karathanos, T.V.; Arevalo, H.J.; Fleischmann, B.K.; Trayanova, N.A.; Sasse, P. Optogenetic defibrillation terminates ventricular arrhythmia in mouse hearts and human simulations. J. Clin. Invest. 2016, 126, 3894–3904. [Google Scholar] [CrossRef] [PubMed]
- Bruegmann, T.; van Bremen, T.; Vogt, C.C.; Send, T.; Fleischmann, B.K.; Sasse, P. Optogenetic control of contractile function in skeletal muscle. Nat. Commun. 2015, 6, 7153. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Ishizuka, T.; Morishima, K.; Yawo, H. Optogenetic induction of contractile ability in immature C2C12 myotubes. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Sebille, S.; Ayad, O.; Chapotte-Baldacci, C.-A.; Cognard, C.; Bois, P.; Chatelier, A. Optogenetic approach for targeted activation of global calcium transients in differentiated C2C12 myotubes. Sci. Rep. 2017, 7, 11108. [Google Scholar] [CrossRef]
- Schobert, B.; Lanyi, J.K. Halorhodopsin is a light-driven chloride pump. J. Biol. Chem. 1982, 257, 10306–10313. [Google Scholar] [PubMed]
- Zhang, F.; Wang, L.-P.; Brauner, M.; Liewald, J.F.; Kay, K.; Watzke, N.; Wood, P.G.; Bamberg, E.; Nagel, G.; Gottschalk, A.; et al. Multimodal fast optical interrogation of neural circuitry. Nature 2007, 446, 633–639. [Google Scholar] [CrossRef]
- Gradinaru, V.; Thompson, K.R.; Deisseroth, K. ENpHR: A Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol. 2008, 36, 129–139. [Google Scholar] [CrossRef]
- Gradinaru, V.; Zhang, F.; Ramakrishnan, C.; Mattis, J.; Prakash, R.; Diester, I.; Goshen, I.; Thompson, K.R.; Deisseroth, K. Molecular and cellular approaches for diversifying and extending optogenetics. Cell 2010, 141, 154–165. [Google Scholar] [CrossRef]
- Wiegert, J.S.; Mahn, M.; Prigge, M.; Printz, Y.; Yizhar, O. Silencing neurons: Tools, applications, and experimental constraints. Neuron 2017, 95, 504–529. [Google Scholar] [CrossRef]
- Fioretti, B.; Pietrangelo, T.; Catacuzzeno, L.; Franciolini, F. Intermediate-conductance Ca2+-activated K+ channel is expressed in C2C12 myoblasts and is downregulated during myogenesis. Am. J. Physiol. Cell Physiol. 2005, 289, C89–C96. [Google Scholar] [CrossRef]
- Leem, Y.-E.; Jeong, H.-J.; Kim, H.-J.; Koh, J.; Kang, K.; Bae, G.-U.; Cho, H.; Kang, J.-S. Cdo Regulates Surface Expression of Kir2.1 K+ Channel in Myoblast Differentiation. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Kunert-Keil, C.; Bisping, F.; Krüger, J.; Brinkmeier, H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genom. 2006, 7, 159. [Google Scholar] [CrossRef] [PubMed]
- Louis, M.; Zanou, N.; Schoor, M.V.; Gailly, P. TRPC1 regulates skeletal myoblast migration and differentiation. J. Cell Sci. 2008, 121, 3951–3959. [Google Scholar] [CrossRef] [PubMed]
- Formigli, L.; Sassoli, C.; Squecco, R.; Bini, F.; Martinesi, M.; Chellini, F.; Luciani, G.; Sbrana, F.; Zecchi-Orlandini, S.; Francini, F.; et al. Regulation of transient receptor potential canonical channel 1 (TRPC1) by sphingosine 1-phosphate in C2C12 myoblasts and its relevance for a role of mechanotransduction in skeletal muscle differentiation. J. Cell Sci. 2009, 122, 1322–1333. [Google Scholar] [CrossRef]
- Kurth, F.; Franco-Obregón, A.; Casarosa, M.; Küster, S.K.; Wuertz-Kozak, K.; Dittrich, P.S. Transient receptor potential vanilloid 2-mediated shear-stress responses in C2C12 myoblasts are regulated by serum and extracellular matrix. FASEB J. 2015, 29, 4726–4737. [Google Scholar] [CrossRef]
- Penna, A.; Juvin, V.; Chemin, J.; Compan, V.; Monet, M.; Rassendren, F.-A. PI3-kinase promotes TRPV2 activity independently of channel translocation to the plasma membrane. Cell Calcium 2006, 39, 495–507. [Google Scholar] [CrossRef]
- Zechel, S.; Werner, S.; Halbach, O.V.B.U. Distribution of TRPC4 in developing and adult murine brain. Cell Tissue Res. 2007, 328, 651–656. [Google Scholar] [CrossRef]
- Constantin, B.; Cognard, C.; Raymond, G. Myoblast fusion requires cytosolic calcium elevation but not activation of voltage-dependent calcium channels. Cell Calcium 1996, 19, 365–374. [Google Scholar] [CrossRef]
- Bidaud, I.; Monteil, A.; Nargeot, J.; Lory, P. Properties and role of voltage-dependent calcium channels during mouse skeletal muscle differentiation. J. Muscle Res. Cell Motil. 2006, 27, 75–81. [Google Scholar] [CrossRef]
- Antigny, F.; Sabourin, J.; Saüc, S.; Bernheim, L.; Koenig, S.; Frieden, M. TRPC1 and TRPC4 channels functionally interact with STIM1L to promote myogenesis and maintain fast repetitive Ca2+ release in human myotubes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, M.; Zhang, Y.-Q.; Mashima, H.; Li, L.; Shibata, H.; Kojima, I. Translocation of a calcium-permeable cation channel induced by insulin-like growth factor-I. Nat. Cell Biol. 1999, 1, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Jahnel, R.; Bender, O.; Münter, L.M.; Dreger, M.; Gillen, C.; Hucho, F. Dual expression of mouse and rat VRL-1 in the dorsal root ganglion derived cell line F-11 and biochemical analysis of VRL-1 after heterologous expression. Eur. J. Biochem. 2003, 270, 4264–4271. [Google Scholar] [CrossRef] [PubMed]
- Monet, M.; Gkika, D.; Lehen’kyi, V.; Pourtier, A.; Vanden Abeele, F.; Bidaux, G.; Juvin, V.; Rassendren, F.; Humez, S.; Prevarsakaya, N. Lysophospholipids stimulate prostate cancer cell migration via TRPV2 channel activation. Biochim. Biophys. Acta 2009, 1793, 528–539. [Google Scholar] [CrossRef]
- Nie, L.; Oishi, Y.; Doi, I.; Shibata, H.; Kojima, I. Inhibition of proliferation of MCF-7 breast cancer cells by a blocker of Ca(2+)-permeable channel. Cell Calcium 1997, 22, 75–82. [Google Scholar] [CrossRef]
- Hisanaga, E.; Nagasawa, M.; Ueki, K.; Kulkarni, R.N.; Mori, M.; Kojima, I. Regulation of calcium-permeable TRPV2 channel by insulin in pancreatic beta-cells. Diabetes 2009, 58, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Ohtake, H.; Suzuki, O.; Matsuda, J.; Komamura, K.; Wakabayashi, S. Blockade of sarcolemmal TRPV2 accumulation inhibits progression of dilated cardiomyopathy. Cardiovasc. Res. 2013, 99, 760–768. [Google Scholar] [CrossRef]
- Lévêque, M.; Penna, A.; Trionnaire, S.L.; Belleguic, C.; Desrues, B.; Brinchault, G.; Jouneau, S.; Lagadic-Gossmann, D.; Martin-Chouly, C. Phagocytosis depends on TRPV2-mediated calcium influx and requires TRPV2 in lipids rafts: Alteration in macrophages from patients with cystic fibrosis. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Iwata, Y.; Katanosaka, Y.; Arai, Y.; Shigekawa, M.; Wakabayashi, S. Dominant-negative inhibition of Ca2+ influx via TRPV2 ameliorates muscular dystrophy in animal models. Hum. Mol. Genet. 2009, 18, 824–834. [Google Scholar] [CrossRef]
- Cohen, M.R.; Johnson, W.M.; Pilat, J.M.; Kiselar, J.; DeFrancesco-Lisowitz, A.; Zigmond, R.E.; Moiseenkova-Bell, V.Y. Nerve growth factor regulates transient receptor potential vanilloid 2 via extracellular signal-regulated kinase signaling to enhance neurite outgrowth in developing neurons. Mol. Cell. Biol. 2015, 35, 4238–4252. [Google Scholar] [CrossRef]
- Oulidi, A.; Bokhobza, A.; Gkika, D.; Vanden Abeele, F.; Lehen’kyi, V.; Ouafik, L.; Mauroy, B.; Prevarskaya, N. TRPV2 mediates adrenomedullin stimulation of prostate and urothelial cancer cell adhesion, migration and invasion. PLoS ONE 2013, 8, e64885. [Google Scholar] [CrossRef]
- Nagel, G.; Brauner, M.; Liewald, J.F.; Adeishvili, N.; Bamberg, E.; Gottschalk, A. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr. Biol. 2005, 15, 2279–2284. [Google Scholar] [CrossRef]
- Canales, J.; Morales, D.; Blanco, C.; Rivas, J.; Díaz, N.; Angelopoulos, I.; Cerda, O. A TR(i)P to cell migration: New roles of TRP channels in mechanotransduction and cancer. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Kiviluoto, S.; Decuypere, J.-P.; De Smedt, H.; Missiaen, L.; Parys, J.B.; Bultynck, G. STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet. Muscle 2011, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Trepat, X.; Chen, Z.; Jacobson, K. Cell migration. Compr. Physiol. 2012, 2, 2369–2392. [Google Scholar] [CrossRef]
- Brundage, R.A.; Fogarty, K.E.; Tuft, R.A.; Fay, F.S. Calcium gradients underlying polarization and chemotaxis of eosinophils. Science 1991, 254, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef]
- Tsai, F.-C.; Meyer, T. Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Curr. Biol. 2012, 22, 837–842. [Google Scholar] [CrossRef]
- Wei, C.; Wang, X.; Zheng, M.; Cheng, H. Calcium gradients underlying cell migration. Curr. Opin. Cell Biol. 2012, 24, 254–261. [Google Scholar] [CrossRef]
- Lee, J.; Ishihara, A.; Oxford, G.; Johnson, B.; Jacobson, K. Regulation of cell movement is mediated by stretch-activated calcium channels. Nature 1999, 400, 382–386. [Google Scholar] [CrossRef]
- Wei, C.; Wang, X.; Chen, M.; Ouyang, K.; Song, L.-S.; Cheng, H. Calcium flickers steer cell migration. Nature 2009, 457, 901–905. [Google Scholar] [CrossRef]
- Monet, M.; Lehen’kyi, V.; Gackiere, F.; Firlej, V.; Vandenberghe, M.; Roudbaraki, M.; Gkika, D.; Pourtier, A.; Bidaux, G.; Slomianny, C.; et al. Role of cationic channel TRPV2 in promoting prostate cancer migration and progression to androgen resistance. Cancer Res. 2010, 70, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, M.; Kojima, I. Translocation of calcium-permeable TRPV2 channel to the podosome: Its role in the regulation of podosome assembly. Cell Calcium 2012, 51, 186–193. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Ref seq | Primers (5′–3′) | Annealing Temperature (°C) | Amplicon (pb) |
|---|---|---|---|---|
| TRPC1 | NM_011643 NM_001311123 | CAAGATTTTGGGAAATTTCTGG TTTATCCTCATGATTTGCTAT | 55 | 371 |
| TRPC4 | NM 016984 | TCTGCAGATATCTCTGGGAAGGATGC AAGCTTTGTTCGAGCAAATTTCCATTC | 57 | 414 |
| TRPM7 | NM_021450 NM_001164325 | TTGGAGCATTTGTGGGACAC ACGGGCTTAAATGGAGAAGCA | 60 | 328 |
| TRPV2 | NM 011706 | AGATGCTTAGAACTAAGGTGGAGG AGAGTCGGTCACGGTCAAAC | 60 | 500 |
| TRPV4 | NM_022017 | GTGGGCAAGAGCTCAGATGG CCGAGGACCAACGATCCCTAC | 60 | 184 |
| 18S mRNA (Mrps6) | NM 080456 | TTTGATTCTGAAAGCCATGCG CCAGTATGTTCTCCACAGCA | 57 | 218 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chapotte-Baldacci, C.-A.; Lizot, G.; Jajkiewicz, C.; Lévêque, M.; Penna, A.; Magaud, C.; Thoreau, V.; Bois, P.; Sebille, S.; Chatelier, A. Fine Tuning of Calcium Constitutive Entry by Optogenetically-Controlled Membrane Polarization: Impact on Cell Migration. Cells 2020, 9, 1684. https://doi.org/10.3390/cells9071684
Chapotte-Baldacci C-A, Lizot G, Jajkiewicz C, Lévêque M, Penna A, Magaud C, Thoreau V, Bois P, Sebille S, Chatelier A. Fine Tuning of Calcium Constitutive Entry by Optogenetically-Controlled Membrane Polarization: Impact on Cell Migration. Cells. 2020; 9(7):1684. https://doi.org/10.3390/cells9071684
Chicago/Turabian StyleChapotte-Baldacci, Charles-Albert, Guénaëlle Lizot, Cyrielle Jajkiewicz, Manuella Lévêque, Aubin Penna, Christophe Magaud, Vincent Thoreau, Patrick Bois, Stéphane Sebille, and Aurélien Chatelier. 2020. "Fine Tuning of Calcium Constitutive Entry by Optogenetically-Controlled Membrane Polarization: Impact on Cell Migration" Cells 9, no. 7: 1684. https://doi.org/10.3390/cells9071684
APA StyleChapotte-Baldacci, C.-A., Lizot, G., Jajkiewicz, C., Lévêque, M., Penna, A., Magaud, C., Thoreau, V., Bois, P., Sebille, S., & Chatelier, A. (2020). Fine Tuning of Calcium Constitutive Entry by Optogenetically-Controlled Membrane Polarization: Impact on Cell Migration. Cells, 9(7), 1684. https://doi.org/10.3390/cells9071684