MRIP Regulates the Myosin IIA Activity and DDR1 Function to Enable Collagen Tractional Remodeling

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cells
2.3. MRIP Deletion by CRISPR/Cas9
2.4. Transfection
2.5. Collagen and Fibronectin Substrates
2.6. Immunofluorescence and Microscopy Analysis
2.7. Immunoblotting
2.8. Immunoprecipitation
2.9. Tandem Mass Tagged Mass Spectrometry
2.10. Wound Healing Assay
2.11. Collagen Substrate Deformation by Cell-Generated Forces
2.12. RhoA Activation Assay
2.13. Isolation of DDR1 Adhesion Complexes
2.14. Fluorescence Resonance Energy Transfer
2.15. Fluorescence Recovery after Photobleaching
2.16. Statistical Analysis
3. Results
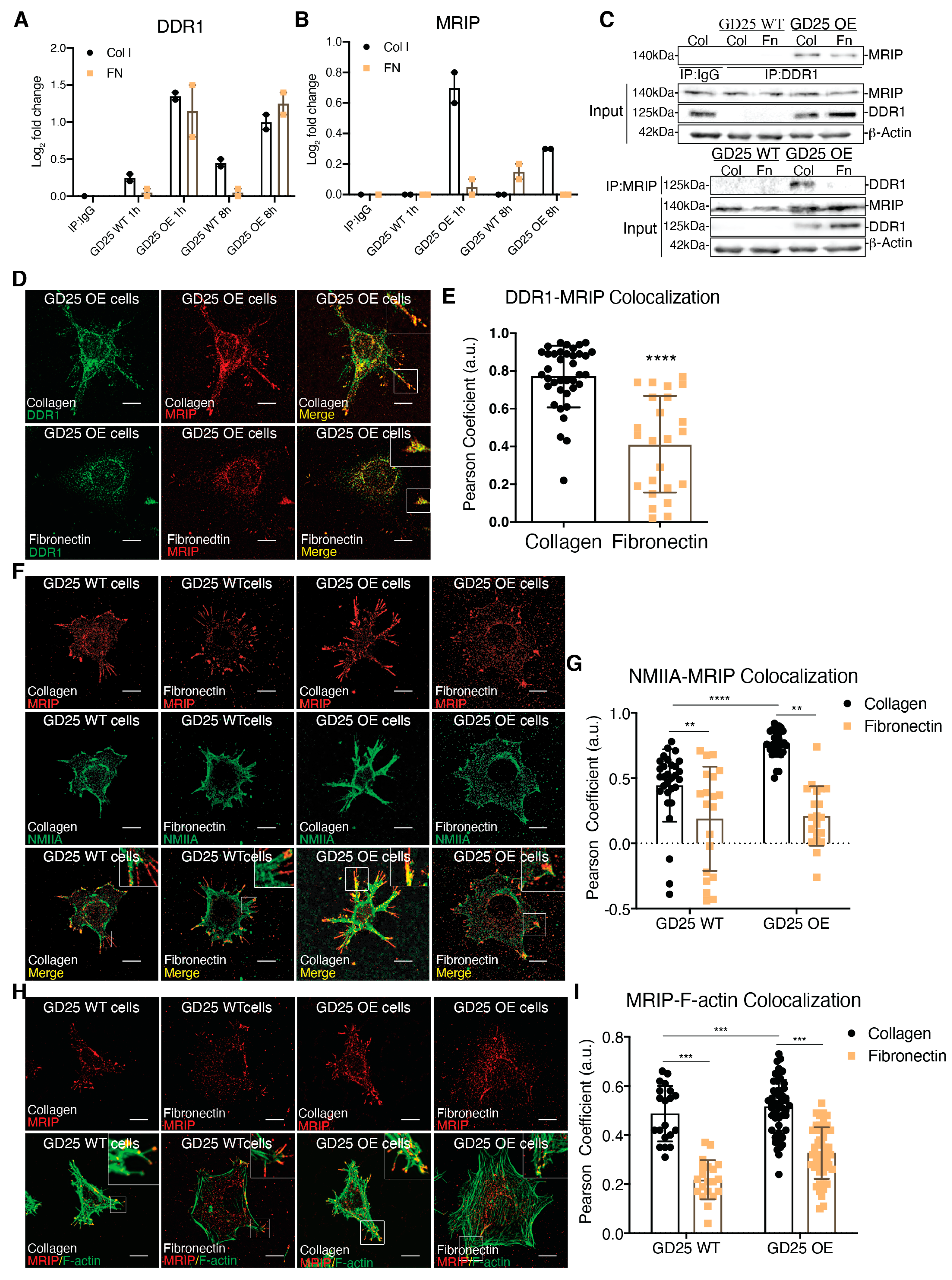
3.1. MRIP is Enriched in DDR1-Collagen-Adhesion Complexes
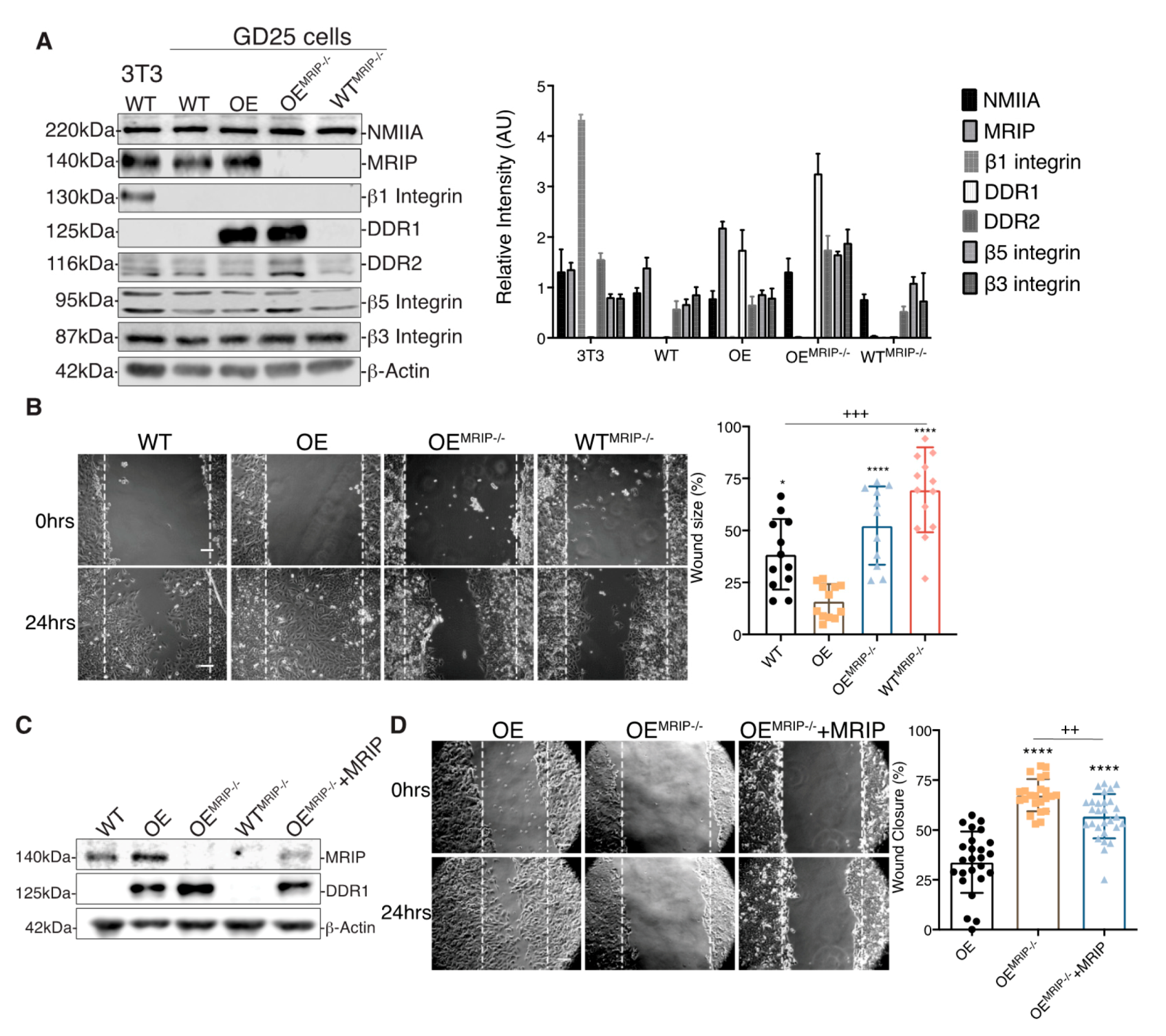
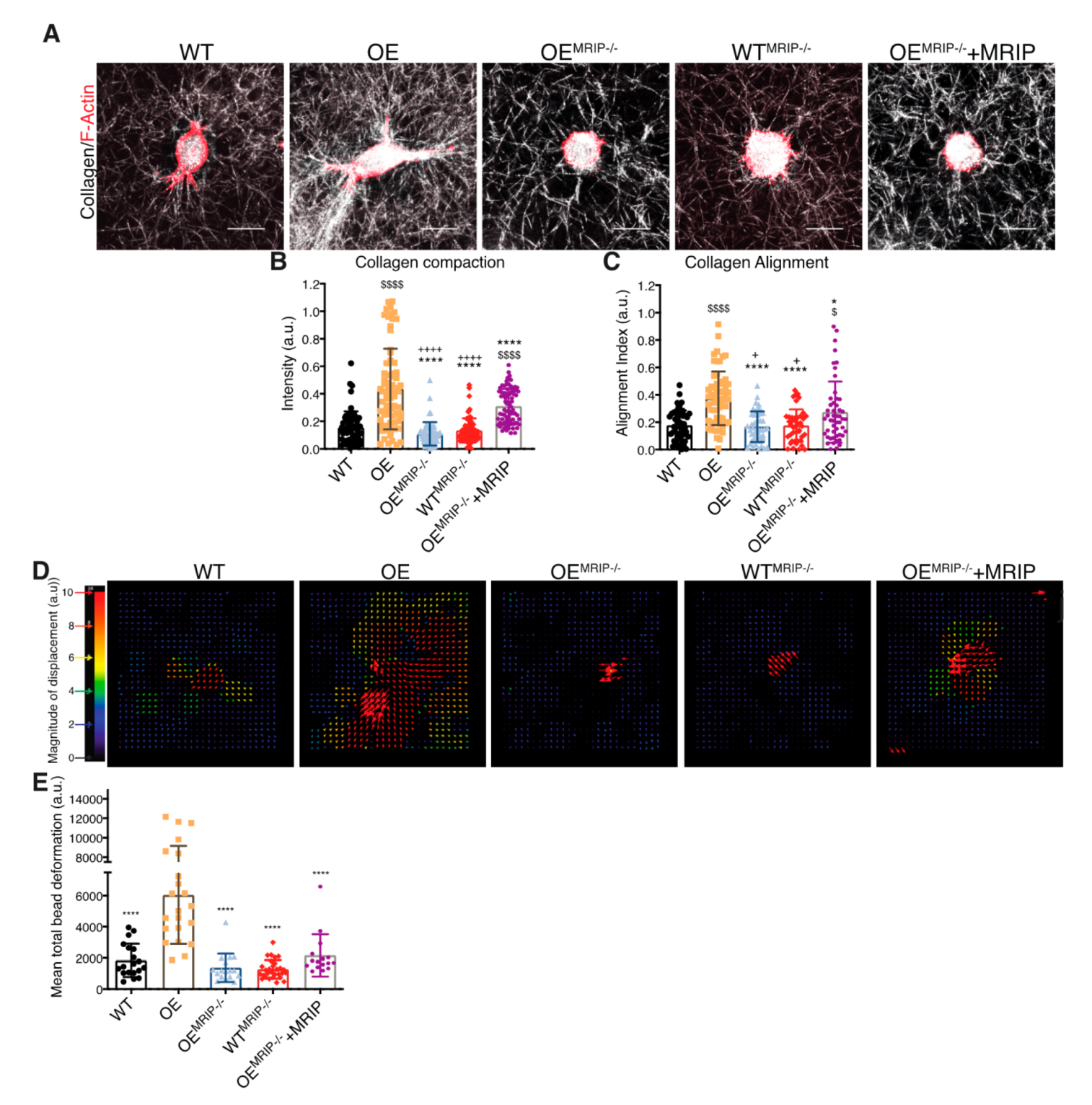
3.2. MRIP Expression Affects Cell Migration and Collagen Tractional Remodeling
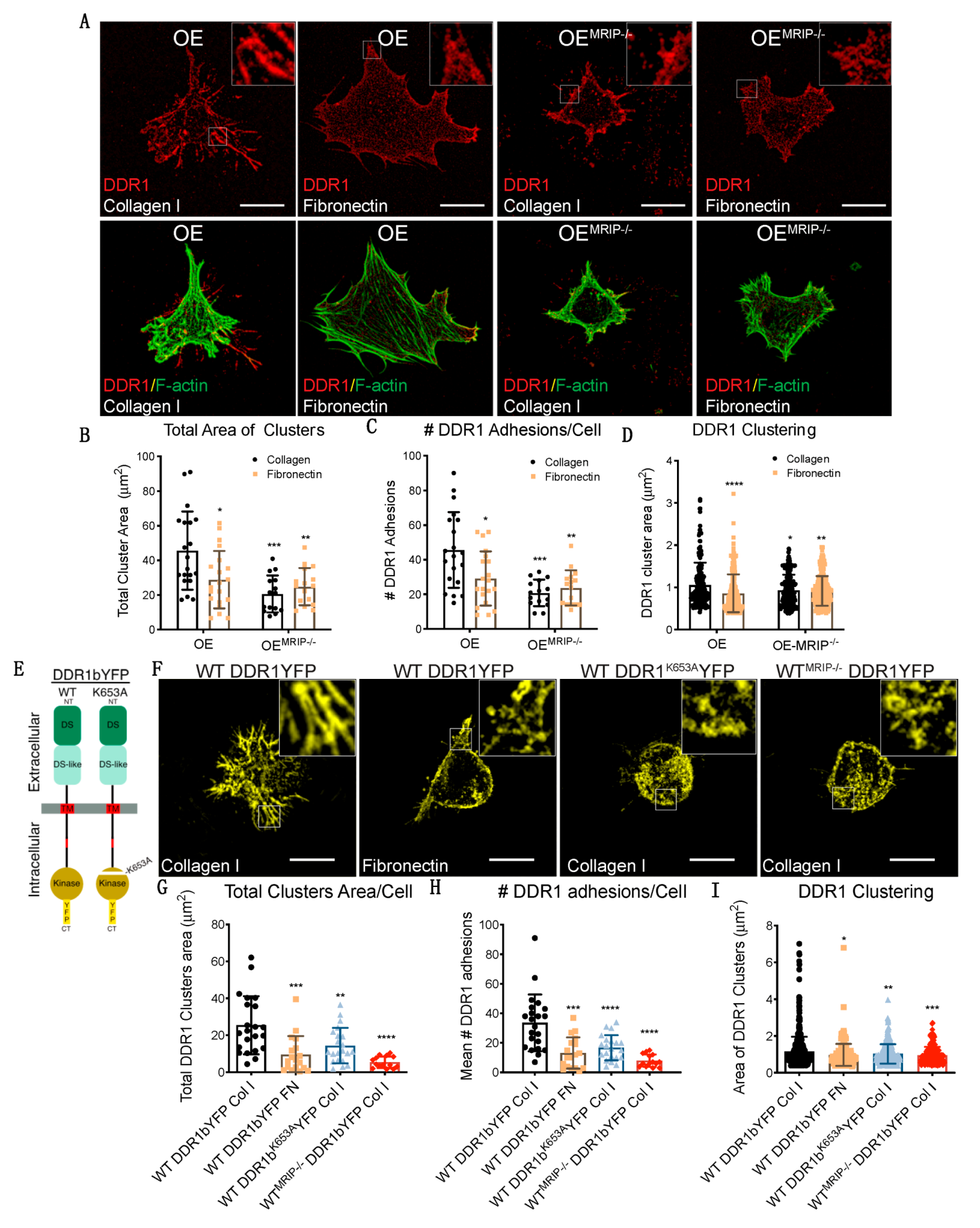
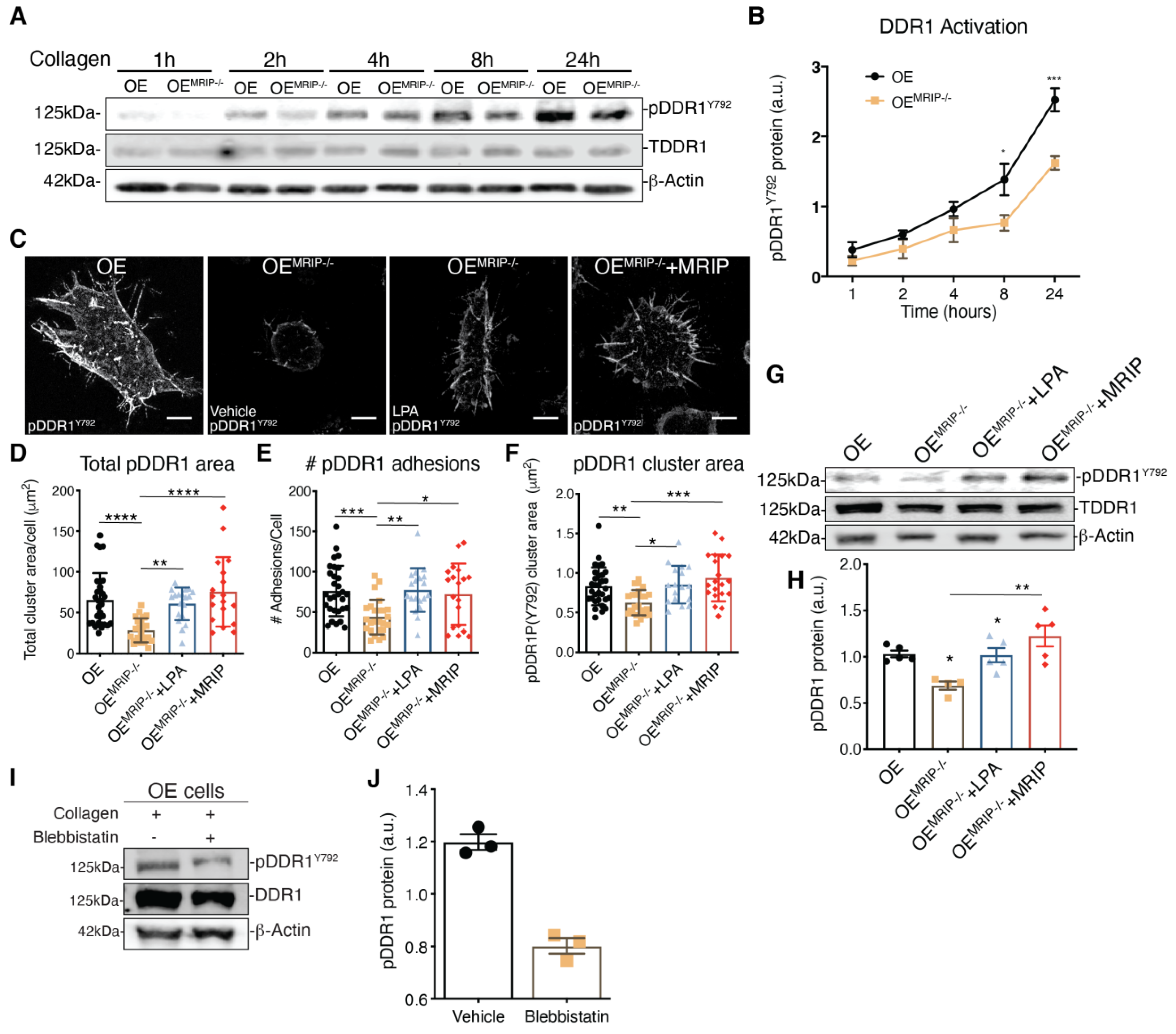
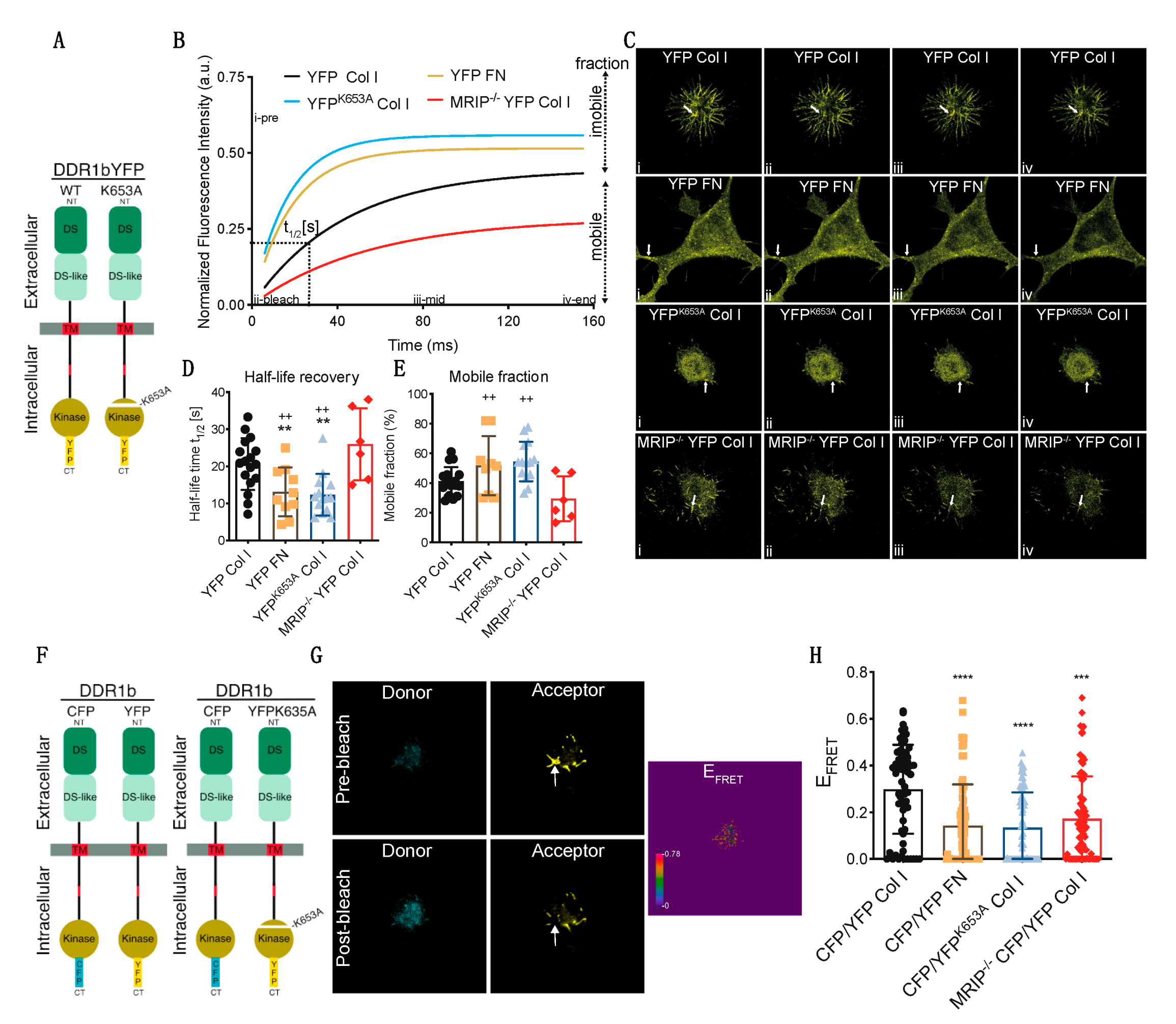
3.3. MRIP Enables DDR1 Cluster Formation and Growth and Stabilizes DDR1 Activation
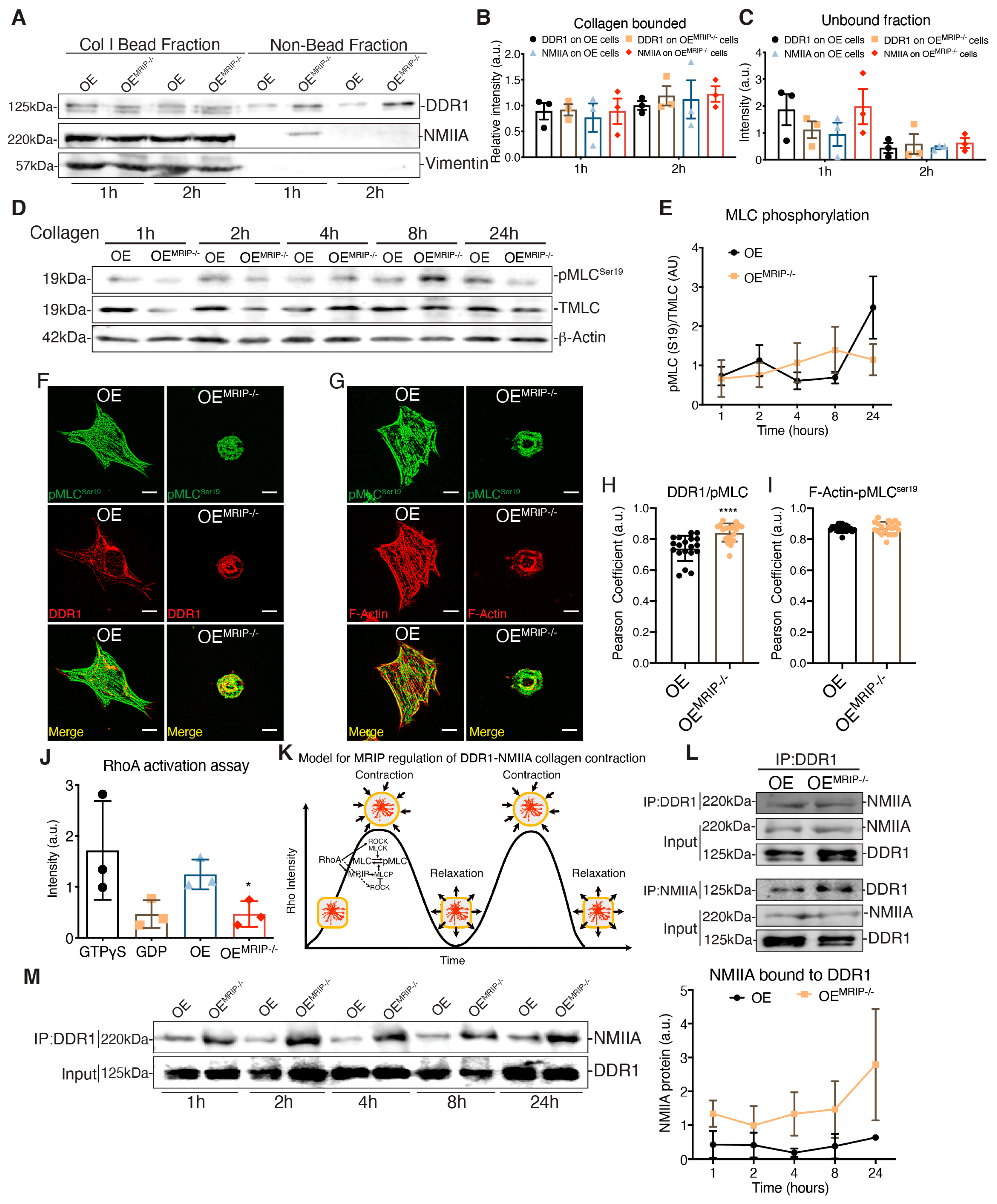
3.4. MRIP Dependent Contraction Cycle Regulates DDR1-NMIIA Association
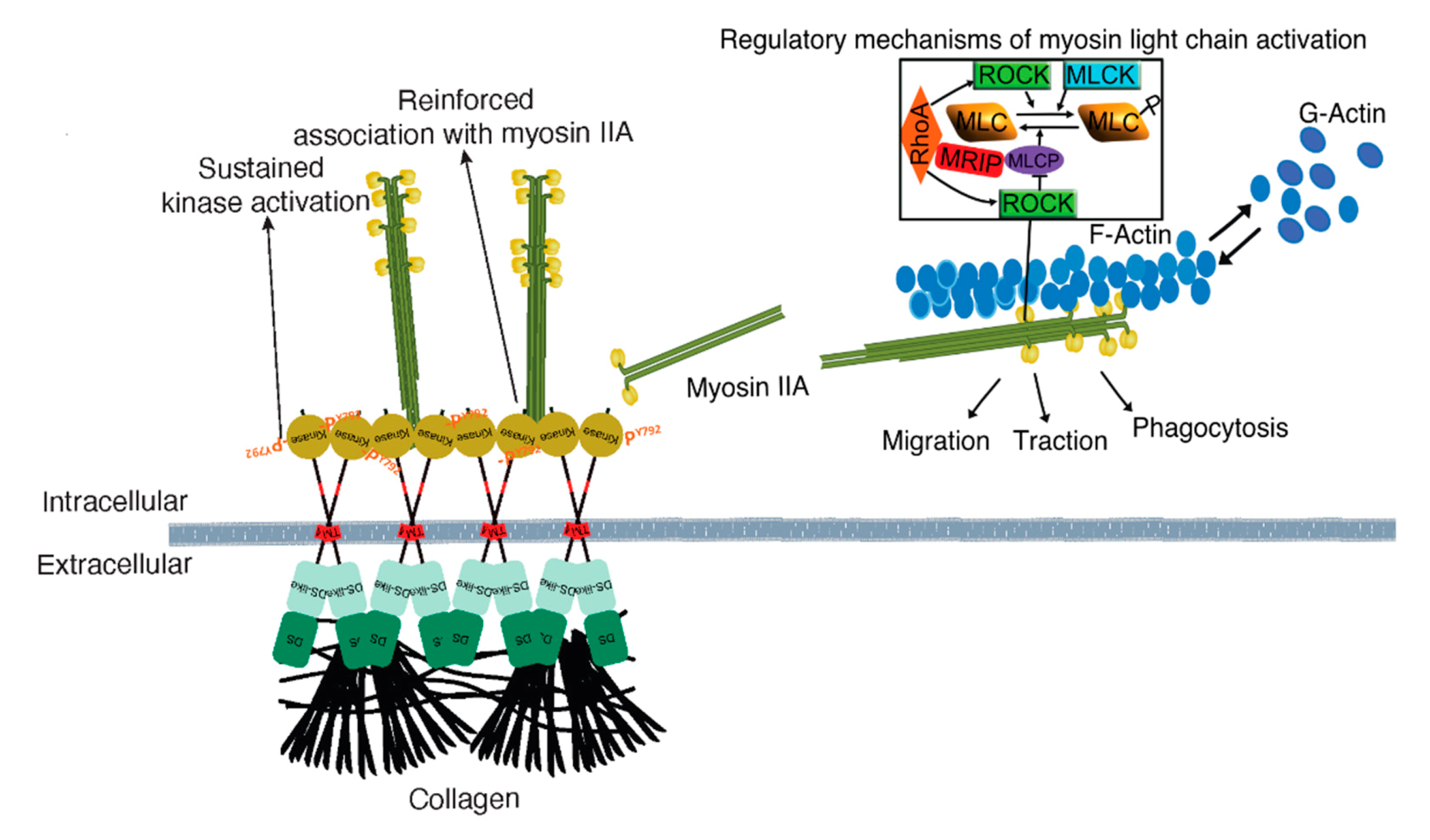
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Vogel, W.; Brakebusch, C.; Fassler, R.; Alves, F.; Ruggiero, F.; Pawson, T. Discoidin domain receptor 1 is activated independently of beta (1) integrin. J. Biol. Chem. 2000, 275, 5779–5784. [Google Scholar] [CrossRef] [PubMed]
- Coelho, N.M.; Arora, P.D.; van Putten, S.; Boo, S.; Petrovic, P.; Lin, A.X.; Hinz, B.; McCulloch, C.A. Discoidin Domain Receptor 1 Mediates Myosin-Dependent Collagen Contraction. Cell Rep. 2017, 18, 1774–1790. [Google Scholar] [CrossRef] [PubMed]
- MacGillivray, M.K.; Cruz, T.F.; McCulloch, C.A.G. The Recruitment of the Interleukin-1 (IL-1) Receptor-associated Kinase (IRAK) into Focal Adhesion Complexes Is Required for IL-1β-induced ERK Activation. J. Biol. Chem. 2000, 275, 23509–23515. [Google Scholar] [CrossRef]
- Fassler, R.; Pfaff, M.; Murphy, J.; Noegel, A.A.; Johansson, S.; Timpl, R.; Albrecht, R. Lack of beta 1 integrin gene in embryonic stem cells affects morphology, adhesion, and migration but not integration into the inner cell mass of blastocysts. J. Cell Biol. 1995, 128, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Leitinger, B. Transmembrane collagen receptors. Annu. Rev. Cell Dev. Biol. 2011, 27, 265–290. [Google Scholar] [CrossRef]
- Mulder, J.; Ariaens, A.; van den Boomen, D.; Moolenaar, W.H. p116Rip targets myosin phosphatase to the actin cytoskeleton and is essential for RhoA/ROCK-regulated neuritogenesis. Mol. Biol. Cell 2004, 15, 5516–5527. [Google Scholar] [CrossRef] [PubMed]
- Vallenius, T.; Vaahtomeri, K.; Kovac, B.; Osiceanu, A.M.; Viljanen, M.; Makela, T.P. An association between NUAK2 and MRIP reveals a novel mechanism for regulation of actin stress fibers. J. Cell Sci. 2011, 124, 384–393. [Google Scholar] [CrossRef]
- Surks, H.K.; Riddick, N.; Ohtani, K. M-RIP targets myosin phosphatase to stress fibers to regulate myosin light chain phosphorylation in vascular smooth muscle cells. J. Biol. Chem. 2005, 280, 42543–42551. [Google Scholar] [CrossRef]
- Huang, Y.; Arora, P.; McCulloch, C.A.; Vogel, W.F. The collagen receptor DDR1 regulates cell spreading and motility by associating with myosin IIA. J. Cell Sci. 2009, 122, 1637–1646. [Google Scholar] [CrossRef]
- Corcoran, D.S.; Juskaite, V.; Xu, Y.; Görlitz, F.; Alexandrov, Y.; Dunsby, C.; French, P.M.W.; Leitinger, B. DDR1 autophosphorylation is a result of aggregation into dense clusters. Sci. Rep. 2019, 9, 17104. [Google Scholar] [CrossRef]
- Juskaite, V.; Corcoran, D.S.; Leitinger, B. Collagen induces activation of DDR1 through lateral dimer association and phosphorylation between dimers. eLife 2017, 6, e25716. [Google Scholar] [CrossRef] [PubMed]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Delcorde, J.; Tang, T.; Downey, G.P.; McCulloch, C.A. Regulation of IL-1 signaling through control of focal adhesion assembly. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 3119–3132. [Google Scholar] [CrossRef] [PubMed]
- Surks, H.K.; Richards, C.T.; Mendelsohn, M.E. Myosin phosphatase-Rho interacting protein. A new member of the myosin phosphatase complex that directly binds RhoA. J. Biol. Chem. 2003, 278, 51484–51493. [Google Scholar] [CrossRef]
- Totsukawa, G.; Yamakita, Y.; Yamashiro, S.; Hartshorne, D.J.; Sasaki, Y.; Matsumura, F. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J. Cell Biol. 2000, 150, 797–806. [Google Scholar] [CrossRef]
- Agarwal, P.; Zaidel-Bar, R. Principles of Actomyosin Regulation In Vivo. Trends Cell Biol. 2019, 29, 150–163. [Google Scholar] [CrossRef]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression predicts clinical outcome in breast cancer. Nature Med. 2008, 14, 518–527. [Google Scholar] [CrossRef]
- Sun, X.; Gupta, K.; Wu, B.; Zhang, D.; Yuan, B.; Zhang, X.; Chiang, H.C.; Zhang, C.; Curiel, T.J.; Bendeck, M.P.; et al. Tumor-extrinsic discoidin domain receptor 1 promotes mammary tumor growth by regulating adipose stromal interleukin 6 production in mice. J. Biol. Chem. 2018, 293, 2841–2849. [Google Scholar] [CrossRef]
- Jin, H.; Ham, I.-H.; Oh, H.J.; Bae, C.A.; Lee, D.; Kim, Y.-B.; Son, S.-Y.; Chwae, Y.-J.; Han, S.-U.; Brekken, R.A.; et al. Inhibition of Discoidin Domain Receptor 1 Prevents Stroma-Induced Peritoneal Metastasis in Gastric Carcinoma. Mol. Cancer Res. 2018, 16, 1590–1600. [Google Scholar] [CrossRef]
- Malik, R.; Lelkes, P.I.; Cukierman, E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015, 33, 230–236. [Google Scholar] [CrossRef]
- Conti, M.A.; Adelstein, R.S. Nonmuscle myosin II moves in new directions. J. Cell Sci. 2008, 121, 11–18. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho GTPases and cell migration. J. Cell Sci. 2001, 114, 2713–2722. [Google Scholar]
- Riddick, N.; Ohtani, K.; Surks, H.K. Targeting by myosin phosphatase-RhoA interacting protein mediates RhoA/ROCK regulation of myosin phosphatase. J. Cell Biochem. 2008, 103, 1158–1170. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nature Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Ikebe, M. p116Rip decreases myosin II phosphorylation by activating myosin light chain phosphatase and by inactivating RhoA. J. Biol. Chem. 2005, 280, 4983–4991. [Google Scholar] [CrossRef]
- Pakshir, P.; Alizadehgiashi, M.; Wong, B.; Coelho, N.M.; Chen, X.; Gong, Z.; Shenoy, V.B.; McCulloch, C.A.; Hinz, B. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat. Commun. 2019, 10, 1850. [Google Scholar] [CrossRef]
- Dembo, M.; Wang, Y.L. Stresses at the cell-to-substrate interface during locomotion of fibroblasts. Biophys. J. 1999, 76, 2307–2316. [Google Scholar] [CrossRef]
- Plotnikov, S.V.; Sabass, B.; Schwarz, U.S.; Waterman, C.M. Chapter 20—High-Resolution Traction Force Microscopy. In Methods in Cell Biology; Waters, J.C., Wittman, T., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 123, pp. 367–394. [Google Scholar]
- Katoh, K.; Kano, Y.; Amano, M.; Kaibuchi, K.; Fujiwara, K. Stress fiber organization regulated by MLCK and Rho-kinase in cultured human fibroblasts. Am. J. Physiol. Cell Physiol. 2001, 280, C1669–C1679. [Google Scholar] [CrossRef]
- Matsumura, F.; Hartshorne, D.J. Myosin phosphatase target subunit: Many roles in cell function. Biochem. Biophys. Res. Commun. 2008, 369, 149–156. [Google Scholar] [CrossRef]
- Clark, K.; Langeslag, M.; Figdor, C.G.; van Leeuwen, F.N. Myosin II and mechanotransduction: A balancing act. Trends Cell Biol. 2007, 17, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Kural, M.H.; Billiar, K.L. Regulating tension in three-dimensional culture environments. Exp. Cell Res. 2013, 319, 2447–2459. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Trier, S.M.; Keely, P.J. Contact guidance mediated three-dimensional cell migration is regulated by Rho/ROCK-dependent matrix reorganization. Biophys J. 2008, 95, 5374–5384. [Google Scholar] [CrossRef]
- Avivi-Green, C.; Singal, M.; Vogel, W.F. Discoidin domain receptor 1-deficient mice are resistant to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Flamant, M.; Placier, S.; Rodenas, A.; Curat, C.A.; Vogel, W.F.; Chatziantoniou, C.; Dussaule, J.C. Discoidin domain receptor 1 null mice are protected against hypertension-induced renal disease. J. Am. Soc. Nephrol. JASN 2006, 17, 3374–3381. [Google Scholar] [CrossRef]
- Ford, C.E.; Lau, S.K.; Zhu, C.Q.; Andersson, T.; Tsao, M.S.; Vogel, W.F. Expression and mutation analysis of the discoidin domain receptors 1 and 2 in non-small cell lung carcinoma. Br. J. Cancer 2007, 96, 808–814. [Google Scholar] [CrossRef]
- Toy, K.A.; Valiathan, R.R.; Nunez, F.; Kidwell, K.M.; Gonzalez, M.E.; Fridman, R.; Kleer, C.G. Tyrosine kinase discoidin domain receptors DDR1 and DDR2 are coordinately deregulated in triple-negative breast cancer. Breast Cancer Res. Treat. 2015, 150, 9–18. [Google Scholar] [CrossRef]
- Staudinger, L.A.; Spano, S.J.; Lee, W.; Coelho, N.; Rajshankar, D.; Bendeck, M.P.; Moriarty, T.; McCulloch, C.A. Interactions between the discoidin domain receptor 1 and beta1 integrin regulate attachment to collagen. Biol. Open 2013, 2, 1148–1159. [Google Scholar] [CrossRef]
- Meyer zum Gottesberge, A.M.; Gross, O.; Becker-Lendzian, U.; Massing, T.; Vogel, W.F. Inner ear defects and hearing loss in mice lacking the collagen receptor DDR1. Lab. Investig. 2008, 88, 27–37. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Vogel, W.; Gish, G.D.; Alves, F.; Pawson, T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1997, 1, 13–23. [Google Scholar] [CrossRef]
- Mihai, C.; Chotani, M.; Elton, T.S.; Agarwal, G. Mapping of DDR1 distribution and oligomerization on the cell surface by FRET microscopy. J. Mol. Biol. 2009, 385, 432–445. [Google Scholar] [CrossRef]
- Yeung, D.; Chmielewski, D.; Mihai, C.; Agarwal, G. Oligomerization of DDR1 ECD affects receptor-ligand binding. J. Struct. Biol. 2013, 183, 495–500. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coelho, N.M.; McCulloch, C.A. Mechanical signaling through the discoidin domain receptor 1 plays a central role in tissue fibrosis. Cell Adhes. Migr. 2018, 12, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, N.M.; Wang, A.; Petrovic, P.; Wang, Y.; Lee, W.; McCulloch, C.A. MRIP Regulates the Myosin IIA Activity and DDR1 Function to Enable Collagen Tractional Remodeling. Cells 2020, 9, 1672. https://doi.org/10.3390/cells9071672
Coelho NM, Wang A, Petrovic P, Wang Y, Lee W, McCulloch CA. MRIP Regulates the Myosin IIA Activity and DDR1 Function to Enable Collagen Tractional Remodeling. Cells. 2020; 9(7):1672. https://doi.org/10.3390/cells9071672
Chicago/Turabian StyleCoelho, Nuno M., Andrew Wang, Petar Petrovic, Yongqiang Wang, Wilson Lee, and Christopher A. McCulloch. 2020. "MRIP Regulates the Myosin IIA Activity and DDR1 Function to Enable Collagen Tractional Remodeling" Cells 9, no. 7: 1672. https://doi.org/10.3390/cells9071672
APA StyleCoelho, N. M., Wang, A., Petrovic, P., Wang, Y., Lee, W., & McCulloch, C. A. (2020). MRIP Regulates the Myosin IIA Activity and DDR1 Function to Enable Collagen Tractional Remodeling. Cells, 9(7), 1672. https://doi.org/10.3390/cells9071672