The GABARAP Co-Secretome Identified by APEX2-GABARAP Proximity Labelling of Extracellular Vesicles

 ,
, 
 and
and
Abstract
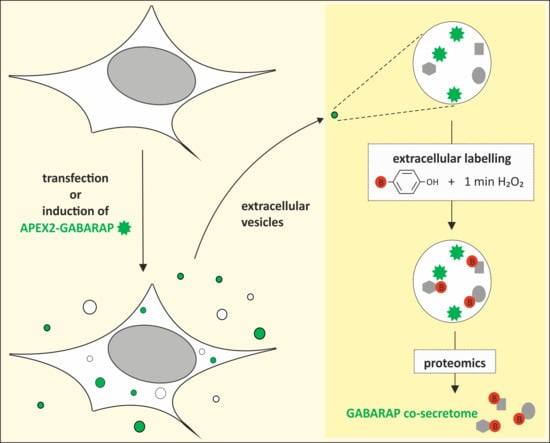
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Cloning
2.3. Cell Culture and Transfection
2.4. Blood Sampling
2.5. Isolation of EVs
2.6. Proximity Labelling
2.7. Immunoblotting (IB)
2.8. Mass Spectrometric Protein Identification and Quantification
2.9. Immunofluorescence (IF)
2.10. Image Acquisition—Laser Scanning Microscopy (LSM)
2.11. Transmission Electron Microscopy (TEM)
2.12. Nanoparticle Tracking Analysis (NTA)
3. Results
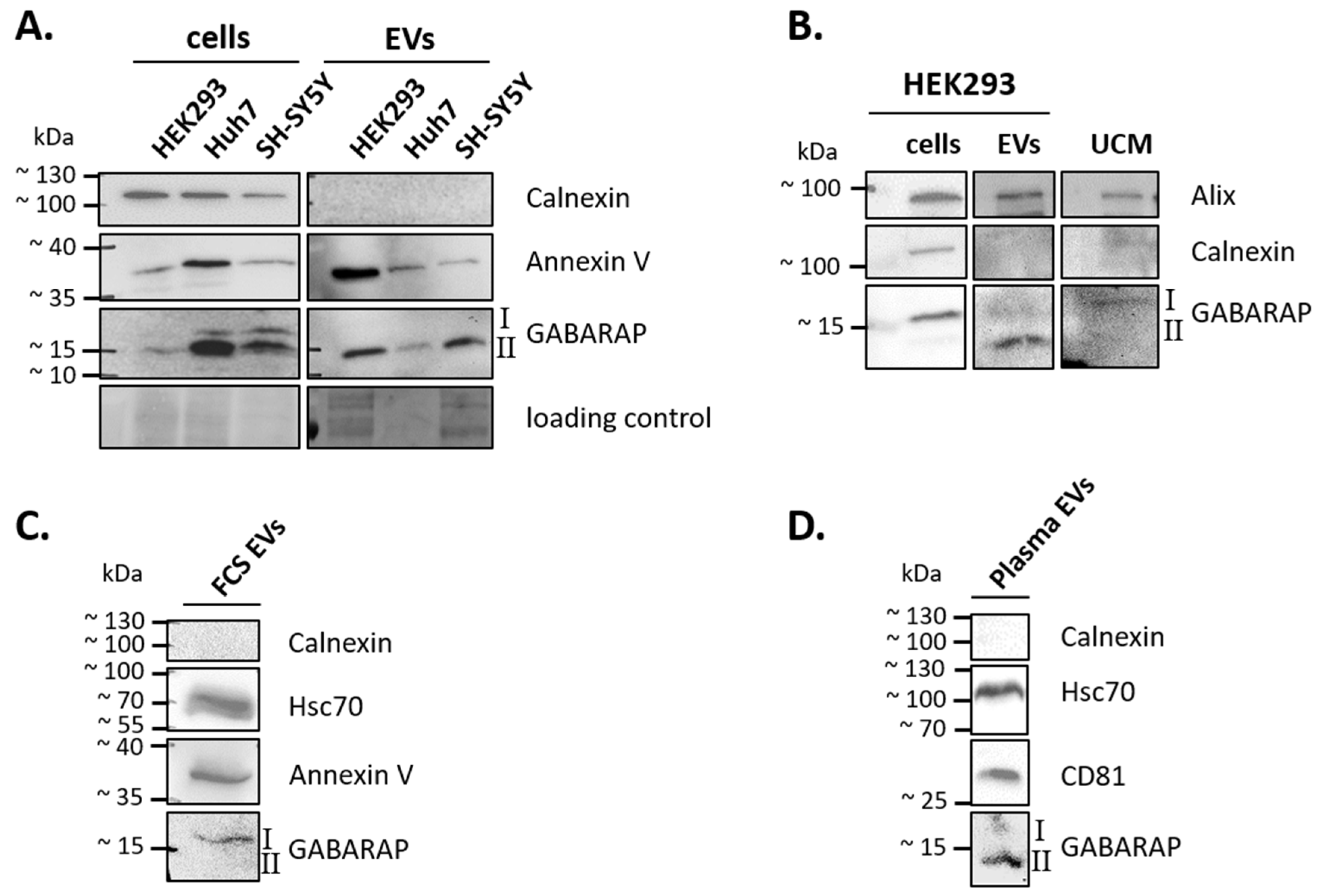
3.1. GABARAP is Secreted in EVs of Different Cell Lines and Sample Types
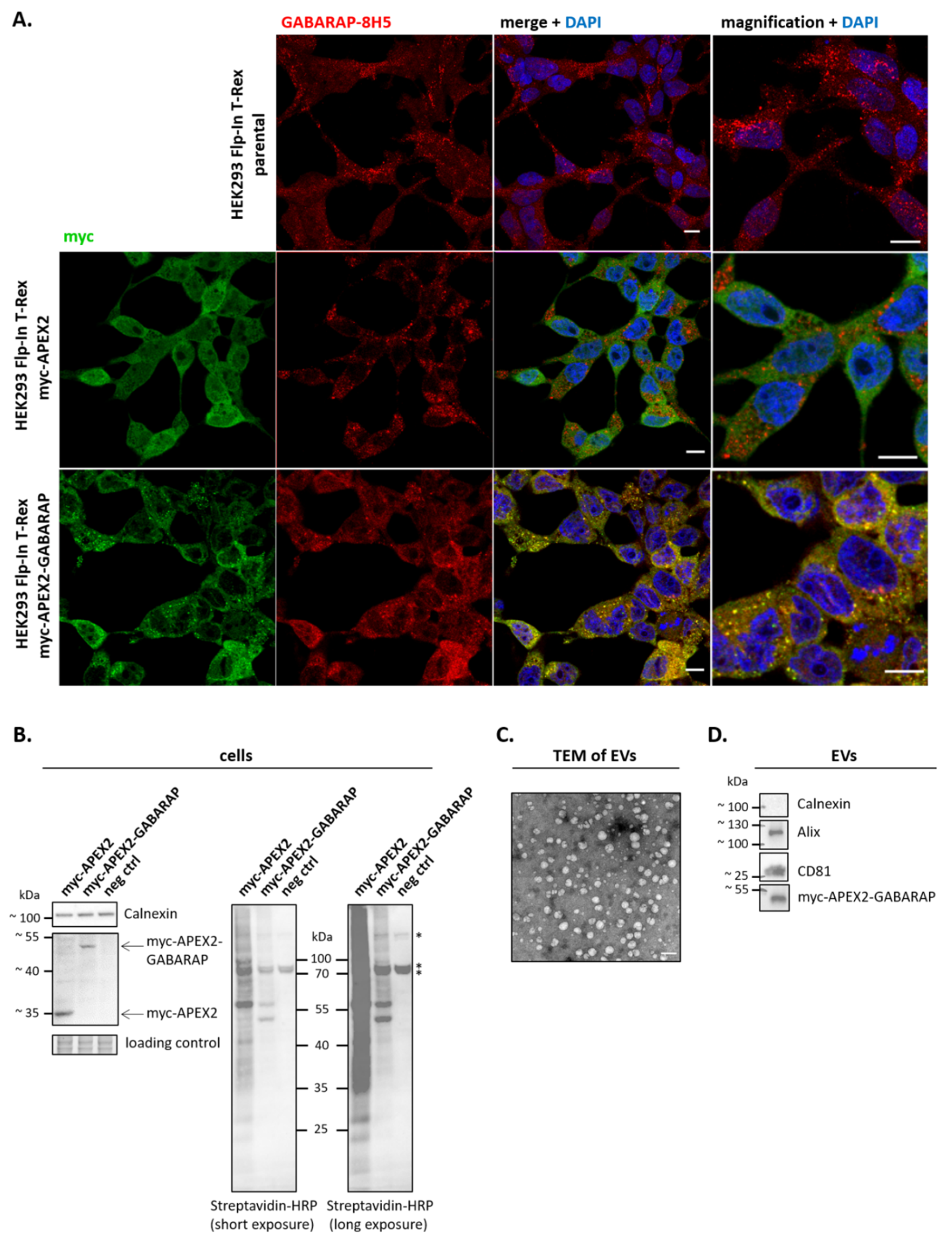
3.2. APEX2-GABARAP Exhibits Different Cellular Localisation Than APEX2’s
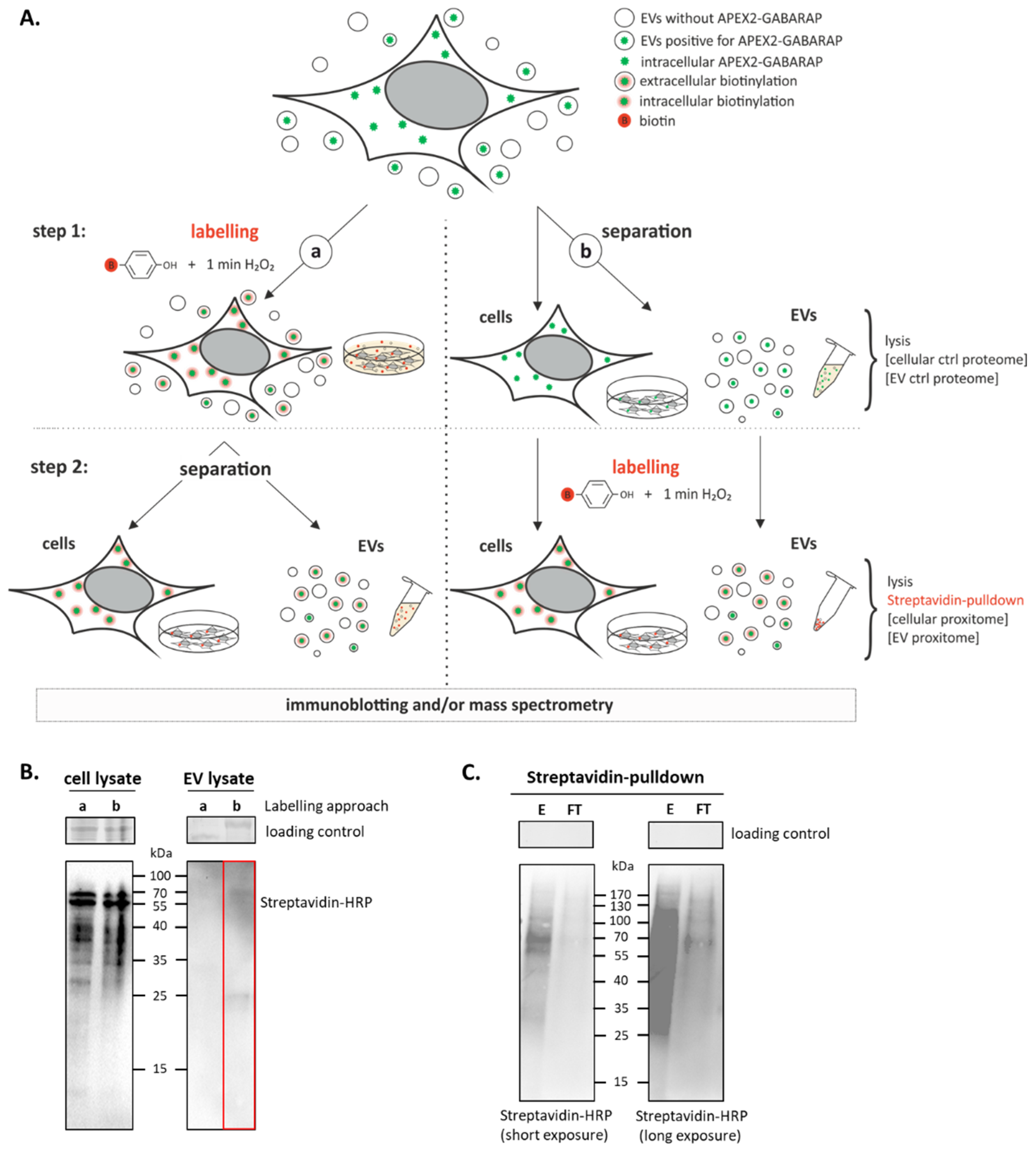
3.3. APEX2-GABARAP Proximity Labelling in EVs
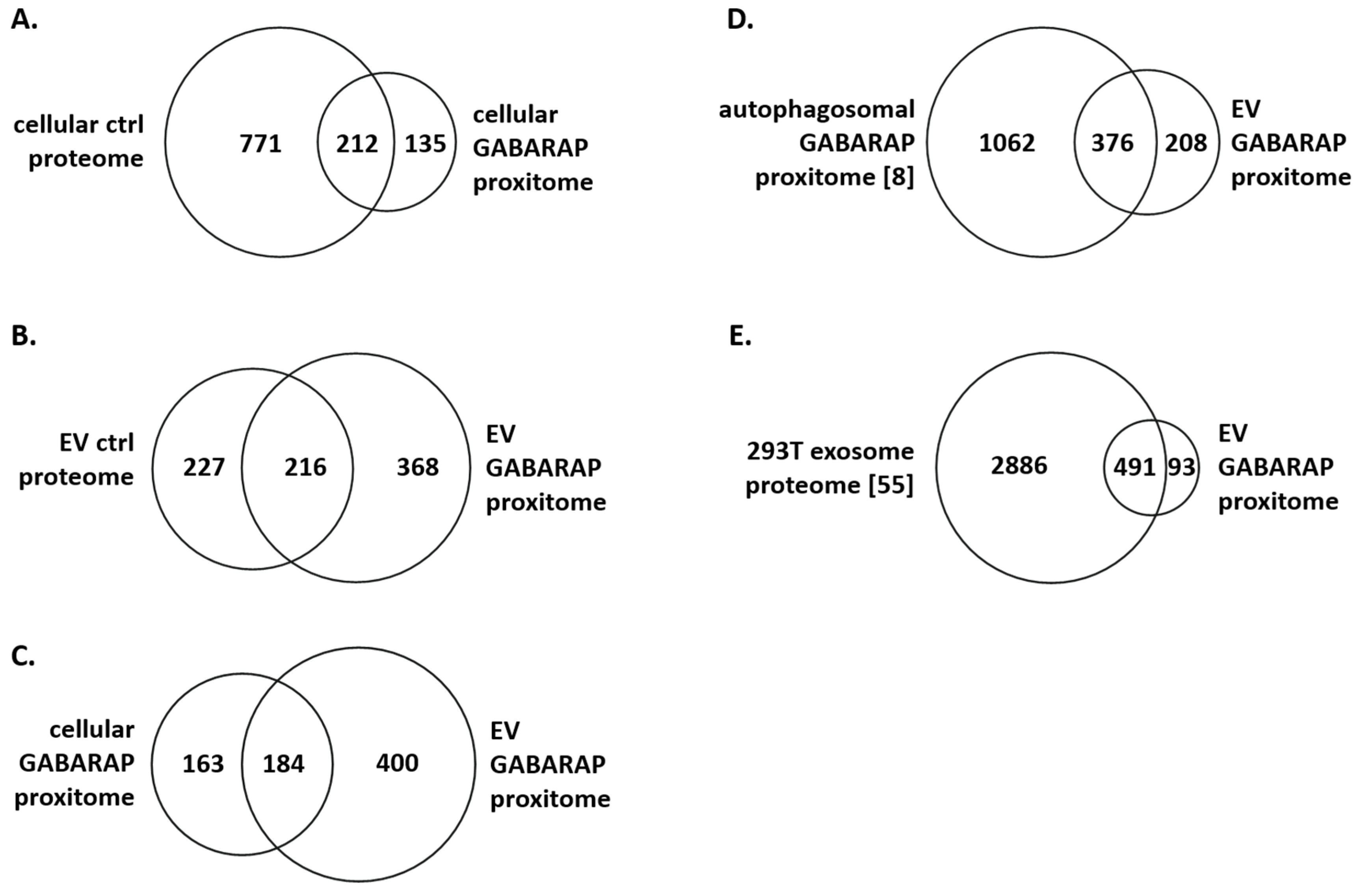
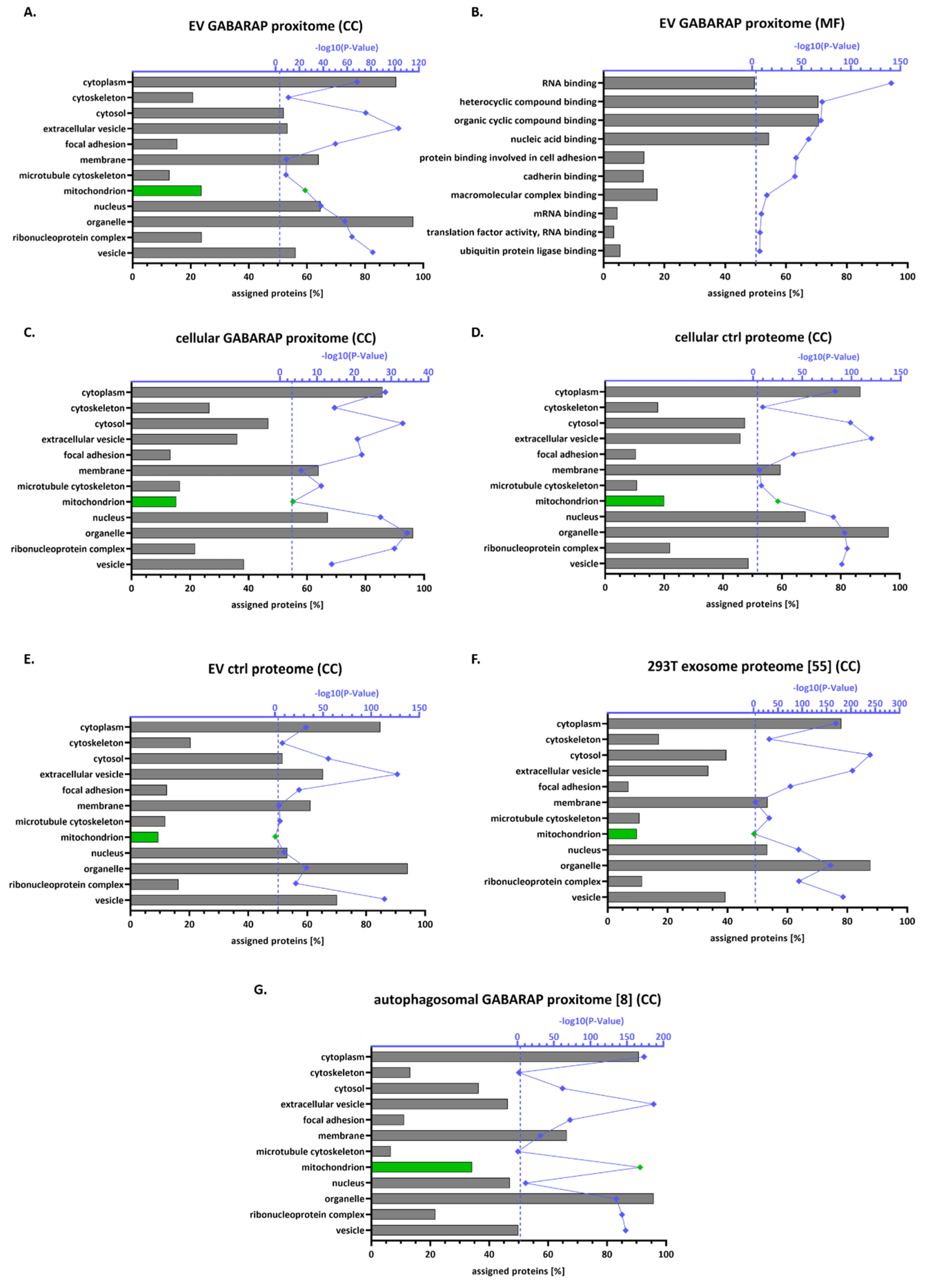
3.4. The GABARAP Co-Secretome as Defined by Proximity Labelling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martell, J.D. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 2012, 30, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.-W. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.S. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Paek, J. Multidimensional tracking of GPCR signaling via peroxidase-catalyzed proximity labeling. Cell 2017, 169, 338–349. [Google Scholar] [CrossRef]
- Zhen, Y. Proximity Labeling by a recombinant APEX2–FGF1 fusion protein reveals interaction of FGF1 with the proteoglycans CD44 and CSPG4. Biochemistry 2018, 57, 3807–3816. [Google Scholar] [CrossRef]
- Del Olmo, T. APEX2-mediated RAB proximity labeling identifies a role for RAB21 in clathrin-independent cargo sorting. EMBO Rep. 2019, 20, e47192. [Google Scholar] [CrossRef]
- Bersuker, K. A Proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Dev. Cell 2018, 44, 97–112. [Google Scholar] [CrossRef]
- Le Guerroué, F. Autophagosomal content profiling reveals an LC3C-Dependent piecemeal mitophagy pathway. Mol. Cell 2017, 68, 786–796. [Google Scholar] [CrossRef]
- Singer-Kruger, B. APEX2-mediated proximity labeling resolves protein networks in Saccharomyces cerevisiae cells. FEBS J. 2020, 287, 325–344. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213. [Google Scholar] [CrossRef]
- Nickel, W. Pathways of unconventional protein secretion. Curr. Opin. Biotechnol. 2010, 21, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M. RNA in extracellular vesicles. Wiley Interdiscip. Rev. RNA 2017, 8, e1413. [Google Scholar] [CrossRef] [PubMed]
- Théry, C. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2013, 24, 9. [Google Scholar] [CrossRef]
- Nguyen, T.N. Atg8 family LC3/GABARAP proteins are crucial for autophagosome–lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef]
- Ichimura, Y. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef]
- Tanida, I. GATE-16 and GABARAP are authentic modifiers mediated by Apg7 and Apg3. Biochem. Biophys. Res. Commun. 2003, 300, 637–644. [Google Scholar] [CrossRef]
- Kabeya, Y. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef]
- Schaaf, M.B.E. LC3/GABARAP family proteins: Autophagy-(un)related functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef] [PubMed]
- Weiergräber, O.H.; Mohrlüder, J.; Willbold, D. Atg8 family proteins—Autophagy and beyond. In Autophagy-A Double-Edged Sword-Cell Survival or Death? Bailly, Y., Ed.; InTech: Rijeka, Croatia, 2013; pp. 13–45. [Google Scholar]
- Chen, Z.-W. C-terminal modification is required for GABARAP-Mediated GABAA receptor trafficking. J. Neurosci. 2007, 27, 6655–6663. [Google Scholar] [CrossRef]
- Wang, H. GABAA-receptor-associated protein links GABAA receptors and the cytoskeleton. Nature 1999, 397, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Green, F. Association of human transferrin receptor with GABARAP. FEBS Lett. 2002, 518, 101–106. [Google Scholar] [CrossRef]
- Cook, J.L. The trafficking protein GABARAP binds to and enhances plasma membrane expression and function of the angiotensin II type 1 receptor. Circ. Res. 2008, 102, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Asano, S. Phospholipase C-related catalytically inactive protein (PRIP) controls KIF5B-mediated insulin secretion. Biol. Open 2014, 3, 463–474. [Google Scholar] [CrossRef]
- Kalra, H. Vesiclepedia: A compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 2012, 10, e1001450. [Google Scholar] [CrossRef]
- Pathan, M. Vesiclepedia 2019: A compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019, 47, D516–D519. [Google Scholar] [CrossRef]
- Skog, J. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Hong, B.S. Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genom. 2009, 10, 556. [Google Scholar] [CrossRef]
- Sinha, A. In-depth proteomic analyses of ovarian cancer cell line exosomes reveals differential enrichment of functional categories compared to the NCI 60 proteome. Biochem. Biophys. Res. Commun. 2014, 445, 694–701. [Google Scholar] [CrossRef]
- Hurwitz, S.N. Proteomic profiling of NCI-60 extracellular vesicles uncovers common protein cargo and cancer type-specific biomarkers. Oncotarget 2016, 7, 86999–87015. [Google Scholar] [CrossRef] [PubMed]
- Steenbeek, S.C. Cancer cells copy migratory behavior and exchange signaling networks via extracellular vesicles. EMBO J. 2018, 37, e98357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Simons, I.M. The highly GABARAP specific rat monoclonal antibody 8H5 visualizes GABARAP in immunofluorescence imaging at endogenous levels. Sci. Rep. 2019, 9, 526. [Google Scholar] [CrossRef] [PubMed]
- Lobb, R.J. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 2015, 4, 27031. [Google Scholar] [CrossRef] [PubMed]
- Hung, V. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc. 2016, 11, 456–475. [Google Scholar] [CrossRef]
- Ladner, C.L. Visible fluorescent detection of proteins in polyacrylamide gels without staining. Anal. Biochem. 2004, 326, 13–20. [Google Scholar] [CrossRef]
- Grube, L. Mining the secretome of C2C12 muscle cells: Data dependent experimental approach to analyze protein secretion using label-free quantification and peptide based analysis. J. Proteome Res. 2018, 17, 879–890. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44. [Google Scholar] [CrossRef]
- Thery, C. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Nath, S. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat. Cell Biol. 2014, 16, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Muratori, C. Massive secretion by T cells is caused by HIV nef in infected cells and by nef transfer to bystander cells. Cell Host Microbe 2009, 6, 218–230. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Dobner, J. Deficiency of GABARAP but not its paralogs causes enhanced EGF-induced EGFR degradation. Cells 2020, 9, 1296. [Google Scholar] [CrossRef]
- Pathan, M. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef] [PubMed]
- Pathan, M. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J. Extracell. Vesicles 2017, 6, 1321455. [Google Scholar] [CrossRef]
- Mukhopadhyay, R. Digitoxin suppresses human cytomegalovirus replication via Na(+), K(+)/ATPase alpha1 subunit-dependent amp-activated protein kinase and autophagy activation. J. Virol. 2018, 92, e01861-17. [Google Scholar] [CrossRef]
- Massey, K.J. Phosphorylation of rat kidney Na-K pump at Ser938 is required for rapid angiotensin II-dependent stimulation of activity and trafficking in proximal tubule cells. Am. J. Physiol. Cell Physiol. 2016, 310, C227–C232. [Google Scholar] [CrossRef]
- Gu, H. Sorting protein VPS33B regulates exosomal autocrine signaling to mediate hematopoiesis and leukemogenesis. J. Clin. Investig. 2016, 126, 4537–4553. [Google Scholar] [CrossRef]
- Brisson, L. Lactate dehydrogenase B controls lysosome activity and autophagy in cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Mullen, L. Cysteine oxidation targets peroxiredoxins 1 and 2 for exosomal release through a novel mechanism of redox-dependent secretion. Mol. Med. 2015, 21, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.K. Uba1 functions in Atg7- and Atg3-independent autophagy. Nat. Cell Biol. 2013, 15, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Identification and characterization of 293T cell-derived exosomes by profiling the protein, mRNA and microrna components. PLoS ONE 2016, 11, e0163043. [Google Scholar] [CrossRef]
- Choi, D.S. Quantitative proteomics of extracellular vesicles derived from human primary and metastatic colorectal cancer cells. J. Extracell. Vesicles 2012, 1, 18704. [Google Scholar] [CrossRef]
- Kim, B.Y. 27-hydroxycholesterol upregulates the production of heat shock protein 60 of monocytic cells. J. Steroid Biochem. Mol. Biol. 2017, 172, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Li, P. Interaction of heat shock protein 90 B1 (Hsp90B1) with liposome reveals its potential role in protection the integrity of lipid membranes. Int. J. Biol. Macromol. 2018, 106, 1250–1257. [Google Scholar] [CrossRef]
- Wang, H.J.; Chen, S.F.; Lo, W.Y. Identification of cofilin-1 induces G0/G1 arrest and autophagy in angiotensin-(1-7)-treated human aortic endothelial cells from itraq quantitative proteomics. Sci. Rep. 2016, 6, 35372. [Google Scholar] [CrossRef]
- Pu, Z. LncRNA MEG3 contributes to adenosine-induced cytotoxicity in hepatoma HepG2 cells by downregulated ILF3 and autophagy inhibition via regulation PI3K-AKT-mTOR and beclin-1 signaling pathway. J. Cell Biochem. 2019, 120, 18172–18185. [Google Scholar] [CrossRef]
- DeJesus, R. Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. Elife 2016, 5, e17290. [Google Scholar] [CrossRef]
- Zhang, W. Poly C binding protein 1 represses autophagy through downregulation of LC3B to promote tumor cell apoptosis in starvation. Int. J. Biochem. Cell Biol. 2016, 73, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y. TRIM28 activates autophagy and promotes cell proliferation in glioblastoma. Onco Targets Ther. 2019, 12, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Li, Z. HnRNPK modulates selective quality-control autophagy by downregulating the expression of HDAC6 in 293 cells. Int. J. Oncol. 2018, 53, 2200–2212. [Google Scholar] [CrossRef] [PubMed]
- Xu, R. Surfaceome of exosomes secreted from the colorectal cancer cell line SW480: Peripheral and integral membrane proteins analyzed by proteolysis and TX114. Proteomics 2019, 19, e1700453. [Google Scholar] [CrossRef]
- Hutagalung, A.H.; Novick, P.J. Role of rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif-crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar]
- Kalvari, I. ILIR: A web resource for prediction of Atg8-family interacting proteins. Autophagy 2014, 10, 913–925. [Google Scholar] [CrossRef]
- de Castro, E. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef]
- Mohrlüder, J. Identification of calreticulin as a ligand of GABARAP by phage display screening of a peptide library. FEBS J. 2007, 274, 5543–5555. [Google Scholar] [CrossRef]
- Mohrlüder, J. Identification of clathrin heavy chain as a direct interaction partner for the γ-Aminobutyric acid type a receptor associated protein. Biochemistry 2007, 46, 14537–14543. [Google Scholar] [CrossRef]
- McKnight, N.C. Genome-wide siRNA screen reveals amino acid starvation-induced autophagy requires SCOC and WAC. EMBO J. 2012, 31, 1931–1946. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Jiang, S.Y.; Dupont, N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 2012, 22, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Manjithaya, R.; Subramani, S. Autophagy: A broad role in unconventional protein secretion? Trends Cell Biol. 2011, 21, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.T. Comparison of isolation methods of exosomes and exosomal RNA from cell culture medium and serum. Int. J. Mol. Med. 2017, 40, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M. The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat. Cell Biol. 2020, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanwald, J.L.; Poschmann, G.; Stühler, K.; Behrends, C.; Hoffmann, S.; Willbold, D. The GABARAP Co-Secretome Identified by APEX2-GABARAP Proximity Labelling of Extracellular Vesicles. Cells 2020, 9, 1468. https://doi.org/10.3390/cells9061468
Sanwald JL, Poschmann G, Stühler K, Behrends C, Hoffmann S, Willbold D. The GABARAP Co-Secretome Identified by APEX2-GABARAP Proximity Labelling of Extracellular Vesicles. Cells. 2020; 9(6):1468. https://doi.org/10.3390/cells9061468
Chicago/Turabian StyleSanwald, Julia L., Gereon Poschmann, Kai Stühler, Christian Behrends, Silke Hoffmann, and Dieter Willbold. 2020. "The GABARAP Co-Secretome Identified by APEX2-GABARAP Proximity Labelling of Extracellular Vesicles" Cells 9, no. 6: 1468. https://doi.org/10.3390/cells9061468
APA StyleSanwald, J. L., Poschmann, G., Stühler, K., Behrends, C., Hoffmann, S., & Willbold, D. (2020). The GABARAP Co-Secretome Identified by APEX2-GABARAP Proximity Labelling of Extracellular Vesicles. Cells, 9(6), 1468. https://doi.org/10.3390/cells9061468